

Abstract
Many studies have indicated that histone deacetylase (HDAC) activity is always increased in a lot of human tumors, and inhibition of HDAC activity is a promising new strategy in the treatment of cancers. Chidamide, a novel HDAC inhibitor of the benzamide class, is currently under clinical trials. In this study, we aimed to investigate the antitumor activity of Chidamide on myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) cell lines and explore the possible mechanism. Chidamide exhibited efficient anti-proliferative activity on MDS and AML cells in a time- and dose-dependent manner, accompanied by cell cycle arrest at G0/G1 phase and cell apoptosis. Importantly, Chidamide possessed potent HDAC inhibition property, as evaluated by HDAC activity analysis and acetylation of histone H3 and H4. Moreover, Chidamide significantly increased the expression of Suppressors of cytokine signaling 3 (SOCS3), reduced the expression of Janus activated kinases 2 (JAK2) and Signal transducer and activator of transcription 3 (STAT3), and inhibited STAT3 downstream genes, including c-Myc, Bcl-xL, and Mcl-1, which are involved in cell cycle progression and anti-apoptosis. Therefore, we demonstrate that Chidamide exhibits potent inhibitory effect on cell viability of MDS and AML cells, and the possible mechanism may lie in the downregulation of JAK2/STAT3 signaling through SOCS3 upregulation. Our data provide rationale for clinical investigations of Chidamide in MDS and AML.
Keywords: Chidamide, myelodysplastic syndromes, acute myeloid leukemia, histone deacetylase inhibitor, STAT3
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic stem cell disorders which are characterized by ineffective hematopoiesis, peripheral blood cytopenias, and high risk of transformation to acute myeloid leukemia (AML) [1]. AML is a myeloid malignancy that involves precursor cells with a reduced capacity to differentiate into more mature cellular elements and with increased capacity of proliferation and self-renewal [2]. Current strategies for the treatment of both MDS and AML are suboptimal. Thus, there is great need to develop new agents to improve treatment of MDS and AML.
Histone deacetylases (HDACs) are a family of enzymes that remove the acetyl group from histone lysine residues, inducing transcriptional repression through chromatin condensation [3]. Since abnormally high expression of HDACs has been implicated in kinds of tumors, inhibition of HDAC activity is a promising new strategy in the treatment of cancers [4]. By inhibiting the activity of HDAC enzymes, HDAC inhibitors may cause re-expression of genes abnormally suppressed in cancer cells, thus potentially reversing the malignant phenotype and inducing growth inhibition, cell cycle arrest, extrinsic and intrinsic apoptosis.
Increasing evidence has shown that JAK2/STAT3 signaling is frequently upregulated in many human cancers, and it can induce cell proliferation, differentiation and anti-apoptosis [5,6]. Therefore, this pathway is considered a target for anticancer therapy in many human cancers. SOCS proteins, the negative regulators of JAK2/STAT3 signaling, have been reported to function as tumor suppressors [7].
Chidamide (CS055/HBI-8000) is a novel HDAC inhibitor of the benzamide class, which specifically inhibits HDAC1, 2, 3, and 10 [8]. In this study, we show that Chidamide possesses potent inhibitory effect against HDACs and induces growth inhibition, cell cycle arrest and apoptosis in MDS and AML cell lines. Furthermore, JAK2/STAT3 signaling is downregulated with the treatment of Chidamide.
Materials and methods
Reagents
Chidamide was supplied by Shenzhen Chipscreen Biosciences Ltd. (Shenzhen, China) and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 50 mM. Suberoylanilide hydroxamic acid (SAHA) was purchased from Sigma (St. Louis, MO, USA) and dissolved in DMSO at a concentration of 50 mM.
Cell culture
MDS cell line SKM-1 was gifted from Professor Xiang Li in Jiangnan University. Acute erythroleukemia cell line HEL was purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured in RPMI-1640 with 10% fetal bovine serum and penicillin (100 units/ml) /streptomycin (100 µg/ml). All cells were maintained in humidified air containing 5% CO2 at 37°C.
Cell growth assay
The cells were plated in 96-well plate (5000 cells/well) and treated with different doses of Chidamide and SAHA for 24, 48 and 72 hours. At different time points, the cell number was measured using Cell-Counting Kit-8 (CCK8) proliferation assay kit (DOJINDO, kamimashiki gun Kumamoto, Japan). 10 µL of CCK-8 solution were added to each well of the plate. After incubation for 2 hours at 37°C, the plates were measured at 450 nm using a microplate reader (Biotech, NY, USA).
Flow cytometry analysis
The proportion of apoptotic cells was quantified by Alexa Fluor 488 Annexin V/propidium iodide (PI) dual staining (Invitrogen, Carlsbad, CA, USA). Following drug treatment, cells were harvested and washed with phosphate-buffered saline (PBS), and re-suspended in 100 μL of binding buffer. Then incubate cells with 5 μL Annexin V and 1 μL PI for 15 minutes in the dark at room temperature. Analyze the stained cells by flow cytometry as soon as possible. For cell cycle analysis, the cells were washed with PBS and then re-suspended in 75% ice-cold ethanol overnight. After that, the cells were harvested and re-suspended in PBS with 100 μg/ml RNase A and 100 mg/ml PI for 30 minutes. Cell cycle distribution was performed using a FACS Calibur system flow cytometer with CellQuest software (Becton-Dickinson, Sunnyvale, CA, USA). The percentage of cells was analyzed using ModFit LT software program.
HDAC fluorescence activity assay
Cells were treated with different doses of Chidamide and SAHA for 24 hours, and then cell lysate was subjected to a HDAC Fluorometric Activity Assay Kit (Biovision, Milpitas, CA, USA) to determine the HDAC activity. 50 µg of cell lysate was added to react with fluorometric substrate and incubated at 37°C for 30 minutes. After the incubation, the lysine developer in this kit was added and the mixture was incubated for another 30 minutes at 37°C. Fluorescence was detected by plate reader paradigm detection platform (Biotech, NY, USA) with Ex=360 nm (excitation) and Em=465 nm (emission).
Real-time PCR
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. cDNA was synthesized using the Revert Aid TM First Strand cDNA Synthesis Kit (Fermentas, Burlington, Canada) according to the manufacturer’s protocol. PCR was performed with Real Master Mix (Takara, Dalian, China) on an ABI 7500 real-time PCR machine (Applied Biosystems, Foster, CA, USA). Conditions were as follows: hold stage was 95°C for 30 s, and cycling was 40 cycles of 95°C for 5 s, 60°C for 30 s, and 72°C for 30 s. The primer sequences are listed in Table 1.
Table 1.
Primer sequences for quantitive real-time PCR
| Genes | Primer sequences (5’ to 3’) |
|---|---|
| SOCS3 | Forward Primer CCTGCGCCTCAAGACCTTC |
| Reverse Primer GTCACTGCGCTCCAGTAGAA | |
| JAK2 | Forward Primer TCTGGGGAGTATGTTGCAGAA |
| Reverse Primer AGACATGGTTGGGTGGATACC | |
| STAT3 | Forward primer CAGCAGCTTGACACACGGTA |
| Reverse primer AAACACCAAAGTGGCATGTGA | |
| p21 | Forward primer ACATCGCCAAGGAAAAACGC |
| Reverse primer GTCTGTTTCGGTACTGTCATCC | |
| c-Myc | Forward primer GGCTCCTGGCAAAAGGTCA |
| Reverse primer CTGCGTAGTTGTGCTGATGT | |
| Bcl-xL | Forward primer GAGCTGGTGGTTGACTTTCTC |
| Reverse primer TCCATCTCCGATTCAGTCCCT | |
| Mcl-1 | Forward primer TGCTTCGGAAACTGGACATCA |
| Reverse primer TAGCCACAAAGGCACCAAAAG | |
| GAPDH | Forward primer TGTGGGCATCAATGGATTTGG |
| Reverse primer ACACCATGTATTCCGGGTCAAT |
Western blot analysis
After treatment of cells with Chidamide and SAHA for 24 hours, cells were washed with ice-cold PBS and lysed with ice-cold lysis buffer. Lysates were centrifuged at 14,000 r/min for 10 minutes at 4°C and the supernatant was collected. Protein concentrations were determined using the bicinchoninic acid (BCA) assay (CWBIO, Beijing, China). 50 μg of protein was electrophoresized on 8 to 15% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel electrophoresis and transferred to poly (vinylidene difluoride) membranes. After incubation at 4°C with the primary antibodies against acetyl-histone H3 (Lys9), acetyl-histone H4 (Lys8), p21, STAT3, p-STAT3, JAK2, p-JAK2, SOCS3, c-Myc, BCL-xL, Mcl-1 and GAPDH (Cell Signaling Technologies, Boston, MA, USA) overnight, the blots were washed, exposed for 1 hour to corresponding HRP-conjugated secondary antibodies, and finally detected by chemiluminescence reagents (Millipore, Billerica, MA, USA).
Statistical analysis
Each experiment was performed at least three times and the data are presented as mean ± SEM for the indicated number of separate experiments. Student’s t-test was used to compare the mean of each group with that of the control group in experiments. All analysis was performed by SPSS 17.0 System. Results were considered to be significant if the P-value was less than 0.05.
Results
Chidamide inhibits HDAC enzyme activity in MDS and AML cell lines
To determine the efficacy of Chidamide on MDS and AML cell lines, we assessed HDAC enzyme activity under treatment of Chidamide or SAHA in SKM-1 and HEL cells through HDAC fluorescence activity assay. As shown in Figure 1A, Chidamide inhibited HDAC enzyme activity in a concentration-dependent manner, which is more potent than inhibitory activity of SAHA. We further evaluated the acetylation of lysine residues on histones H3 and H4. Western blot analysis demonstrated that acetylation of histone H3 at Lys9 and the acetylation of histone H4 at Lys8 were significantly increased after administration of Chidamide and SAHA for 24 hours (Figure 1B). These results demonstrate that Chidamide has potent inhibitory effects on HDAC enzyme activity, further inducing acetylation of histone H3 and H4 lysine residues in SKM-1 and HEL cells.
Figure 1.
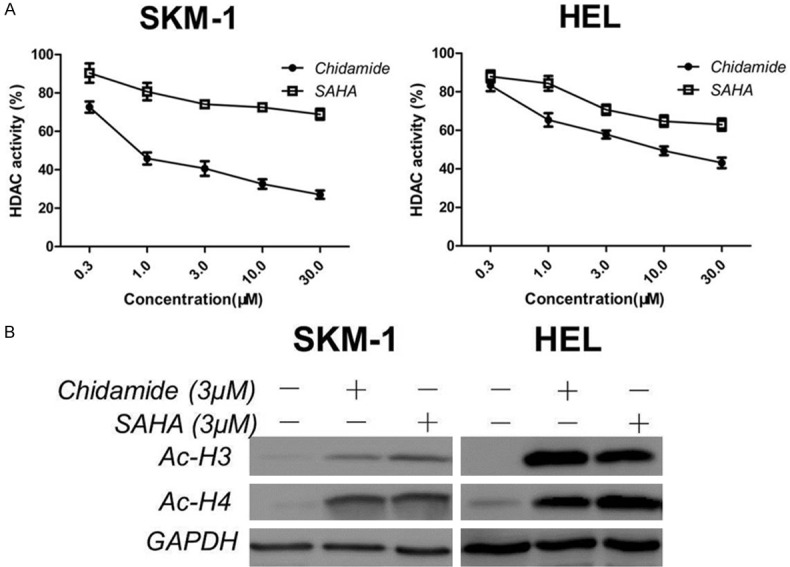
Effect of Chidamide on HDAC enzyme activity and protein expression in MDS and AML cells. A. Effect of Chidamide on HDAC enzyme activity. SKM-1 and HEL cells were treated with different concentrations of Chidamide or SAHA for 24 h and then HDAC activities were detected as described in Materials and Methods. Data represent mean ± SEM from at least three independent experiments. B. Effect of Chidamide on HDAC inhibition markers. Cells were treated with Chidamide or SAHA for 24 h and then harvested for HDAC inhibition marker (acetyl-histone 3, acetyl-histone 4) detection using western blotting.
Chidamide induces growth inhibition, cell cycle arrest and apoptosis
To investigate the effects of Chidamide on cell proliferation in comparison with SAHA in MDS and AML, SKM-1 and HEL cells were exposed to Chidamide or SAHA at concentrations ranging from 0.3 to 30 μM for 24, 48 or 72 hours. As shown in Figure 2A, Chidamide exhibited anti-proliferation potency in both cell lines examined, which is similar with SAHA. Moreover, cell growth was inhibited in a time- and dose-dependent manner. Cell cycle analysis showed that Chidamide induced G0/G1 phase arrest in SKM-1 and HEL cells. As indicated in Figure 2B and 2C, the cell proportion at G0/G1 phase increased from (38.0±1.5)% to (84.7.5±4.8)% in SKM-1 cells and increased from (39.0±0.4)% to (54.2±4.6)% in HEL cells respectively, after exposure to 3 μM of Chidamide for 24 hours. At the same time, proportion of S phase cells decreased dramatically. In addition, the cell cycle arrest effects of Chidamide were similar to SAHA. Apoptosis analysis further confirmed that Chidamide induced significant apoptosis in both cell lines. In SKM-1 cells, treatment with 3 μM of Chidamide for 48 hours killed nearly half of cells with levels slightly lower than that of SAHA; while in HEL cells, majority of cells died with 10 μM of Chidamide. Moreover, this apoptosis inducing effect was also dose-dependent (Figure 2D and 2E). Taken together, these results demonstrate that Chidamide inhibits cell proliferation and induces G0/G1 phase arrest and cell apoptosis in SKM-1 and HEL cells.
Figure 2.

Chidamide reduces the cell viability of MDS and AML cells. (A) Time and dose-dependent effect of Chidamide and SAHA on the inhibition of cell growth in MDS and AML cells lines. SKM-1 and HEL cells were treated with different concentrations of Chidamide or SAHA for 24 h, 48 h and 72 h, and then cell viability was determined by CCK-8 assay. (B, C) Effect of Chidamide and SAHA on the progression of cell cycle. SKM-1 and HEL cells were treated with 3 μM of Chidamide or SAHA for 24 h, and then cell cycles were determined by flow cytometry (B). The cell cycle distribution was quantified in Chidamide- or SAHA-treated and untreated cells (C). (D, E) Effect of Chidamide and SAHA on cell apoptosis. SKM-1 and HEL cells were treated with different concentrations of Chidamide or SAHA for 48 h, and then cell apoptosis was determined by AnnexinV-FITC/PI staining. The quantitative data for the percentage of apoptotic cells were pooled from three independent experiments (D). Dot plots of Annexin V-FITC/PI-stained SKM-1 cells upon different concentrations of Chidamide (E). Data represent mean ± SEM from at least three independent experiments. *P<0.05; compared with the respective control group.
Chidamide downregulates levels of c-Myc, Bcl-xL and Mcl-1 while upregulates p21
To gain some insight into the underlying molecular mechanism of the anti-proliferative and pro-apoptotic activity of Chidamide in SKM-1 and HEL cells, we evaluated the mRNA and protein expression of p21, c-Myc, Bcl-xL and Mcl-1, which play important roles in regulating cell growth and apoptosis. Real-time PCR analysis demonstrated that the mRNA level of p21 was obviously upregulated, while the expression of c-Myc, Bcl-xL and Mcl-1 was downregulated significantly after exposure to 3 μM of Chidamide for 24 hours in both cell lines (Figure 3A). Western blot analysis further confirmed that Chidamide increased the protein expression of p21 and decreased that of c-Myc, Bcl-xL and Mcl-1, with the similar effects to SAHA (Figure 3B). These results indicate that Chidamide may induce G0/G1 phase arrest and cell apoptosis through upregulation of p21 and downregulation of c-Myc, Bcl-xL and Mcl-1 in SKM-1 and HEL cells.
Figure 3.
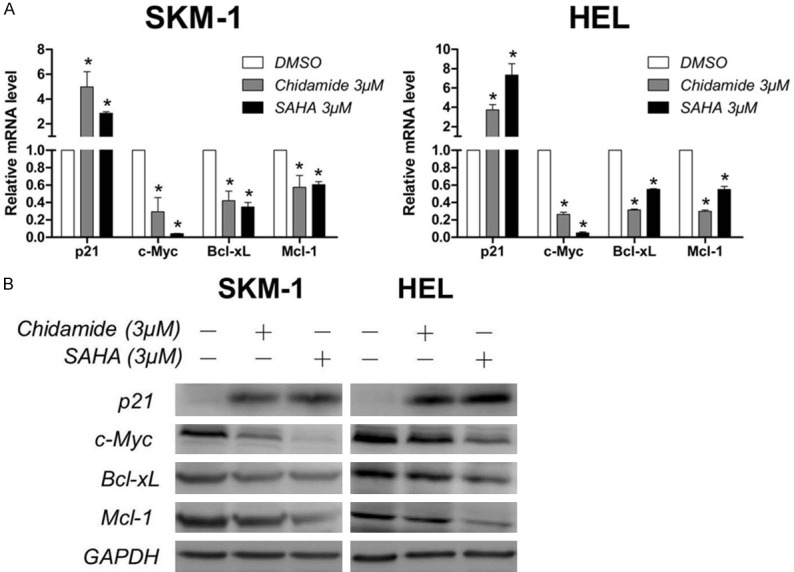
Chidamide downregulates c-Myc, Bcl-xL and Mcl-1 while upregulates p21. A. Effect of Chidamide on the mRNA expression of p21, c-Myc, Bcl-xL and Mcl-1. SKM-1 and HEL cells were treated with 3 μM of Chidamide or SAHA for 24 h, and then harvested for mRNA expression analysis using quantitative real-time PCR. B. Effect of Chidamide on the protein expresson of p21, c-Myc, Bcl-xL and Mcl-1. Cells were treated with Chidamide or SAHA for 24 h and then harvested for western blot analysis. Data represent mean ± SEM from at least three independent experiments. *P<0.05; compared with the respective control group.
Chidamide suppresses JAK2/STAT3 signaling
The JAK2/STAT3 signaling pathway is an important cell-growth promoting pathway which is aberrantly activated in most cancers. Pharmacological inhibition of JAK2/STAT3 signaling could induce cell cycle arrest and cell apoptosis. C-Myc, Mcl-1, and Bcl-xL are the downstream targets of JAK2/STAT3 signaling. Our investigation that Chidamide and SAHA treatment results in G1 phase arrest and induction of apoptosis, accompanied by the downregulation of c-Myc, Mcl-1 and Bcl-xL, suggests that inhibition of HDACs by Chidamide or SAHA may suppress the JAK2/STAT3 signaling. Indeed, after incubation with Chidamide or SAHA for 24 hours, the mRNA levels of JAK2 and STAT3 decreased dramatically in SKM-1 and HEL cells (Figure 4A). In addition, western blot analysis showed that the protein levels of phosphorylated JAK2 and STAT3 as well as total JAK2 and STAT3 were diminished in Chidamide- or SAHA-treated cells (Figure 4B). Importantly, we found that SOCS3, the negative regulator of JAK2/STAT3 signaling, was dramatically upregulated in both mRNA and protein levels with the treatment of Chidamide or SAHA, which may account for their inhibitory effect on JAK2/STAT3 signaling. Therefore, these results indicate that Chidamide upregulates the expression of SOCS3 and thus suppresses JAK2/STAT3 signaling in SKM-1 and HEL cells.
Figure 4.
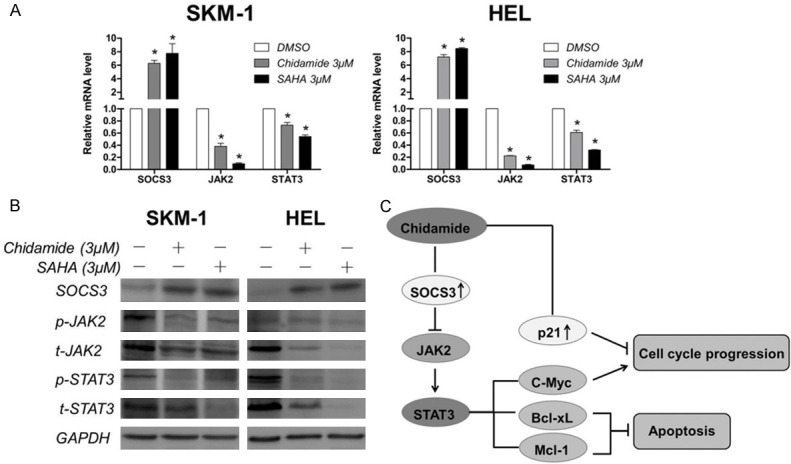
Effect of Chidamide on JAK2/STAT3 signaling. A. Effect of Chidamide on the mRNA expression of SOCS3, JAK2, and STAT3. SKM-1 and HEL cells were treated with 3 μM of Chidamide or SAHA for 24 h, and then harvested for mRNA expression analysis using quantitative real-time PCR. B. Effect of Chidamide on the protein expresson of SOCS3, p-JAK2, total-JAK2, p-STAT3, total-STAT3. Cells were treated with 3 μM of Chidamide or SAHA for 24 h, and then harvested for western blot analysis. C. The possible mechanism of cell cycle arrest and apoptosis-inducing effect of Chidamide on MDS and AML cells. HDAC inhibition leads to upregulation of SOCS3, and then downregulation of JAK2/STAT3 signaling. Some of the downstream targets are associated with cell cycle arrest and anti-apoptosis. Upregulation of p21 by Chidamide is also involved in cell cycle arrest. Data represent mean ± SEM from at least three independent experiments. *P<0.05; compared with the respective control group.
Discussion
In this study, we have shown that Chidamide possesses potent HDAC inhibition property and exhibits efficient anti-proliferative activity on MDS and AML cells, accompanied by cell cycle arrest at G0/G1 phase and cell apoptosis. Moreover, Chidamide significantly upregulates the expression of SOCS3 and induced downregulation of JAK2/STAT3 signaling, including some downstream targets, c-Myc, Bcl-xL, and Mcl-1, which are involved in cell cycle progression and anti-apoptosis.
Recently, it has been recognized that epigenetic changes are playing critical roles in the pathogenesis of MDS and AML. Thus, epigenetic modification is identified as a therapeutic strategy, and histone acetylation alteration is one of these approaches. Indeed, many HDAC inhibitors are in clinical trials of patients with MDS and AML, and the overall response rates generally range from 10% to 30% [9-12]. Combination of HDAC inhibitors and hypomethylating agents suggests significant activity in patients with advanced, relapsed-refractory AML and MDS [13-15]. Thus, HDAC inhibitors have become promising agents in recent years. Considering the adverse events of pan-class HDAC inhibitors such as fatigue, nausea, anorexia, diarrhea, thrombus and thrombocytopenia, selective HDAC inhibitors targeting specific isoforms may reduce toxicity [16,17]. HDAC1, 2 and 3 are most frequently overexpressed in human tumors, and knockdown of HDAC1, 2 or 3 is sufficient to inhibit tumor growth in vivo [18,19]. Particularly, the expression of HDAC2 was possibly increased in MDS and AML [20]. Thus, targeting of Class I isoforms has been a promising approach. In this study, we explored the effects of a novel Class I selective HDAC inhibitor Chidamide which has shown potent inhibitory effects on many cancer cells [8,21], but few studies have been reported in MDS cells. In present study, we chose MDS cell line SKM-1 cells as the target and proved that Chidamide could play a role in inhibiting MDS cells, in addition to AML cells.
P21, also known as cyclin-dependent kinase (CDK) inhibitor 1A, functions as a negative regulator of cell cycle progression at G1, which plays a critical role in the suppression of tumor cell proliferation [22]. The expression of p21 is always regulated by HDAC inhibitors [23-25]. C-Myc is reported to induce the expression of several positive regulators of the cell cycle such as cyclins, CDKs and E2F transcription factors, and to repress cell cycle inhibitors such as p15, p16, p21 or p27, thus stimulating the cell cycle progression and the cellular proliferation [26]. Our data showed that Chidamide significantly elevated the expression of p21 while decreased the level of c-Myc, suggesting that cycle arrest at G0/G1 phrase induced by Chidamide may be put down to its promotion of p21 and suppression of c-Myc. Induction of apoptosis upon HDAC inhibitors treatment has previously been shown in various tumors [21,27,28]. We found that Chidamide induced apoptosis in SKM-1 and HEL cells in a dose-dependent manner, along with the reduced expression of anti-apoptotic proteins, Bcl-xL and Mcl-1, indicating that downregulation of anti- apoptotic proteins may be one of the mechanisms of apoptosis induction upon Chidamide treatment. Thus, these data together indicate that Chidamide inhibits cell growth by cell cycle arrest at G0/G1 phase and apoptosis induction in MDS and AML cells.
The aberrantly activated JAK2/STAT3 signaling is critical in malignant progression by promoting cell growth. Moreover, SOCS3, the negative regulator of the JAK2/STAT3 signaling, is proved to have tumor suppressor functions [7]. In current study, our data indicated that Chidamide elevated the mRNA and protein levels of SOCS3, and decreased the expression of JAK2, pJAK2, STAT3, and pSTAT3. Furthermore, c-Myc, Bcl-xL and Mcl-1, known as downstream targets of JAK2/STAT3 signaling, were also downregulated. Thus, we postulated that Chidamide-induced cell cycle arrest and apoptosis can be attributed to the downregulation of JAK2/STAT3 signaling (Figure 4C). In agreement with our study, J Minami et al [19] demonstrated that phosphorylation of STAT3 was downregulated upon HDAC3 inhibition in multiple myeloma. HDAC inhibitors are always associated with re-expression of genes abnormally suppressed in cancer cells by restoring acetylation status of histones and relaxing the chromatin structure. HDAC inhibitor TSA was reported to increase in the acetylation of H3 and H4 histone proteins associated with the promoter regions of SOCS3 in colorectal cancer cells [28]. In this study, acetylation of histone H3 and H4 in SKM-1 and HEL cells was induced upon Chidamide treatment. Thus we speculate that the mechanism of Chidamide-induced upregulation of SOCS3 in SKM-1 and HEL cells may also due to the accumulation of acetylated H3 and H4 in its promotor regions, which may need to be further elucidated.
Therefore, we demonstrate that Chidamide inhibit the viability of MDS and AML cells, causing G0/G1 phase arrest and apoptosis, and the possible mechanism may lie in the downregulation of JAK2/STAT3 signaling through SOCS3 upregulation apart from the upregulation of p21 (Figure 4C). Thus, with comparable anti-tumor effort to SAHA and possibly more favorable tolerability, Chidamide is a potential candidate agent in MDS and AML.
Acknowledgements
This study was supported in part by the National Natural Science Foundation of China (NNSFC81570108; NNSFC81170463).
Disclosure of conflict of interest
None.
References
- 1.Heaney ML, Golde DW. Myelodysplasia. N Engl J Med. 1999;340:1649–1660. doi: 10.1056/NEJM199905273402107. [DOI] [PubMed] [Google Scholar]
- 2.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellstrom-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 3.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res. 2015;38:933–949. doi: 10.1007/s12272-015-0571-1. [DOI] [PubMed] [Google Scholar]
- 5.Qi QR, Yang ZM. Regulation and function of signal transducer and activator of transcription 3. World J Biol Chem. 2014;5:231–239. doi: 10.4331/wjbc.v5.i2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–5029. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He B, You L, Uematsu K, Zang K, Xu Z, Lee AY, Costello JF, McCormick F, Jablons DM. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100:14133–14138. doi: 10.1073/pnas.2232790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X, Lu XP. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69:901–909. doi: 10.1007/s00280-011-1766-x. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, Hardwick JS, Reilly JF, Chen C, Ricker JL, Secrist JP, Richon VM, Frankel SR, Kantarjian HM. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA] ) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 10.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, Gore SD, Sausville EA, Zwiebel J, Karp JE. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781–2790. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112:981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuendgen A, Strupp C, Aivado M, Bernhardt A, Hildebrandt B, Haas R, Germing U, Gattermann N. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 2004;104:1266–1269. doi: 10.1182/blood-2003-12-4333. [DOI] [PubMed] [Google Scholar]
- 13.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’Brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirschbaum M, Gojo I, Goldberg SL, Bredeson C, Kujawski LA, Yang A, Marks P, Frankel P, Sun X, Tosolini A, Eid JE, Lubiniecki GM, Issa JP. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br J Haematol. 2014;167:185–193. doi: 10.1111/bjh.13016. [DOI] [PubMed] [Google Scholar]
- 16.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett. 2009;280:211–221. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Ononye SN, van Heyst M, Falcone EM, Anderson AC, Wright DL. Toward isozyme-selective inhibitors of histone deacetylase as therapeutic agents for the treatment of cancer. Pharm Pat Anal. 2012;1:207–221. doi: 10.4155/ppa.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minami J, Suzuki R, Mazitschek R, Gorgun G, Ghosh B, Cirstea D, Hu Y, Mimura N, Ohguchi H, Cottini F, Jakubikova J, Munshi NC, Haggarty SJ, Richardson PG, Hideshima T, Anderson KC. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia. 2014;28:680–689. doi: 10.1038/leu.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H, Maddipoti S, Quesada A, Bohannan Z, Cabrero Calvo M, Colla S, Wei Y, Estecio M, Wierda W, Bueso-Ramos C, Garcia-Manero G. Analysis of class I and II histone deacetylase gene expression in human leukemia. Leuk Lymphoma. 2015;56:3426–3433. doi: 10.3109/10428194.2015.1034705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443:735–746. doi: 10.1042/BJ20111685. [DOI] [PubMed] [Google Scholar]
- 22.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frys S, Simons Z, Hu Q, Barth MJ, Gu JJ, Mavis C, Skitzki J, Song L, Czuczman MS, Hernandez-Ilizaliturri FJ. Entinostat, a novel histone deacetylase inhibitor is active in B-cell lymphoma and enhances the anti-tumour activity of rituximab and chemotherapy agents. Br J Haematol. 2015;169:506–519. doi: 10.1111/bjh.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao B, He T. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep. 2015;33:304–310. doi: 10.3892/or.2014.3595. [DOI] [PubMed] [Google Scholar]
- 26.Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849:506–516. doi: 10.1016/j.bbagrm.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Chen B, Qin S, Li S, He X, Qiu S, Zhao W, Zhao H. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun. 2010;392:190–195. doi: 10.1016/j.bbrc.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Zhang P, Guo Z, Wu Y, Hu R, Du J, He X, Jiao X, Zhu X. Histone Deacetylase Inhibitors Inhibit the Proliferation of Gallbladder Carcinoma Cells by Suppressing AKT/mTOR Signaling. PLoS One. 2015;10:e0136193. doi: 10.1371/journal.pone.0136193. [DOI] [PMC free article] [PubMed] [Google Scholar]