

Abstract
Previous studies indicate that microRNA-122 (miR-122) is down-regulated in several cancer cells and regulates cell apoptosis, proliferation, metastasis, and tumor angiogenesis. However, the mount of miR-122 in bladder cancer and the pivotal molecular mechanisms of miR-122 used to regulate bladder carcinogenesis and angiogenesis remain to be clarified. Here, we reveal that miR-122 expression is down-regulated in human bladder cancer tissues and cell lines. MiR-122 represses vascular endothelial growth factor C (VEGFC) post-transcriptional expression by directly binding to its 3’-UTR. The protein kinase B (AKT) and mammalian target of rapamycin (mTOR), which are the most important downstream molecules of VEGFC, are also decreased in bladder cancer cell after miR-122 overexpression. Furthermore, miR-122 over-expression decreases bladder cancer cell migration, invasion, colony formation in vitro and slow bladder cancer growth and angiogenesis in vivo. Finally, miR-122 sensitizes bladder cancer cells to cisplatin-induced apoptosis. Taken together, these studies suggest that miR-122 serves as a tumor suppressor and down-regulating VEGFC expression, leading to the inhibition of bladder cancer growth and angiogenesis.
Keywords: MiR-122, VEGFC, bladder cancer, angiogenesis
Introduction
MicroRNAs are a class of small, endogenous, noncoding RNAs with single-stranded RNA that that have been identified to regulate gene expression by combine with messenger RNAs (mRNAs) and inhibiting mRNA translation or promoting it degradation [1]. Accumulated evidence has confirmed the important roles of miRNAs in regulating various cellular functions such as neural development, stem cell differentiation, and miRNAs have been found to play crucial roles in many cancer cellular processes, such as proliferation and apoptosis [2]. The deregulation of this microRNA-122 (miR-122) was demonstrated in hepatobiliary cancer, gastric carcinoma, gastric carcinoma and non-small-cell lung cancer (NSCLC) [3]. Recently, miRNAs are found to play pivotal roles in tumorigenesis, cancer migration, invasion and metastasis. MiR-122 boosts the inhibitory activity of ionizing radiation (IR) on cancer cell anchor-independent growth and invasion [4]. Moreover, miR-122 reduced the expression of its targeted genes related to tumor-survival or cellular stress response. The down-regulation of miR-122 has been associated with hepatocellular carcinoma (HCC) development and progression and is closely related to poor prognosis of HCC [5]. Increasing evidence indicates that miR-122 modulate the chemo-sensitivity of HCC cells. Ectopic expression of miR-122 in HCC cells inhibits tumorigenic properties, such as growth, invasion, and tumor formation in nude mice [6]. Furthermore, high miR-122 levels in the circulation are associated with metastasis in breast cancer patients and cancer-cell-secreted miR-122 facilitates metastasis by increasing nutrient availability in the pre-metastatic niche [7]. In short, miR-122 has been proven to act as a vital tumor suppressor in hepatocellular carcinoma, NSCLC, gallbladder carcinoma, and its deregulation is correlated with clinic-pathologic features and prognostic potential in several cancers.
Angiogenesis is a process by which new micro-vessels sprout from existing vessels and is vital for tumorigenesis and tumor development [8]. Since angiogenesis is deemed necessary for tumor progression, it is assumed that anti-angiogenic therapy should be of benefit for patients with cancer. Among all the angiogenic factors, the most potent one is vascular endothelial growth factor (VEGF), which is responsible for the growth and permeability of vascular endothelial cells, vasculature and tumor angiogenesis [9]. VEGF promotes endothelial cell proliferation and migration, increases vascular permeability and inhibits apoptosis of endothelial cells lining newly formed vessels. There are numerous splice isoforms of VEGF that bind with varying degrees of affinity to VEGF receptors (VEGFR) on the surface of endothelial cells. Most of the angiogenic effects attributed to VEGF are a result of activation of VEGFR-2, which signals through the phosphatidyl-inositol 3 kinase/protein kinase B (PI3K/AKT) pathway [10]. Hypox ia-inducible factor 1 (HIF-1) is a heterodimeric transcription factor composed of HIF-1α and HIF-1β subunits, and is a major regulator of VEGF, in response to hypoxia, by binding to the promoter of VEGF [11]. The mammalian target of rapamycin/protein kinase B (mTOR/AKT) is an important signaling pathway in regulating cellular functions. The activation of mTOR enhances the translation of mRNAs that bear a 5’ terminal oligopyrimidine tract, which encodes proteins related with the translational apparatus like ribosomal proteins, elongation factors eEF1A, eEF2 and the poly A-binding protein [12]. Given the important roles of these proteins in cancerous characteristics such as the cell cycle, cell apoptosis, cell growth and proliferation, mTOR/AKT is proven to be one of the most important targets in cancer therapy, and its inhibitors exhibit encouraging effects in animal experiments and clinical trials. It has been demonstrated that miR-21, is able to control the proliferation of renal cell carcinoma cells by regulating the PI3K/AKT pathway [13]. MiR-122 also functions as a tumor suppressor and plays an important role in inhibiting the breast tumorigenesis through targeting IGF1R and regulating PI3K/AKT/mTOR/p70S6K pathway [14].
Bladder cancers are the most commonly diagnosed types of cancer and the leading cause of cancer-related death. Growing interest is focused on the role of microRNAs in tumor progression and miR-122 has been identified to participate in the regulation of tumor progress as tumor-suppressors. However, the role and the underlying mechanisms of miR-122 in bladder cancer largely remain unclear. In this study, we demonstrate that: (i) the level of miR-122 is decreased in bladder cancer cell lines as well as in clinical cancer tissues; (ii) VEGFC is a potential target of miR-122; (iii) miR-122 suppresses bladder cancer growth and angiogenesis by inhibiting AKT/mTOR signaling through targeting VEGF-C. These results demonstrate for the first time that VEGFC is a direct target of miR-122 in the regulation of bladder cancer growth and angiogenesis.
Materials and methods
Cell culture and tissue samples
The human bladder cancer cells BIU-87, T24, SW780, HT1376, 5637, RT4 and normal bladder epithelial cell line (SV-HUC-1) (American Type Culture Collection, Manassas, VA, USA) were cultured in a RPMI 1640 or DMEM medium that contained a 10% heat-inactivated fetal bovine serum, 100 units/ml, penicillin G and 100 mg/ml streptomycin in a 5% CO2 incubator at 37°C. Fifty-four pairs of human bladder cancer samples, including some primary bladder cancer tissues and paired adjacent normal bladder tissues, were obtained from the Second Hospital of Tianjin Medical University, China. In all the cases, the diagnoses and grading were confirmed by two experienced pathologists, which were done in accordance to the principles laid down in the latest World Health Organization Classification. Informed consent was also obtained from all patients, and the study was approved by Tianjin Medical University ethics committee.
Lentivirus packaging and stable cell lines
The lentiviral packaging kit was purchased from OriGene Technologies (OriGene, Beijing, China). Lentivirus carrying miR-122 or miR-negative control (miR-scr) was packaged following the manufacturer’s manual. Lentivirus were packaged in HEK-293T cells and collected from the medium supernatant. Stable cell lines were established by infecting lentivirus into HT1376 cells and selected by 1.5 µg/ml puromycin over a period of four weeks.
Cell proliferation assay
Briefly, cells (3 × 104 cells per well) were seeded in 96-well plates after infection and cell viability was measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Each well was added 20 µl MTT (5 mg/mL) (Sigma) and incubated for 4 hours at 37°C, followed by adding 200 µl DMSO. The absorbance of each well at 490 nm was measured by the Microplate Reader (Epoch, Winooski, USA). Three independent experiments with triplicate were carried out.
Cell death analysis
To measure cell death, a FITC Annexin-V apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA) was used according to the manufacturer’s instructions. Briefly, the cells were harvested and washed with using ice-cold PBS, stained with Annexin V/propidium iodide (PI) at room temperature in the dark and then detected with a flow cytometer (FACSCalibur; BD Biosciences). All flow cytometry data were analysed with the FlowJO software program.
Wound healing assay
Briefly, HT1376 cells were seeded in 60 mm dishes and cultured at 37°C overnight to produce a confluent monolayer. After starvation in serum-free medium for 24 hours, a wound was created by scratching the monolayer with a 200 µl sterile pipette tip. The wounded monolayer was then washed three to remove cell debris and incubated with fresh medium. The area of cell-free scratch was photographed at 0 h and 24 h after scratching respectively [15].
Invasion assay
HT1376 cells were transfected with miR-122 or a scrambled control precursors. Forty-eight hours after transfection, cells (5 × 104) were plated in the top chamber with polymerized collagen-coated membrane (24-well insert; pore size, 8 μm) in medium with 0.1% FBS. Complete medium was used as a chemoattractant in the lower chamber. The cells were left to incubate for 24 h, and unattached cells were removed cautiously by cotton swab. Cells that had invaded the lower surface of the membrane were stained with 1% crystal violet. Cell counting was then carried out by photographing the membrane through the microscope. Five random fields under microscope were taken and migration cells number were quantified [16].
Colony formation assay
Cells were plated onto a 6-well tissue culture plate in complete medium at the density of 500 cells per well and incubated at 37°C and were allowed to grow in complete medium for 14 days. Then cells were washed with PBS twice, fixed with absolute methanol for 15 min. Then, fixed cells were stained with 1% crystal violet for 20 min. After cleaning with PBS, colonies were counted under light microscope [17].
Construction of VEGFC 3’-UTR luciferase plasmids and reporter assays
A length of 510 bp PCR product of VEGFC (nt724-1233) containing its 3’-UTR was amplified by the following primers: 5’-CGGGGATCCGGGTGGACCTGGGGTTTATTT-3’ (sense) and 5’-CCCCTCGAGTTCATCAAAAGGCCATCAAAT-3’ (antisense). The PCR product was then cloned into a pcDNA6.2 vector. After identifying the orientation, the correct 3’-UTR of VEGFC (Wt) was cut from pcDNA6.2 and cloned into pMIR-REPORTER (ambion, TX, USA). Site-directed mutagenesis of the miR-122 seed sequence in VEGFC-3’-UTR (Mut) was performed by using a Quick change-mutagenesis kit (Stratagene, CA, USA), with pcDNA6.2-VEGFC Wt as the template. The primers for site-directed mutagenesis were as follows: 5’-GCAGTACTGCTATGTGCTAAGCTTAACTCCAAGCCTTGGAATGGG-3’ (sense), 5’-CCCATTCCAAGGCTTGGAGTTAAGCTTAGCACATAGTACTGC-3’ (antisense). The mutant VEGFC-3’-UTR was then cloned into pMIR-REPORTER. For reporter assays, human 293 cells were transiently transfected with Wt or Mut reporter plasmid, pCMV β-galactosidase plasmid, and miRNA-122 plasmid (Open Biosystems) using lipofectamine 2000. Luciferase (Luc) activity was measured 24 h after transfection by using the Luc assay system.
ELISA
HT1376 cells were transfected with miR-122 or a scrambled control precursors. Cells (7 × 105) were plated in six-well dishes and growth for 24 h. Cell culture supernatants of bladder cancer cells transfected with the miRNA mimics or the negative control were collected and following filtration with a 0.22 µm filter. ELISA for VEGFC secretion levels was performed with a Quantikine immunoassay kit (DVE00; R&D Systems) following the manufacturer’s instructions.
Chorioallantoic membrane (CAM) assay
Fertilized white leghorn chicken embryos were incubated for 3 days at 37°C and 70% humidity. A small hole was made over the air sac at the end of the egg, and a second hole was made directly over the embryonic CAM. After 10 d, supernatant from HT136 miR-122 overexpression cells or control cells were dropped onto the CAM to form a plug. After 48 h, CAMs were fixed with PBS solution/3.7% paraformaldehyde for 10 min at room temperature, and images were taken with a Nikon digital color camera [18]. Hemoglobin in plug was quantified using the QuantiChrom Hemoglobin Assay Kit (Bioassay, Hayward, CA) following the manufacturer’s instructions.
Quantitative real-time PCR
A quantitative real-time RT-PCR was used to determine the expression levels of miR-122 and VEGFC in cells and tumor tissues. To measure the miR-122 levels, total RNAs were extracted from cultured cells and tumor tissues using Trizol (Invitrogen, Carlsbad, CA, USA). The quantitative real-time RT-PCR analysis of miR-122 levels was performed using the TaqMan Reverse Transcription Kit and TaqMan MicroRNA Assays Kit (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturers’ instructions. The expression levels of miR-122 were calculated using the delta-delta Ct method with U6 as an internal control. Real-time PCR was performed to detect the VEGF mRNA levels using SYBR Green. Reverse transcription reactions were performed using High Capacity RNA-to-cDNA Kit according to the manufacturer’s instructions. The 100 ng of RT product was used for the PCR reaction using Power SYBR Green PCR Master Mix. The reaction contained the following: 10 µl of 2 × PCR Master Mix, 1 µl of forward primer, 1 µl of reverse primer, 1 µl of cDNA template, and 7 µl of H2O. The reaction program was set at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 30 s, and the melt curve was included. The primers of VEGFC are the forward primer, 5’-CGAGGGCCTGGAGTGTG-3’ and the reverse primer, 5’-CCGCATAATCTGCATGGTGAT-3’. The primers of GAPDH are the forward primer, 5’-ATGGGTGTGAACCATGAGAAGTATG-3’ and the reverse primer, 5’-GGTGCAGGAGGCATTGCT-3’.
Adenovirus transduction
Recombinant adenoviruses were made using the AdEasy system [19]. The VEGFC cDNA was subcloned into the vector pAdTrack-CMV and then transferred to AdEasy-1 plasmid through homologous recombination. The viral vectors were then transfected into Ad-293 cells in order to generate viruses. A control virus carrying the green fluorescent protein (Ad-GFP) was derived from the same vector system. The viruses were titered using the BD Adeno-X Rapid Titer kit (BD Biosciences Clontech) according to the manufacturer’s manual. 20 multiplicity infections of Ad-VEGFC and Ad-GFP were used to infect HT1376 cells.
Western blotting
Cells were washed with ice-cold PBS buffer, scraped from the dishes, and centrifuged at 15,000 rpm, 4°C for 15 min. Cell lysates were prepared using RIPA buffer supplemented with protease inhibitors (100 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 1% deoxycholate acid, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 2 mM DTT, 2 mM leupeptin, 2 mM pepstatin). The supernatants were collected and protein concentration was determined using BCA assay (Beyotime Institute of Biotechnology, China). Tumor tissues from nude mice were grinded into powder in liquid nitrogen with RIPA buffer, and the total tissue proteins were extracted as described above. Aliquots of protein lysates were fractionated by SDS-PAGE, transferred to a PVDF membrane (Roche, Switzerland), and membranes were blocked with 5% non-fat dry milk for 2 h and incubated with primary antibodies. ECL Detection System (Thermo Scientific, Rockford, IL, USA) was used for signal detection.
In vivo tumor growth assay
All animal procedures were approved by the ethical commission of the Tianjin Medical University. HT1376 were transfected with miR-122 or a scrambled control precursors. 8 × 106 HT1376 cells per mouse were injected s.c. into the right posterior flanks of 7-wk-old immunodeficient NOD/SCID female mice (6 mice per group). Tumor volume and mice body weight were measured every 3 days. Tumor volume was calculated as mm3 = 0.5 × length (mm)3 width (mm)2. After sacrificing mice on day 25, deparaffinized tumor sections were stained with specific antibodies including VEGFC (Abbiotec). Detection was done with avidin-biotin-HRP complex (Thermo scientific) and diaminobenzidine as chromogen. Nuclei were counterstained with hematoxylin [20]. All animal experiments were carried out in compliance with the Guidelines for the Tianjin Medical University.
Statistical analysis
Numerical results were analyzed using independent mean T-test and expressed in mean ± standard deviation (SD). Statistical analysis was performed using post hoc testing using Bonferroni’s method. Differences were considered statistically significant at P < 0.05.
Results
MiR-122 is down-regulated in bladder cancer
To determine whether or not miR-122 is dys-regulated in bladder neoplasms tissues, miR-122 expression levels from bladder cancer tissues and their parallel adjacent normal tissues were analyzed by qRT-PCR analysis. As shown in Figure 1A, the mRNA expression levels of miR-122 in bladder cancer tissues were much lower than those in the adjacent normal tissues. Further experiments were performed by using normal bladder epithelial cell line (SV-HUC-1) and several different bladder cancer cell lines to show that miR-122 expression was very low in bladder cancer cell lines, including BIU-87, T24, SW780, HT1376, 5637 and RT4 cells in contrast with normal cells SV-HUC-1 (Figure 1B). Taken together, these results indicate that miR-122 was down-regulated in both human bladder cancer tissues and bladder cancer cell lines.
Figure 1.
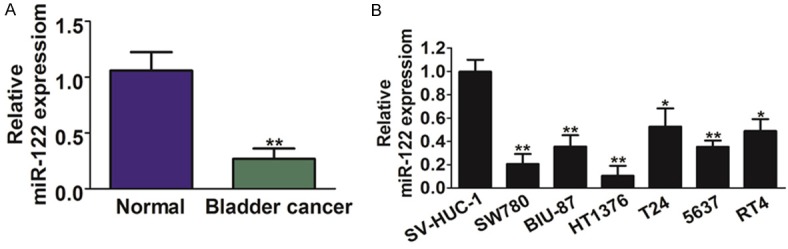
The expression of miR-122 in bladder cancer tissues and cell lines. A. The expression levels of miR-122 were analyzed in 54 pairs of bladder cancer tissues and its adjacent normal tissues by qRT-PCR. GAPDH levels were used as an internal control. B. The expression levels of miR-122 were analyzed by qRT-PCR in normal epithelial cell line (SV-HUC-1), bladder cancer cell lines including BIU-87, T24, SW780, HT1376, 5637 and RT4. Results represent the mean ratio between miR-122 and GAPDH from three experiments. The data are presented as mean ± SD. For indicated comparisons,*P < 0.05 and **P < 0.01.
MiR-122 over-expression inhibits bladder cancer cell proliferation, migration, invasion and colony formation
To test the direct role of miR-122 in bladder cancer cells, we established stable cell lines by infecting HT1376 cells with lentivirus carrying miR-122 or the control scramble (miR-src). High level of miR-122 expression was confirmed in the HT1376/miR-122 stable cell line (Supplementary Figure 1). Cell proliferation analysis indicated that over-expressing miR-122 suppressed HT1376 cells growth (Figure 2A). Given the important role of cell migration in tumor progression, miR-122-overexpressing HT1376 cells were used to analyze cell mobility. The results from wound healing assay showed that cell migration was attenuated in HT1376 cells over-expressing miR-122 compared with cells expressing miR-src (Figure 2B). Since invasion is also the key characteristic of malignant tumor, we next investigated the role of miR-122 on HT1376 cell invasion in vitro. As expected, increasing miR-122 expression dramatically inhibited the invasive capacity of HT1376 cells (Figure 2C). Similarly, miR-122-overexpressing inhibited the ability of HT1376 cells to form colonies in soft-agar (Figure 2D). Thus, the impact of miR-122 on cell proliferation, migration, invasion and colony growth in vitro suggests it was likely that miR-122 had an important function in the development of bladder cancer.
Figure 2.
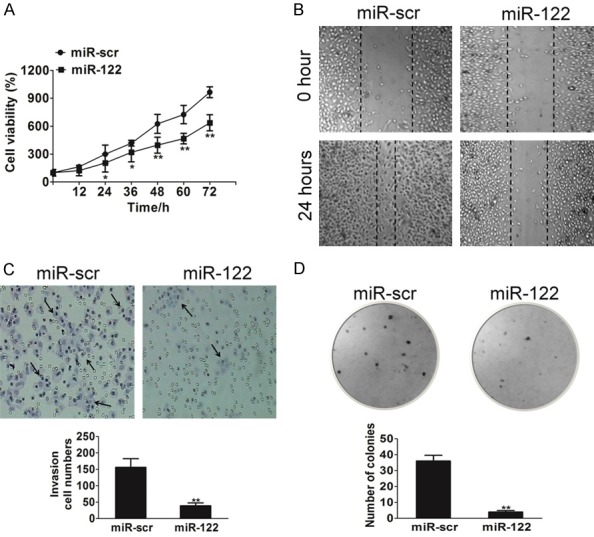
Overexpression of miR-122 delays tumor malignant. A. Overexpression of miR-122 arrested HT1376 cell proliferation. Cell proliferation rates of HT1376 cells infected with lentivirus miR-scr or lentivirus miR-122 were determined by MTT assay. The data are presented as mean ± SD. The values are expressed as percentage of viable cells normalized to percentage of viable cells in 0.5% DMSO-treated HT1376 cells. For indicated comparisons, *P < 0.05 and **P < 0.01. B. Wound healing assay of HT1376 cells transfected with miR-scr or miR-122. A significant decrease was observed in miR-122-transfected cells. C. miR-122 overexpression decreased cell invasion in HT1376 cells. Cells were transfected with miR-122 or miR-scr. All cells were subjected to a Matrigel invasion assay. The data are presented as mean ± SD. For indicated comparisons, *P < 0.05 and **P < 0.01. D. Anchorage-independent colonies growth of HT1376 cells stably transfected with miR-scr or miR-122 was shown. Cells were plated in culture plate. Two weeks later, colonies were stained with 1% crystal violet and scored. The data are presented as mean ± SD. For indicated comparisons, *P < 0.05, **P < 0.01.
MiR-122 inhibits AKT and mTOR pathways via targeting VEGFC
To further understand the potential targets of miR-122, we found a putative miR-122 binding site located in the 3’-UTR of VEGFC using several bioinformatic databases, including TargetScan, PicTar and miTarget (Figure 3A). To validate the miR-122 target, we cloned the 3’-UTR region of VEGFC with wild type or mutant miR-122 binding sites, respectively, into pMIR-REPORTER vector. Both luciferase activity assays and immunoblotting analysis in HT1376 cells following lentivirus-miR-122 infection showed that overexpression of miR-122 repressed VEGFC, indicating miR-122 directly suppresses VEGFC. However, VEGFC wild-type but not mutant reporter activity was affected by the transfection of miR-122 (Figure 3B). The expression levels of VEGFC in HT1376/miR-122 stable cell line were also decreased as confirmed by western blotting assay (Figure 3C) and ELISA analysis (Figure 3D). The AKT and mTOR pathways act as major downstream of VEGFC signaling, and several downstream factors such as hypoxia-inducible factor 1α (HIF-1α) has been linked to the AKT and mTOR pathway. To test whether or not the overexpression of miR-122 in bladder cells will have any effect on AKT and mTOR pathways, HT1376-miR-122 and HT1376-miR-src cells were used to test the levels of AKT and mTOR expression. Cellular levels of phosphorylated AKT and phosphorylated mTOR were significantly decreased in HT1376 cells stably expressing miR-122 compared with miR-src, while no statistically significant reduction of AKT and mTOR was detected (Figure 3C).
Figure 3.
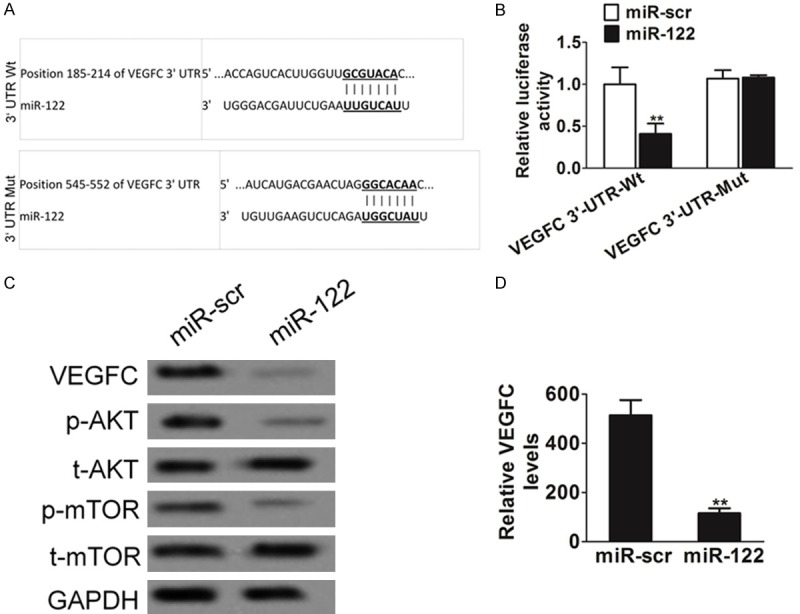
MiR-122 targeting VEGFC. A. Putative seed-matching sites or mutant sites between miR-122 and 3’-UTR of VEGFC. B. HT1376 cells were transfected with 0.2 μg of Wt or Mut reporter, 0.3 μg of pLE-miR-122, and 0.1 μg of β-galactosidase plasmids. The relative Luc activity was assayed and calculated by the ratio of luc/β-galactosidase activity, which was normalized to that of the control. Results were presented as mean ± SD from three duplicates. Double asterisks indicated significant difference when compared to HT1376 cells transfected with miR-scr (P < 0.01). C. HT1376 cells were transfected with miR-122 or miR-scr. Total cellular proteins were collected and subjected to western blot for measuring protein levels of VEGFC, phosphor-AKT (Ser9), t-AKT, phosphor-mTOR (Ser2448) and t-mTOR, respectively. D. HT1376 cells overexpressing miR-122 were analyzed for VEGF secretion by ELISA. The data are presented as mean ± SD. Asterisk indicates significant difference when compared to cells transfected with miR-scr (**P < 0.01).
MiR-122 inhibits tumor angiogenesis and tumor growth
Science VEGFC has essential roles in tumor angiogenesis; we employed the chorioallantoic membrane (CAM) system to test the effects of miR-122 on angiogenesis. Hemoglobin quantification assay was future employed for evaluating angiogenesis in CAM assay. As shown in Figure 4A, forced expression of miR-122 in HT1376 cells significantly suppressed the angiogenesis when compared with the negative control (P < 0.01). To test the effect of miR-122 on bladder cancer growth, HT1376 cells overexpressing miR-122 or miR-src were subcutaneously injected into posterior flank of nude mice (n = 6). Xenograft tumor volumes were measured every 3 d when they were palpable. Nude mice were sacrificed on day 25 after implantation, and xenografts were collected and weighed. On day 25 after implantation, tumors from cells overexpressing miR-122 were significantly smaller than those from control cells (Figure 4B and 4C). It was encouraging that miR-122 decreased HT1376 xenograft tumor growth without significant toxicity (Figure 4D). Furthermore, the protein levels of VEGFC in xenografts from miR-122-expressing cells were much lower than that from miR-scr control cells (Figure 4E). Total proteins from representative tumor samples were analyzed by western blotting, and it was determined that miR-122 suppressed its target VEGFC expression and AKT/mTOR signaling pathway in vivo (Figure 4F).
Figure 4.
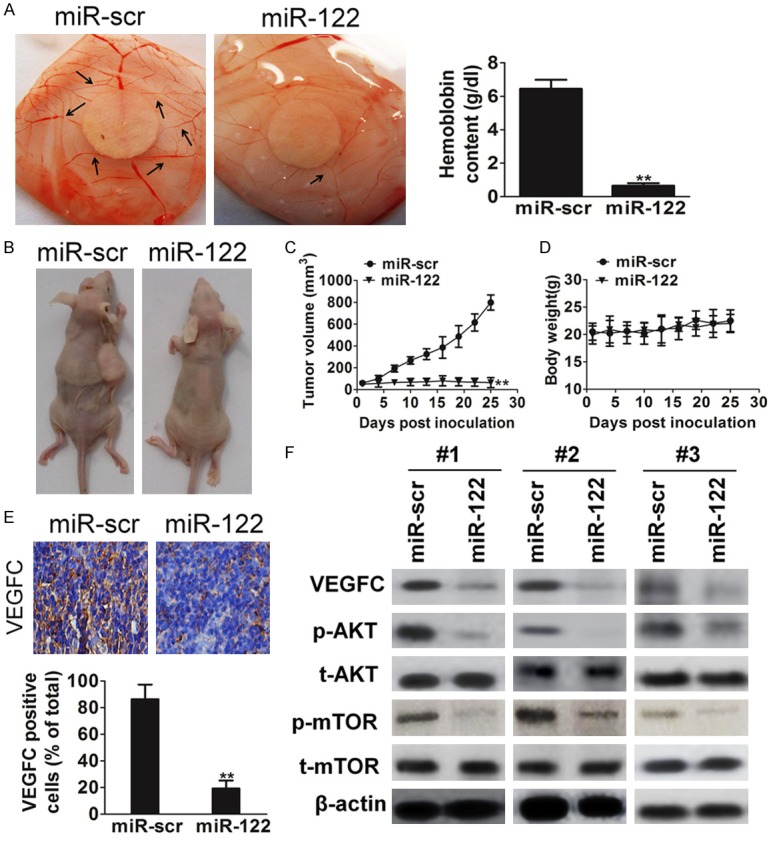
MiR-122 inhibits angiogenesis and tumorigenesis in vivo. A. Angiogenesis assay by chorioallantoic membrane (CAM) model as described in Methods. The data represented as mean ± SD of hemoglobin in CAM were normalized to those of miR-scr control cells. B. Tumor growth assay in nude mice (n = 6). Representative pictures of xenograft tumors were shown. C. Tumor growth curve upon implantation. Data were means ± SD. Asterisk indicated significant difference when compared to the control (**P < 0.01). D. Body weight was measured once every three days. Error bars represent mean ± SD (n = 6). E. Protein levels of VEGFC in xenograft tumors assayed by immunohistochemistry. Data were presented as means ± SD *P < 0.05 indicated significant difference comparing HT1376 cells transfected with miR-122 and cells transfected with miR-scr (control). F. Western blot analysis of VEGF, AKT, and mTOR levels in xenograft tumors.
MiR-122 inhibits cell proliferation and sensitizes bladder cancer cells to cisplatin treatment through VEGFC
To investigate whether VEGFC, the target gene of miR-122, was involved in miR-122-regulated bladder cancer cells proliferation, we overexpressed VEGFC protein levels in HT1376/miR-122 cells by infecting cells with lentivirus carrying VEGFC. As expected, over-expression of VEGFC reversed miR-122-mediated suppression of HT1376 cell growth (Figure 5A). To explore the role of miR-122 in chemotherapy, we treated bladder cancer cells HT1376 with different concentrations of cisplatin, a leading chemo-drug used for treatment of bladder cancer. As shown in Figure 5B, overexpression of miR-122 in bladder cancer cells significantly increased chemo-sensitivity to cisplatin treatment, while VEGFC overexpression reversed the chemo-sensitivity process. In order to explore whether miR-122 played a role in HT1376 cell apoptosis, cell apoptosis rates in the presence of cisplatin were assayed by FACS analysis at indicated time points. Overexpression of miR-122 in the presence of cisplatin treatment significantly induced cell apoptosis, when compared with HT1376 cells overexpressing miR-src (Figure 5C). While VEGFC over-expression neutralized HT1376 cell apoptosis induce by miR-122. These results indicated that miR-122 induced bladder cancer chemo-sensitivity to cisplatin treatment in VEGFC-dependent manner.
Figure 5.
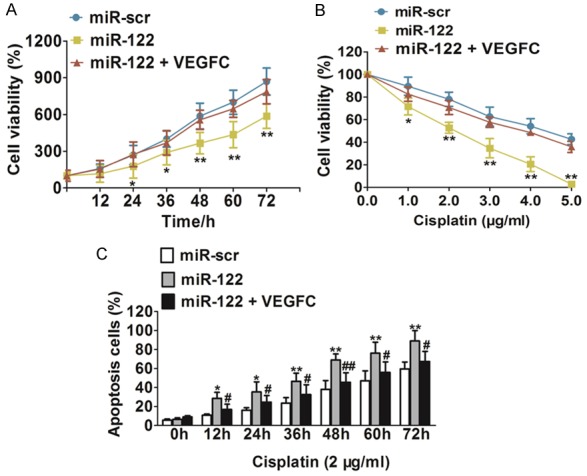
MiR-122 increases chemosensitivity of bladder cancer cells to cisplatin treatment dependent on VEGFC. A. HT1376 cells transfected with miR-scr, miR-122 or miR-122 + VEGFC, respectively. Cell viability was evaluated for indicated times. Data were means ± SD. For indicated comparisons, *P < 0.05, **P < 0.01. B. Cell viability was evaluated in cells stably expressing miR-scr, miR-122 or miR-122 + VEGFC, respectively with the cisplatin treatments at different doses. Data were means ± SD. For indicated comparisons, *P < 0.05, **P < 0.01. C. Cells stably expressing miR-scr, miR-122 or miR-122 + VEGFC were treated with cisplatin (2 ug/ml) at indicated time points and subjected to apoptosis analysis by flow cytometry. Data were means ± SD, *P < 0.05, **P < 0.01 indicated significance between group of miR-122 and control group miR-scr, #P < 0.05 and ##P < 0.01 represented significance between group of miR-122 and group of miR-122 + VEGFC.
Discussion
In addition to regulating cytoskeletal dynamics, cell reprogramming, neural development and smooth muscle cell fate and plasticity, varieties of powerful evidence revealed that miRNAs have been reported to be frequently down-regulated in various kinds of cancers [21]. MiRNAs bind to partially or fully complementary sequences of mRNA in the 3’-untranslated region (3’-UTR) to induce mRNA degradation or repress mRNA translation, then influencing the output of many protein-coding cancer genes and are important regulators of tumor progression. Recently, an important discovery indicated that the crucial role of miR-122 in carcinogenesis and cancer development [22]. This study clearly demonstrates the inhibitory role of miR-122 in bladder cancer angiogenesis and growth both in vitro and in vivo. Our results show that compared with adjacent normal bladder tissues, the miR-122 levels in bladder cancer from clinical samples was down-regulated. Consistently, we found that miR-122 expression was down-regulation in several bladder cancer cell lines. The growth of new blood vessels from an existing vasculature is a main driving force in cancer and targeting these processes has been demonstrated to be an effective approach for human cancer treatment. Here, we reported that miR-122 functioned as an anti-angiogenic regulator in bladder cancer. Overexpression of miR-122 in bladder carcinoma cell line led to reduced amount of microvessels in a CAM model and impaired tumor growth in a xenograft model of nude mice. The invasion capability and migration capabilities of tumor cells are important process to tumor angiogenesis. Further analyses in vitro revealed that miR-122 over-expression inhibited bladder carcinoma cell proliferation, migration, invasion, and colony forming ability.
To date, several targets of miR-122 have been identified, such as ADAM10, SRF, MAP3K12, Ndrg3, AldoA, Bckdk and CD320 [23]. To further understand molecular mechanism of miR-122 in inhibiting angiogenesis, we further predicted the additional miR-122 target, VEGFC on the basis of bioinformatics analysis. There were several lines of evidence to support this. Firstly, miR-122 overexpression significantly down-regulated VEGFC by directly targeting the 3’UTR of VEGFC mRNA confirmed using luciferase-reporter-gene assay. This effect was largely eliminated when the sites in VEGFC 3’UTR targeted by miR-122 were mutated. Moreover, overexpression of miR-122 in human bladder cancer cell lines HT1376 inhibited VEGFC expression at protein level as well as decreased VEGFC secretion, further confirming that miR-122 targeted VEGFC.
The mTOR is a ubiquitously expressed serine/threonine (Ser/Thr) kinase that is a crucial regulator of cell proliferation, metabolism, and other biological progresses in a lot of tumors [24]. Inhibitors of mTOR and block the AKT/mTOR signaling pathway and suppress tumor cells growth in many malignant tumors. AKT, one of the downstream targets of mTOR, functions as a key regulator in various cellular functions such as cell proliferation, cell cycle and cell apoptosis [25]. The activation and/or overexpression of AKT are a hallmark of cancer. Consistent with the inhibitory effect on VEGFC, the overexpression of miR-122 also suppressed AKT and mTOR phosphorylation activation in this study. To confirm the data obtained from in vitro studies, we then subcutaneously injected HT1376 cells into nude mice. In vivo studies showed that miR-122 inhibited xenograft tumor growth and angiogenesis. Consistent with our prediction, miR-122 expression inhibited angiogenesis by decreasing VEGFC, AKT and mTOR levels in tumor tissues. Taken together, these in vitro and in vivo results suggest that miR-122 was a tumor suppressor, which inhibited tumor growth and angiogenesis through the inhibition of VEGFC expression and blocking the AKT/mTOR signaling pathway. Previous studies showed the apoptosis-inducing properties of miR-122 in a variety of cancer cell models, including colorectal cancer, esophageal cancer, bladder cancer and other tumors. In the present study, miR-122 induced bladder cancer cell chemo-sensitivity to cisplatin treatment in a VEGFC-dependent manner. In addition, re-expression of VEGFC also rescued HT1376 cell growth, which was suppressed by miR-122 overexpression as assessed by cell proliferation assay. As expected, VEGFC over-expression could resistance to apoptosis in HT1376 cells induced by miR-122.
In summary, we identified a link between miR-122 and VEGFC that was a novel constituent of bladder cancer tumorigenesis. As a tumor suppressor in bladder cancer, overexpression of miR-122 inhibited bladder cancer cell growth. Our results show that miR-122 regulated cell proliferation through the VEGFC/AKT/mTOR signaling pathway and exogenous overexpression of miR-122 may represent a promising approach for targeted bladder cancer therapies. To the best of our knowledge, this study is the first to directly link miR-122 to bladder cancer angiogenesis and growth and indicates that miR-122 is of considerable therapeutic value in bladder cancer.
Acknowledgements
Tianjin Research Program of Application Foundation and Advanced Technology (14JCQNJC12700).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, Li D, Lai L, Jiang BH. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2012;40:761–774. doi: 10.1093/nar/gkr730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gong A, Ge N, Yao W, Lu L, Liang H. Aplysin enhances temozolomide sensitivity in glioma cells by increasing miR-181 level. Cancer Chemother Pharmacol. 2014;74:531–538. doi: 10.1007/s00280-014-2534-5. [DOI] [PubMed] [Google Scholar]
- 3.Qiu Y, Yang J, Bian S, Chen G, Yu J. PPARgamma suppresses the proliferation of cardiac myxoma cells through downregulation of MEF2D in a miR-122-dependent manner. Biochem Biophys Res Commun. 2016;474:560–565. doi: 10.1016/j.bbrc.2016.04.112. [DOI] [PubMed] [Google Scholar]
- 4.Zhang HS, Zhang FJ, Li H, Liu Y, Du GY, Huang YH. Tanshinone A inhibits human esophageal cancer cell growth through miR-122-mediated PKM2 down-regulation. Arch Biochem Biophys. 2016;598:50–56. doi: 10.1016/j.abb.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 5.Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122--a key factor and therapeutic target in liver disease. J Hepatol. 2015;62:448–457. doi: 10.1016/j.jhep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Basu S, Bhattacharyya SN. Insulin-like growth factor-1 prevents miR-122 production in neighbouring cells to curtail its intercellular transfer to ensure proliferation of human hepatoma cells. Nucleic Acids Res. 2014;42:7170–7185. doi: 10.1093/nar/gku346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venkatadri R, Muni T, Iyer AK, Yakisich JS, Azad N. Role of apoptosis-related miRNAs in resveratrol-induced breast cancer cell death. Cell Death Dis. 2016;7:e2104. doi: 10.1038/cddis.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Z, Zhao C, Wang L, Cao X, Li J, Huang R, Lao Q, Yu H, Li Y, Du H, Qu L, Shou C. A VEGFR1 antagonistic peptide inhibits tumor growth and metastasis through VEGFR1-PI3K-AKT signaling pathway inhibition. Am J Cancer Res. 2015;5:3149–3161. [PMC free article] [PubMed] [Google Scholar]
- 9.Wang ZD, Wei SQ, Wang QY. Targeting oncogenic KRAS in non-small cell lung cancer cells by phenformin inhibits growth and angiogenesis. Am J Cancer Res. 2015;5:3339–3349. [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y, Yu J, Pei CG, Li YY, Tu P, Gao GP, Shao Y. Xanthatin, a novel potent inhibitor of VEGFR2 signaling, inhibits angiogenesis and tumor growth in breast cancer cells. Int J Clin Exp Pathol. 2015;8:10355–10364. [PMC free article] [PubMed] [Google Scholar]
- 11.Tang Y, Lv P, Sun Z, Han L, Luo B, Zhou W. 14-3-3zeta up-regulates hypoxia-inducible factor-1alpha in hepatocellular carcinoma via activation of PI3K/Akt/NF-small ka, CyrillicB signal transduction pathway. Int J Clin Exp Pathol. 2015;8:15845–15853. [PMC free article] [PubMed] [Google Scholar]
- 12.Dey N, De P, Brian LJ. Evading anti-angiogenic therapy: resistance to anti-angiogenic therapy in solid tumors. Am J Transl Res. 2015;7:1675–1698. [PMC free article] [PubMed] [Google Scholar]
- 13.Huang JJ, Shi YQ, Li RL, Hu A, Lu ZY, Weng L, Wang SQ, Han YP, Zhang L, Li B, Hao CN, Duan JL. Angiogenesis effect of therapeutic ultrasound on HUVECs through activation of the PI3K-Akt-eNOS signal pathway. Am J Transl Res. 2015;7:1106–1115. [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B, Wang H, Yang Z. MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS One. 2012;7:e47053. doi: 10.1371/journal.pone.0047053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang HL, Jiang Y, Wang YH, Chen T, He HJ, Liu T, Yang T, Yang LW, Chen J, Song ZQ, Yao W, Wu B, Liu G. FBXO31 promotes cell proliferation, metastasis and invasion in lung cancer. Am J Cancer Res. 2015;5:1814–1822. [PMC free article] [PubMed] [Google Scholar]
- 16.Liu XY, Liu ZJ, He H, Zhang C, Wang YL. MicroRNA-101-3p suppresses cell proliferation, invasion and enhances chemotherapeutic sensitivity in salivary gland adenoid cystic carcinoma by targeting Pim-1. Am J Cancer Res. 2015;5:3015–3029. [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao X, He W, Li J, Huang S, Wan X, Luo H, Wu D. MiRNA-125b inhibits proliferation and migration by targeting SphK1 in bladder cancer. Am J Transl Res. 2015;7:2346–2354. [PMC free article] [PubMed] [Google Scholar]
- 18.Rudy SF, Brenner JC, Harris JL, Liu J, Che J, Scott MV, Owen JH, Komarck CM, Graham MP, Bellile EL, Bradford CR, Prince ME, Carey TE. In vivo Wnt pathway inhibition of human squamous cell carcinoma growth and metastasis in the chick chorioallantoic model. J Otolaryngol Head Neck Surg. 2016;45:26. doi: 10.1186/s40463-016-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo XW, Liu K, Chen Z, Zhao M, Han XW, Bai YG, Feng G. Adenovirus-mediated GDF-5 promotes the extracellular matrix expression in degenerative nucleus pulposus cells. J Zhejiang Univ Sci B. 2016;17:30–42. doi: 10.1631/jzus.B1500182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng X, Jiang Y, Xie L, Jiang L, Li J, Sun C, Xu H, Wang R, Zhou M, Zhou Y, Dan H, Wang Z, Ji N, Deng P, Liao G, Geng N, Wang Y, Zhang D, Lin Y, Ye L, Liang X, Li L, Luo G, Feng M, Fang J, Zeng X, Wang Z, Chen Q. Overexpression of proteasomal activator PA28alpha serves as a prognostic factor in oral squamous cell carcinoma. J Exp Clin Cancer Res. 2016;35:35. doi: 10.1186/s13046-016-0309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez YG, Lucas AL. MicroRNA in pancreatic ductal adenocarcinoma and its precursor lesions. World J Gastrointest Oncol. 2016;8:18–29. doi: 10.4251/wjgo.v8.i1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang SC, Lin XL, Li J, Zhang TT, Wang HY, Shi JW, Yang S, Zhao WT, Xie RY, Wei F, Qin YJ, Chen L, Yang J, Yao KT, Xiao D. MicroRNA-122 triggers mesenchymal-epithelial transition and suppresses hepatocellular carcinoma cell motility and invasion by targeting RhoA. PLoS One. 2014;9:e101330. doi: 10.1371/journal.pone.0101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Zhu J, Fu H, Wan J, Hu Z, Liu S, Li J, Tie Y, Xing R, Zhu J, Sun Z, Zheng X. Hepato-specific microRNA-122 facilitates accumulation of newly synthesized miRNA through regulating PRKRA. Nucleic Acids Res. 2012;40:884–891. doi: 10.1093/nar/gkr715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnaud-Batista FJ, Peruchetti DB, Abreu TP, do Nascimento NR, Malnic G, Fonteles MC, Caruso-Neves C. Uroguanylin modulates (Na+K)ATPase in a proximal tubule cell line: Interactions among the cGMP/protein kinase G, cAMP/protein kinase A, and mTOR pathways. Biochim Biophys Acta. 2016;1860:1431–1438. doi: 10.1016/j.bbagen.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Chen YC, Chien LH, Huang BM, Chia YC, Chiu HF. Aqueous Extracts of Toona sinensis Leaves Inhibit Renal Carcinoma Cell Growth and Migration Through JAK2/stat3, Akt, MEK/ERK, and mTOR/HIF-2alpha Pathways. Nutr Cancer. 2016:1–13. doi: 10.1080/01635581.2016.1158292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.