

Abstract
Patients with esophageal squamous cell carcinoma (ESCC) have an overall poor prognosis due to invasion and metastasis. Although it has been studied extensively, the metastatic mechanisms of ESCC remains largely unclear. Here, we evaluated microRNA expression in ESCC cell sublines with distinct motility and found that microRNA-17 and microRNA-20a (miR-17/20a) dramatically impeded cell migration and invasion of ESCC in vitro and decreased pulmonary arrest in vivo. Furthermore, we identified that TGF-β receptor 2 (TGFBR2) and Smad anchor for receptor activation (SARA) served as genuine miR-17/20a targets, which are both implicated in TGF-β pathway. TGF-β treatment promoted the motility of ESCC cells, and miR-17/20a could attenuate the activation of TGF-β pathway by weakening the phosphorylation of Smad2/3 to reduce the expression of ITGB6, which was crucial in migration and invasion of ESCC cells. Moreover, evaluation of ESCC specimens revealed a close correlation between miR-17/20a, TGFBR2, SARA and lymph node metastasis. Together, our findings demonstrate that miR-17/20a suppresses cell migration and invasion of ESCC by modulating TGF-β/ITGB6 pathway, suggesting a promising strategy for diagnosis and therapy of ESCC invasion and metastasis.
Keywords: Esophageal cancer, miR-17/20a, TGF-β, ITGB6, migration, invasion
Introduction
As one of the most common digestive tumors, esophageal cancer ranks the 6th lethal cancer around the world [1]. Furthermore, esophageal squamous cell carcinoma (ESCC) accounts for over 90% of esophageal cancer diagnosed worldwide, whose overall 5-year survival rate is less than 25% [2]. Local invasion and distant metastasis are the leading cause of poor prognosis in patients with ESCC [3]. However, the underlying molecular mechanisms of ESCC metastasis remain largely unknown. Invasion-metastasis cascade of malignant tumors is a complex process [4,5], regulated by multiple molecules including microRNAs [6]. In this study, we aimed to characterize functional microRNAs related to metastasis and explore their mechanism in ESCC metastasis.
MicroRNAs, a class of small noncoding RNA molecules, play a critical role in metastasis of multiple tumors, including ESCC. For example, through repressing insulin-like growth factor 1 receptor, MicroRNA-375 could inhibit tumor growth and metastasis in ESCC [7]. Moreover, Hiyoshi Y et al. found that MicroRNA-21 promotes ESCC cell proliferation and invasion by targeting programmed cell death 4 [8]. We previously established a screening model and found that miR-92b repressed invasion-metastasis cascade of ESCC by targeting ITGAV [9]. Besides, based on the same screening model, we also revealed that miR-17 and miR-20a expressed differentially among cell lines with distinct motility. With the same seed sequence, miR-17 and miR-20a are both located in miR-17-92 cluster [10].
MiR-17-92 cluster, which aberrantly expresses in multiple tumors, is reported to function as an “onco-mir” and indicates poor prognosis [11,12]. Once regulated by MYC, miR-17-92 cluster suppresses specific target genes (Sin3b, Hbp1 and Bim) to maintain survival, autonomous proliferation, and a neoplastic state [13]. Silencing of miR-17-92 cluster expression could inhibit progression of medulloblastoma [14]. It is worth noting that, as two mature microRNAs encoded from miR-17-92 cluster, miR-17 and miR-20a promote or inhibit tumor progression depending on cellular contexts. One side, miR-17/20a enhances cell proliferation and invasion by targeting ZBTB4 in breast cancer [15]. On the other side, miR-17/20a suppresses ITGB8 expression to hamper cell motility of oral squamous cell carcinoma and correlates negatively with lymph node metastasis [16]. However, the function and mechanism of these two microRNAs in ESCC are rarely reported.
Here, we demonstrated that miR-17/20a dramatically impeded migration and invasion of ESCC cells while exerted little influence on proliferation and apoptosis. Moreover, miR-17/20a decreased pulmonary arrest of ESCC cells in an experimental model of lung metastasis, which further confirmed their negative impacts on ESCC metastasis. As the genuine targets of miR-17/20a, Silence of TGFBR2 and SARA hampered ESCC cell migration and invasion, and their expression correlated positively with lymph node metastasis. TGFBR2 and SARA are both implicated in TGF-β pathway, and TGF-β treatment could promote ESCC cell motility. Moreover, we found that miR-17/20a could weaken the phosphorylation of Smad2/3, which are critical in the TGF-β signaling. Furthermore, miR-17/20a attenuated ITGB6 expression to inhibit cell migration and invasion of ESCC via TGF-β pathway. Together, these results indicate that miR-17/20a serve as key regulators of ESCC cell motility.
Materials and methods
Cell culture
ESCC cell lines (KYSE30/180) were kindly provided by Dr. Y. Shimada (Kyoto University, Kyoto, Japan) [17]. Derived sublines (30-U/D and 180-U/D) were established previously [9] and cultured in RPMI1640 supplemented with 10% fetal bovine serum, under humidified condition (37°C, 5% CO2).
Plasmids construction
TGFBR2 or SARA was cloned into pcDNA3-myc respectively. Potential binding sites of miR-17/20a (same seed sequence) in 3’ UTR of the predicted genes were cloned into pIS0 luciferase plasmid. The mutant TGFBR2 and SARA 3’ UTR sites were generated using a KOD-plus-Mutagenesis kit (Toyobo, Osaka, Japan). The primers are provided in Supplementary Table 1.
Transfection
MiR-17/20a mimics, specific miR-17/20a inhibitors and negative control oligos (NC for mimics, NC-i for inhibitors) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). siRNAs for TGFBR2, SARA, ITGB6 and relevant negative control si-NC were ordered from Qiagen (Germantown, MD, USA). HiperFect (Qiagen) or Lipofectamine 2000 (Invitrogen, Camarillo, CA, USA) was used for Oligonucleotide or plasmid transfection according to the manufacturers’ recommendations respectively. Owing to the same seed sequence, mimics or inhibitors of miR-17 and miR-20a were co-transfected into ESCC cells. The sequences of oligonucleotides are shown in Supplementary Table 1.
Viral infection
30-D cells were seeded into a 6-well plate and infected by lentivirus (Genechem, shanghai, China) with polybrene (5 μg/ml), further screened in complete medium containing puromycin (1 μg/ml). Stable 30-D clones expressing miR-17, miR-20a or control were labeled as V-17, V-20a or V-Ctrl, respectively.
Flow cytometry
For detecting cell cycle, cells were fixed by 75% ethanol and stored at 4°C. Then, cells suspended in 400 μl PBS were incubated with 10 μl propidium iodide (PI) (1 mg/ml) and 0.5 μl RNase (100 mg/ml) at 37°C for 30 min in the dark. Apoptosis assay was performed according to manufacturer instruction of Annexin V/PI Apoptosis Detection kit (BD, Franklin Lakes, NJ, USA). All experiments were manipulated on the same LSR II (BD).
qPCR
Total RNA was extracted according to standard Trizol method (Invitrogen), then reversely transcribed to cDNA using Quantscript RT kit (Tiangen, Beijing, China). U6 and mRNAs were converted using random hexamers, while miR-17 and miR-20a were reversely transcribed using gene specific primer. qPCR was performed in triplicates using SYBR Premix Ex TaqTM II (TaKaRa, Japan) on Step-one plus real-time PCR system (Applied Biosystems, Foster City, CA, USA). The primers are listed in Supplementary Table 1. GAPDH (for mRNAs) and U6 (for microRNAs) were used as internal controls for normalization and gene expression level was calculated using 2-ΔΔCt method.
Immunoblotting assay
Cells were lysed in CSH buffer (0.1 M Tris-HCl, 0.5 M NaCl, 50 mM EDTA, 0.1% Triton X-100, and 1× protease inhibitor mix) and 15-30 μg of total protein quantified were performed for immunoblotting assay according to standard procedure. The membranes were incubated with specific primary antibodies at 4°C overnight: TGFBR2 (1:1000), p-Smad2 (1:1000), t-Smad2 (1:1000), p-Smad3 (1:1000), t-Smad3 (1:1000, all purchased from Cell Signaling Technology, Danvers, MA, USA), SARA (1:1000, abcam, Cambridge, UK) and GAPDH (1:5000, Sigma-Aldrich, Missouri, USA). Antibodies used in immunoblotting assay are listed in Supplementary Table 2. All images were acquired by ImageQuant LAS-4000 System (GE, Fairfield, Connecticut, USA).
Transwell assay
In migration or invasion assay, cells were plated in the upper chambers (4×104 cells/chamber for 30-D, 8×104 cells/chamber for 180-U) and cultured at 37°C for 24 hours. Then, the cells penetrated onto lower side of membrane were fixed and stained. Cells from at least three randomly selected fields were counted and data were represented as mean ± SD.
Dual-luciferase reporter assay
The pIS0 plasmid containing wild type or mutant miR-17/20a recognized sites, Renilla luciferase vector and miR-17/20a mimics were co-transfected into 30-D or 180-U cells in quadruplicate respectively. After 24 hrs, luciferase activities were detected using Dual-Luciferase Reporter Assay (Promega, Madison, WI, USA) according to manufacturer’s instruction.
Growth assay in vivo
In growth assay in vivo, the control cells (V-Ctrl) and treated cells (V-17 or V-20a) were injected subcutaneously in the same immunocompromised mice, then the tumors were removed and weighed after 4 weeks.
Pulmonary arrest assay
For pulmonary arrest assay, 30-D cells stably expressing luciferase were delivered with miR-17/20a mimics and negative control oligos respectively and 5×105 cells were injected into tail veins of mice. Within 24 hrs, in vivo fluorescence imaging was performed as previously described [9]. The animal experiments were approved by the ethical committee of the Chinese Academy of Medical Sciences Cancer Institute.
In situ hybridization (ISH)
The paraffin-embedded ESCC tissue array (HEso-Squ127lym-01) was purchased from Shanghai Outdo Biotech Co., Itd (Shanghai, China). MiR-17 and miR-20a LNATM probes were purchased from Exiqon (Vedbaek, Denmark) respectively, and in situ hybridization was performed according to manufacturer’s recommendations under RNase-free condition.
Immunohistochemistry (IHC)
Immunohistochemical staining (IHC) was performed as described previously [18], with a specific primary antibody against TGFBR2 (1:100) or SARA (1:200) respectively. Images were visualized and analyzed by ImageScope software (Leica Biosystems, Nussloch, Germany). The experiments on tissue specimens were approved by the ethical committee of the Chinese Academy of Medical Sciences Cancer Institute.
Statistical analysis
All experiments other than histological and in vivo animal assays were repeated at least three times. All data are presented as mean ± SD, unless otherwise stated. The results were analyzed by using two-tailed or paired Student’s t-test unless stated particularly. Graphs were illustrated using GraphPad Prism 6 (La Jolla, USA), in which *, **, and *** indicate P<0.05, 0.01 and 0.001, respectively.
Results
MiR-17/20a expression correlates negatively with ESCC cell motility and lymph node metastasis
30-U/D and 180-U/D cell sublines were derived from KYSE30 and KYSE180 ESCC cell lines respectively, based on in vitro chemotaxis model we established as previously described [9]. “D” sublines possessed stronger motility capacity than “U” sublines in vitro. Analysis of differentially expressed microRNAs (GSE67510) between 30/180-U and 30/180-D cells revealed that miR-17 and miR-20a were down-regulated in 30/180-D cells compared with 30/180-U cells respectively (Figure 1A), which was confirmed by qPCR assay to assess the expression of two microRNAs in four cell sublines (Figure 1B). These results suggest that miR-17/20a may be implicated in invasion and even metastasis of ESCC. Then we detected the expression of miR-17 and miR-20a in an ESCC tissue array to verify our hypothesis. According to the outcome of in situ hybridization (ISH) experiment in 40 pairs of primary tumors and positive lymph nodes, we verified that miR-17 and miR-20a correlated inversely with lymph node metastasis (Figure 1C, P<0.0001).
Figure 1.

MiR-17/20a expression correlates inversely with cell motility of ESCC. A. Differentially expressed microRNAs between 30-U and 30-D sublines (left), and 180-U and 180-D sublines (right) are shown. B. The levels of miR-17/20a in four cell sublines were analyzed by qPCR. Data, mean ± SD, *P<0.05, **P<0.01, ***P<0.001. C. Representative images of ISH results of miR-17 and miR-20a in an ESCC tissue array (HEso-Squ127lym-01). MiR-17 and miR-20a in primary tumors were both higher relative to that in positive lymph node (n=40, P<0.0001). Scale bars, 100 μm.
MiR-17/20a inhibits migration and invasion of ESCC cells in vitro
To determine the role of miR-17/20a in ESCC, we firstly employed transient transfection method to mimic or antagonize the function of miR-17/20a in ESCC cells. One hand, the mimics of miR-17 and miR-20a were both delivered into 30-D cells, which owned strongest motility of four cell sublines aforementioned, to increase the level of these two microRNAs (Figure 2A). Using the transwell assay, we found that the migration and invasion of 30-D cells were dramatically impeded (Figure 2B). On the other hand, we verified that reduction of miR-17/20a by specific inhibitors in 180-U cells (weakest motility ability) led to a significant increase in their ability of migration and invasion (Figure 2C, 2D). Collectively, miR-17/20a could considerably attenuate ESCC cell motility and invasiveness in vitro.
Figure 2.

MiR-17/20a suppresses motility of ESCC cells. A. qPCR results of enforced expression of miR-17 and miR-20a in 30-D cells. NC, negative control oligos for mimics. B. qPCR results of reduced expression of miR-17 and miR-20a in 180-U cells. NC-i, negative control oligos for inhibitors; miR-17/20a-i, miR-17/20a specific inhibitor. C. Transient expression miR-17/20a mimics impeded migration and invasion of 30-D cells. D. Transient expression miR-17/20a inhibitor (miR-17/20a-i) enhanced motility and invasiveness of 180-U cells. Scale bars, 400 μm. E. Enforced expression of miR-17 and miR-20a in 30-D cells stably expressing luciferase by transient transfection. F. Images revealed that increased level of miR-17/20a impaired arrest of 30-D cells to microvasculature in lungs (P=0.0062). Cells were injected via tail veins, fluorescence was detected within 24 hrs. Data in this figure, mean ± SD, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
MiR-17/20a suppresses pulmonary arrest of ESCC cells in vivo
Metastasis of ESCC cells in vivo is a complex process composed of multiple events, including arresting at distant organs, which plays a fundamental role in metastatic tumor formation. We injected mimics transfected or control 30-D cells into immunocompromised mice via tail veins to determine whether miR-17/20a impaired pulmonary arrest of ESCC cells (Figure 2E). After 24 hours, fluorescence imaging of two cell populations indicated that miR-17/20a could attenuate attachment between malignant cells surviving in the circulation and vascular endothelia (Figure 2F, P=0.0062). And the alteration of transfected cell size was not detected by flow cytometry (Supplementary Figure 1), suggesting that miR-17/20a hampered pulmonary arrest was mainly ascribed to changes in adhesion molecules on the cytomembrane.
MiR-17/20a does not influence on proliferation and apoptosis of ESCC cells
Previous studies revealed that miR-17/20a promoted proliferation and suppressed apoptosis of multiple tumors [19,20]. However, miR-17/20a did not affect cell cycle or apoptosis of these transfected ESCC cells in vitro (Figure 3A, 3B). Moreover, growth assay in vivo indicated that overexpression of miR-17/20a had little influence on proliferation in ESCC cells (Figure 3C, 3D). Together, these results showed that miR-17/20a did not impair cellular viability to attenuate ESCC motility.
Figure 3.

MiR-17/20a shows little influence on proliferation and apoptosis of ESCC cells. A. Increased and decreased expression of miR-17/20a failed to alter cell cycle progression of 30-D and 180-U cells, respectively. B. Flow cytometry results indicated that manipulation of miR-17/20a expression in 30-D and 180-U cells did not affect apoptosis of these transfected cells. C. Stable expression of miR-17 or miR-20a was engineered in 30-D cells via lentivirus-based system. D. Representative pictures of xenograft tumor formed in the subcutaneous tissue (left) and the weight of them (right, n=10).
TGFBR2 and SARA are the bona fide targets of miR-17/20a
Potent effects of miR-17/20a in suppressing the migration and invasion of ESCC cells prompted us to detect the downstream effectors of miR-17/20a. We adopted two widely used online algorithms (Targetscan and Pictar) to explore the potential downstream targets regulated by miR-17/20a. Then, several candidates involved in invasion-metastasis cascade were chosen to perform the luciferase reporter assay initially (Supplementary Figure 2). And we found that TGF-β receptor 2 (TGFBR2) and Smad anchor for receptor activation (SARA), two key proteins implicated in TGF-β signaling, seemed to be potential targets of miR-17/20a (Supplementary Figure 2). Ensuing studies showed that increased miR-17/20a in 30-D and 180-U cells reduced TGFBR2 and SARA at protein level, while endogenous expression of TGFBR2 and SARA enhanced significantly upon the transfection of miR-17/20a inhibitors (Figure 4A, 4B). Subsequently, we constructed mutant sequences (Mut) into pIS0 comparing with wild-type sequences (WT), where miR-17/20a combined in the 3’ UTR of TGFBR2 and SARA, respectively (Figure 4C). Luciferase reporter assay showed that miR-17 and miR-20a both decrease luciferase activity of WT dramatically in 30-D and 180-U cells, but not that of Mutant. Moreover, inhibition of endogenous miR-17 or miR-20a led to a gain of luciferase activity of WT (Figure 4D, 4E), further verifying that miR-17/20a suppressed the expression of TGFBR2 and SARA by directly binding to their 3’ UTR, respectively.
Figure 4.
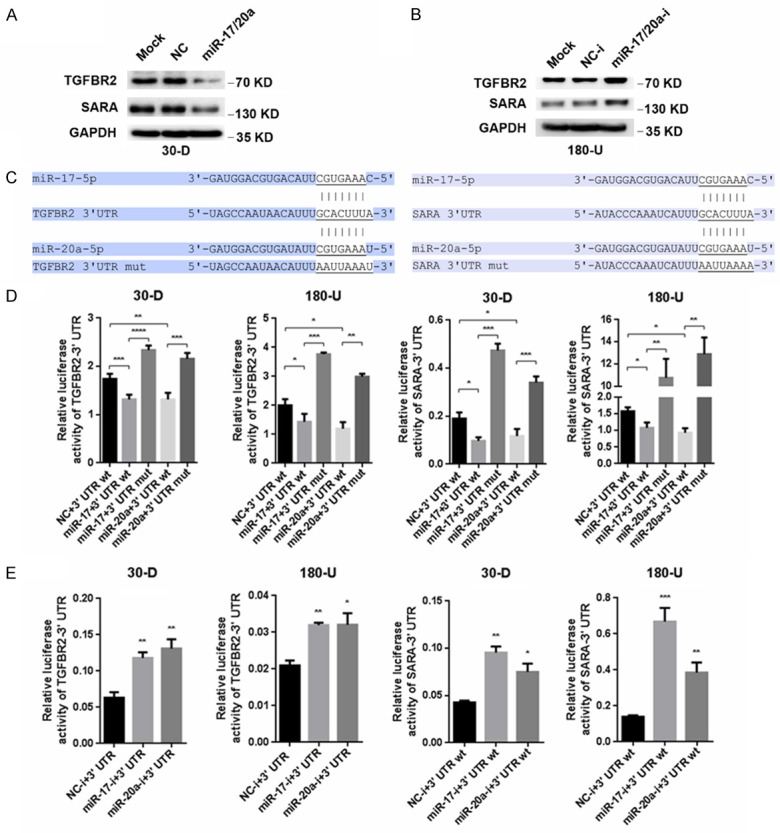
TGFBR2 and SARA are genuine targets of miR-17/20a. A. Elevated expression of miR-17/20a in 30-D cells reduced TGFBR2 and SARA at protein level. B. Inhibition of miR-17/20a in 180-U cells led to increased expression of TGFBR2 and SARA. C. Illustration of wild type and mutated binding sites of miR-17/20a located in 3’ UTR of TGFBR2 (left) and SARA (right). D. After co-transfection of pIS0-3’ UTR wt or pIS0-3’ UTR mut with miR-17/20a or control oligos in 30-D and 180-U cells respectively, the results of luciferase assay showed that miR-17/20a directly bounds to TGFBR2 and SARA 3’ UTR. E. Suppression of endogenous miR-17/20a increased luciferase activity compared with negative controls.
Repression of TGFBR2 and SARA expression is required for miR-17/20a to impair the migration and invasion of ESCC cells
Since TGFBR2 and SARA were both genuine targets of miR-17/20a, then we explored whether these two proteins were implicated in miR-17/20a-mediated suppression of ESCC cell motility. Consistent with their status as targets of miR-17/20a, their knockdown could recapitulate phenotypes of miR-17/20a overexpression in 30-D cells, as demonstrated by the compromised cell migration and invasion in vitro (Figure 5A, 5B). And TGFBR2 and SARA re-expression rescued miR-17/20a-impaired motility of 30-D cells, respectively (Figure 5C). Furthermore, the expression of TGFBR2 and SARA in an ESCC tissue array indicated that these two proteins correlated positively with lymph node metastasis (Figure 5D). To sum up, these results verified that miR-17/20a attenuated migration and invasion of ESCC cells through suppressing TGFBR2 and SARA.
Figure 5.

Repression of TGFBR2 and SARA expression is required for the function of miR-17/20a in ESCC cells. (A) Specific siRNAs against TGFBR2 (50 nM) hampered migration and invasion of 30-D cells in vitro. (B) Specific siRNAs against SARA (50 nM) attenuated motility and invasiveness of 30-D cells in vitro. (C) Transwell assays were performed in miR-17/20a mimics transfected 30-D cells with TGFBR2 or SARA overexpression as indicated. Scale bars in (A-C), 400 μm. (D) TGFBR2 and SARA expression correlated positively with lymph node metastasis of ESCC. The expression of these two protein was analyzed using ImageScope (N0, negative lymph node metastasis; NX, positive lymph node metastasis). Scale bars in (D), 100 μm.
MiR-17/20a impedes TGF-β/ITGB6 pathway
Having validated the roles of TGFBR2 and SARA in ESCC cell motility, we turned to delineate signaling pathways influenced by miR-17/20a. We mainly focused on TGF-β signaling, since both TGFBR2 and SARA are involved in. Once treated with TGF-β, Smad2/3 (critical hub of TGF-β pathway) are activated under the help of TGFBR2 and SARA, to further regulate gene transcription [21]. Consistent with previous studies [22], TGF-β treatment stimulated migration and invasion of 30-D and 180-U cells respectively (Figure 6A), and inhibition of Smad2 reduced motility and invasiveness of ESCC cells (Figure 6B), suggesting that TGF-β pathway was crucial for ESCC cell motility. Subsequently, we detected the level of phosphorylated Smad2/3 at indicated time points, showing that elevated miR-17/20a in 30-D cells reduced the abundance of p-Smad2 and p-Smad3 (Figure 6C). On account of the role of integrins in ESCC invasion-metastasis cascade [9], we speculated whether miR-17/20a could affect the expression of integrins by regulating TGF-β pathway. Among the candidates, the mRNA level of integrin β6 (ITGB6) increased most (about three times) under the stimuli of TGF-β, while miR-17/20a reversed the TGF-β-mediated alteration of ITGB6 (Figure 6D). Simultaneously, 30-D cells transfected with ITGB6 siRNA had weaker motility (Figure 6E). In conclusion, miR-17/20a hampered motility of ESCC cells by attenuating TGF-β/ITGB6 pathway.
Figure 6.

MiR-17/20a impairs ESCC cell motility by regulating TGF-β/ITGB6 pathway. (A) The motility capacity of 30-D (left) and 180-U (right) cells was increased after the treatment of TGF-β. (B) Transwell assays were performed in 30-D cells with Smad2 knockdown or negative control. (C) Under TGF-β treatment, transfection of miR-17/20a in 30-D cells reduces level of phosphorylated Smad2 and Smad3 at the indicated time points. (D) miR-17/20a reversed the TGF-β-mediated elevation of ITGB6. (E) Transwell assays were performed in 30-D cells with ITGB6 knockdown or negative control. Scale bars in (A, B and E), 400 μm.
Discussion
Metastasis, a leading cause of poor prognosis and death in patients suffered from cancer [23], is a complex process consisting of multiple events, including primary tumor formation, local invasion, intravasation, survival in the circulation, arrest at a distant organ site, extravasation, micrometastasis formation and metastatic colonization [24]. Especially, cell directed migration is critical in almost all steps mentioned above [25]. Therefore, we firstly established a directed migration screening model to explore mechanism of ESCC metastasis. Then, we found that despite of dissimilar genetic background, miR-17 and miR-20a were both upregulated in two kinds of ESCC cells with weaker motility, suggesting these two microRNAs may be implicated in regulating migration and even metastasis of ESCC.
miR-17 and miR-20a, encoded by miR-17-92 cluster, are involved in progression of multiple tumors. In gastric cancer cells, miR-17/20a promotes tumor progression by negatively regulating FBXO31 [26], while they inhibit cellular invasion and metastasis by heterotypic secreted signaling in breast cancer [27]. In ESCC, miR-17/20a dramatically attenuates cell migration and invasion while exert little influence on proliferation and apoptosis. These results reconfirm that one microRNA performs different functions by regulating various targets in distinct contexts.
In human genome, miR-17-92 cluster encodes 6 mature microRNAs, including miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92-1 [10]. MiR-17-92 cluster, which is located on chromosome 13q31, aberrantly expresses in various tumors. Previous study indicated that miR-17-92 cluster could accelerate tumor progression in a mouse B-cell lymphoma model to act as a potential oncogene [28]. Strangely, 6 mature microRNAs from the cluster promote or inhibit tumor development, respectively. The increasing evidences demonstrate that miR-19 acts as a key component in mediating the oncogenic activity of miR-17-92. By repressing PTEN and activating Akt-mTOR pathway, miR-19 is proved to promote tumor cell survival compared with other members of miR-17-92 cluster [29]. Together, these findings indicate that the onco-mir feature of miR-17-92 cluster may be a mix among its members as either tumor promoters or suppressors.
TGF-β pathway controls a plethora of cellular responses and plays a crucial role in tumorigenesis as either tumor suppressor or promoter [30]. For tumor suppression, treatment with TGF-β inhibits proliferation of cancer cells. However, cancers are often refractile to this influence because of genetic loss of TGF-β pathway components or downstream perturbation of the signaling pathway [31]. For example, in the gastrointestinal tract and pancreatic tumors, the mutations of TGFBR2 and Smad4 lead to inactivation of TGF-β pathway, which breaks its anti-proliferative effect [32,33]. Similarly, we found that miR-17/20a influenced little on proliferation and apoptosis of ESCC cells even though they suppressed the activation of TGF-β pathway. For tumor promoting effect, TGF-β pathway could stimulate cell invasion by inducing epithelial-mesenchymal transition (EMT), and even promote metastasis eventually, which was thoroughly studied in multiple tumors [34-36]. In ESCC, we demonstrated that cell motility was significantly increased with TGF-β treatment. In general, as a double-edged sword, TGF-β pathway functions differentially depending on various contexts.
TGF-β binding to TGFBR2 leads to phosphorylation and activation of TGFBR1 by TGFBR2. With the help of SARA, Smad2/3 are phosphorylated by TGFBR1. Then, they form a heterotrimeric complex with Smad4 and translocate into nuclear to regulate gene transcription [37]. In ESCC, miR-17/20a could reduce the expression of TGFBR2 and SARA, then inhibit the phosphorylation of Smad2/3 to attenuate activation of TGF-β pathway, which hampered TGF-β induced migration and invasion of ESCC cells. We previously found that miR-92b could inhibit cell motility, pulmonary arrest and lung metastasis of ESCC by suppressing Integrin αV (ITGAV). In view of the key role integrins playing in pulmonary arrest and miR-17/20a mediated suppression of this process, we speculated that whether TGF-β increased expression of Integrins and miR-17/20a could impair the above effect. Then, qPCR results revealed that ITGB6 expression showed the most remarkable increasing by TGF-β stimulation, while enforced expression of miR-17/20a restrained the increasing. Since there are no binding sites of miR-17/20a on 3’ UTR of ITGB6, we identified that miR-17/20a suppressed ITGB6 via regulating TGF-β pathway. Previous study showed that Integrin αVβ6 mediated the activation of TGF-β [38], so miR-17/20a may break the positive feedback loop as shown in Figure 7.
Figure 7.

Diagram of miR-17/20a mediated regulation of TGF-β/ITGB6 pathway. Having activated from latent form by Integrin αVβ6, active TGF-β binds to TGFBR2, which leads to phosphorylation and activation of TGFBR1 by TGFBR2. Subsequently, Smad2/3 were transferred to SARA and then phosphorylated by TGFBR1. Phosphorylated Smad2/3 consist of a heterotrimeric complex with Smad4 and move into the nucleus. In the nucleus, the Smad complex regulates transcription of target genes such as ITGB6, though cooperation with DNA-binding cofactor and co-activator or co-repressor. Once recognizing and binding to the seed sequence of 3’ UTR, miR-17/20a reduces TGFBR2 and SARA expression, attenuate the ability of TGF-β pathway though dephosphorylation of Smad2/3 and then decrease the expression of ITGB6, suggesting that miR-17/20a breaks the positive feedback loop of TGF-β/ITGB6 pathway.
In conclusion, we demonstrate that miR-17/20a impedes cell migration and invasion of ESCC via targeting TGFBR2 and SARA and attenuating the activation of TGF-β/ITGB6 pathway. Furthermore, the expression of miR-17/20a correlates negatively with lymph node metastasis of ESCC. These findings reveal the critical role of miR-17/20a-TGF-β-ITGB6 axis in ESCC invasion-metastasis cascade and provide a candidate for diagnosis and therapy of ESCC.
Acknowledgements
This work was supported by Joint NSFC-ISF Research Program, jointly funded by the National Natural Science Foundation of China and the Israel Science Foundation (81461148025); National Natural Science Foundation of China (81130043, 81472660) and National Basic Research Program of China (2013CB911004).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 2.Glickman JN. Section II: pathology and pathologic staging of esophageal cancer. Semin Thorac Cardiovasc Surg. 2003;15:167–79. [PubMed] [Google Scholar]
- 3.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–12. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19:1450–64. doi: 10.1038/nm.3391. [DOI] [PubMed] [Google Scholar]
- 6.Dykxhoorn DM. MicroRNAs and metastasis: little RNAs go a long way. Cancer Res. 2010;70:6401–6. doi: 10.1158/0008-5472.CAN-10-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong KL, Kwong DL, Chan TH, Law SY, Chen L, Li Y, Qin YR, Guan XY. MicroRNA-375 inhibits tumour growth and metastasis in oesophageal squamous cell carcinoma through repressing insulin-like growth factor 1 receptor. Gut. 2012;61:33–42. doi: 10.1136/gutjnl-2011-300178. [DOI] [PubMed] [Google Scholar]
- 8.Hiyoshi Y, Kamohara H, Karashima R, Sato N, Imamura Y, Nagai Y, Yoshida N, Toyama E, Hayashi N, Watanabe M, Baba H. MicroRNA-21 regulates the proliferation and invasion in esophageal squamous cell carcinoma. Clin Cancer Res. 2009;15:1915–22. doi: 10.1158/1078-0432.CCR-08-2545. [DOI] [PubMed] [Google Scholar]
- 9.Ma G, Jing C, Li L, Huang F, Ding F, Wang B, Lin D, Luo A, Liu Z. MicroRNA-92b represses invasion-metastasis cascade of esophageal squamous cell carcinoma. Oncotarget. 2016;7:20209–22. doi: 10.18632/oncotarget.7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knuutila S, Bjorkqvist AM, Autio K, Tarkkanen M, Wolf M, Monni O, Szymanska J, Larramendy ML, Tapper J, Pere H, El-Rifai W, Hemmer S, Wasenius VM, Vidgren V, Zhu Y. DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol. 1998;152:1107–23. [PMC free article] [PubMed] [Google Scholar]
- 12.Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–95. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26:262–72. doi: 10.1016/j.ccr.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy BL, Obad S, Bihannic L, Ayrault O, Zindy F, Kauppinen S, Roussel MF. Silencing of the miR-17~92 cluster family inhibits medulloblastoma progression. Cancer Res. 2013;73:7068–78. doi: 10.1158/0008-5472.CAN-13-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Chadalapaka G, Lee SO, Yamada D, Sastre-Garau X, Defossez PA, Park YY, Lee JS, Safe S. Identification of oncogenic microRNA-17-92/ZBTB4/specificity protein axis in breast cancer. Oncogene. 2012;31:1034–44. doi: 10.1038/onc.2011.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CC, Yang YJ, Li YJ, Chen ST, Lin BR, Wu TS, Lin SK, Kuo MY, Tan CT. MicroRNA-17/20a functions to inhibit cell migration and can be used a prognostic marker in oral squamous cell carcinoma. Oral Oncol. 2013;49:923–31. doi: 10.1016/j.oraloncology.2013.03.430. [DOI] [PubMed] [Google Scholar]
- 17.Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69:277–84. doi: 10.1002/1097-0142(19920115)69:2<277::aid-cncr2820690202>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 18.He H, Li S, Hong Y, Zou H, Chen H, Ding F, Wan Y, Liu Z. Kruppel-like Factor 4 Promotes Esophageal Squamous Cell Carcinoma Differentiation by Up-regulating Keratin 13 Expression. J Biol Chem. 2015;290:13567–77. doi: 10.1074/jbc.M114.629717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang G, Nishimoto K, Zhou Z, Hughes D, Kleinerman ES. miR-20a encoded by the miR-17-92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression. Cancer Res. 2012;72:908–16. doi: 10.1158/0008-5472.CAN-11-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Q, Luo G, Yang Z, Zhu F, An Y, Shi Y, Fan D. miR-17-5p promotes proliferation by targeting SOCS6 in gastric cancer cells. FEBS Lett. 2014;588:2055–62. doi: 10.1016/j.febslet.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 21.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 22.Yuan Y, Chen H, Ma G, Cao X, Liu Z. Reelin is involved in transforming growth factor-beta1-induced cell migration in esophageal carcinoma cells. PLoS One. 2012;7:e31802. doi: 10.1371/journal.pone.0031802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011;11:573–87. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Kong Y, Xu X, Xing H, Zhang Y, Han F, Li W, Yang Q, Zeng J, Jia J, Liu Z. F-box protein FBXO31 is down-regulated in gastric cancer and negatively regulated by miR-17 and miR-20a. Oncotarget. 2014;5:6178–90. doi: 10.18632/oncotarget.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Z, Willmarth NE, Zhou J, Katiyar S, Wang M, Liu Y, McCue PA, Quong AA, Lisanti MP, Pestell RG. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci U S A. 2010;107:8231–6. doi: 10.1073/pnas.1002080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23:2839–49. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 31.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massague J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102:13909–14. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, Duong V, Dunn LK, Mauviel A, Guise TA. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175–84. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19:192–205. doi: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 38.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.