

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic lung disease involving pulmonary injury associated with tissue repair, dysfunction and fibrosis. Recent studies indicate that some microRNAs (miRNAs) may play critical roles in the pathogenesis of pulmonary fibrosis. In this study, we aim to investigate whether miR-338* (miR-338-5p), which has been found to be associated with tumor progression, is associated with pathological process of pulmonary fibrosis. Balb/c mice were treated with bleomycin (BLM) to establish IPF models. Targtscan was used to predict the downstream target of miR-338*. Morphological changes were observed with light microscope and epithelial to mesenchymal transition (EMT) markers were detected by western blot. The expression of miR-338* or downstream target SMO was analyzed by real-time quantitative RT-PCR, northern blot or western blot. MiR-338* was down-regulated in the lung tissue from mice with bleomycin-induced pulmonary fibrosis. The smoothened (SMO) is a direct target of miR-338*, and knocking-down the expression of SMO could partially rescue the fibrotic phenotype of TGF-β-induced NuLi-1 cells. Over-expression of SMO led to the fibrotic phenotype of NuLi-1 cells even without TGF-β treatment. These findings showed that the over-expression of SMO contributed to the fibrotic phenotype of NuLi-1 cells by affecting the epithelial-to-mesenchymal transition (EMT) procedure. Furthermore, in vivo, lentivirus-mediated over-expression of miR-338* can alleviate lung fibrosis induced by bleomycin in mice. In conclusion, our results suggest that miR-338* can target SMO to reduce the EMT procedure and thus postpone the development of pulmonary fibrosis.
Keywords: Idiopathic pulmonary fibrosis, miR-338-5p, SMO, EMT, pathogenesis
Introduction
IPF is defined as a type of chronic fibrosing interstitial pneumonia of unknown cause and is associated with a histologic pattern of usual interstitial pneumonia (UIP) by a consensus statement of the American Thoracic Society (ATS) and the European Respiratory Society (ERS) [1]. The annual incidence of IPF is rising and is estimated to be 4.6-16.3 per 100,000 people; the prevalence is 13 to 20 cases per 100,000 [1,2]. The course of this disease generally involves the progressive deterioration of lung functions, with a median survival time of 3 years from the time of diagnosis [2]. The prognosis of IPF patients is very poor and many patients die of respiratory failure. To date, no randomized controlled trials support any therapy being identified for IPF.
The pathological process of IPF remains largely unknown. However, important evidences indicate that miRNAs play important roles during the pathological process [4-6]. MiRNAs, a class of small RNA molecules, have been shown to be involved in various pathological processes including cellular proliferation, tissue development and repair [7-9]. Therefore, they are increasingly being recognized as the key regulators of tissue phenotypes and potential targets for therapeutic interventions in various diseases. Of these, miR-338* (miR-338-5p) belongs to the miR-338 family and its expression might be involved in the regulation of tumor progression, including hepatocellular carcinoma and metastatic colorectal carcinoma tissues [10,11]. Besides, in the rat model of bleomycin-induced IPF, the enhanced expression of miR-338* may target the downstream effector LPA1 and thus significantly inhibit the proliferation of pulmonary fibroblasts [12].
Smoothened (SMO), a protein related to G-protein-coupled receptors, is the key activator of the Hedgehog (Hh) signaling pathway [13], which is considered to be involved in the development of tumors [14,15]. Constitutive activation of SMO promotes the clono-genicity and progression of human small cell lung cancer (SCLC) both in vitro and in vivo. Conversely, the deletion of SMO in Rb1 and Tp-53-mutant lung epithelial cells protects mice from SCLC initiation and progression [14]. In liver cancer cells, the expression of SMO is controlled by miR-338-3p, and its over-expression leads to invasion and metastasis in the tumor tissue [16]. In addition, the inhibition of Hh signaling results in the attenuation of cancer stem cell markers and the up-regulation of miR-200b and let-7c, indicating a connection between Hh signaling and miRNAs during drug resistance in lung cancer [17]. In the tumorigenesis mechanism, molecules in the Hh signaling pathway, including SMO, are activated in IPF lungs and Hh may contribute to IPF pathogenesis by increasing the proliferation, migration, extracellular matrix production, and survival of fibroblasts [18].
These findings indicate that miR-338* and the Hh signaling pathway might be involved in the pathogenesis of IPF, and that the Hh signaling pathway is closely regulated by the molecule of the miR-338 family. However, there is no evidence to reveal whether there is a relationship between miR-338* and SMO during the development of IPF. It has been increasingly realized that epithelial-mesenchymal transition (EMT) plays an important role in the repair and scar formation after epithelial injury during pulmonary fibrosis. Of those, some miRNAs are supposed to act as regulators during EMT procedure [19-21]. Therefore, we hypothesize that miR-338* affects EMT procedure by regulating SMO expression and thus participates in the development of lung fibrosis.
Materials and methods
Cell line and the treatment
The normal human bronchial epithelium cell line (NuLi-1) was provided by the Shanghai Cell Biology Institute (China). The cell line was maintained in RPMI-1640 medium (Sigma-Aldrich Co. Ltd, Irvine, CA) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin/streptomycin at 37°C with 5% CO2 and 100% humidity. TGF-β was bought from Life Technologies and dissolved in PBS. Briefly, cells at a density of 1×105 cells/well were seeded into 6-well plates in RPMI-1640 supplemented with 10% calf serum and were cultured for 24 h. The cells were then treated with PBS control, TGF-β (10 ng/ml), or TGF-β (10 ng/ml) plus siSMO (0.05 μM) for 72 hours. The treatment was terminated at a specific time point, and cell lysates were extracted for the next experiments. For the transfection experiment, NuLi-1 cells were transiently transfected with pcDNA3.1 or pcDNA3.1-SMO plasmids using the Lipofectamine 3000 reagent (Life Technologies, USA) for the indicated time points according to the manufacturer’s instructions. Briefly, 1×105 cells were seeded into six-well plates containing an antibiotic-free medium and incubated overnight. For each well, 2 μg DNA (pcDNA3.1 or pcDNA3.1-SMO) was mixed with 95 μl Opti-MEM. The mixture was then combined with a solution of 5 μl Lipofectamine 3000 reagent (Life Technologies) in 95 μl Opti-MEM. After a 20-min incubation period at room temperature, the mixture was applied to the cells in an appropriate volume of culture medium to achieve a final volume of 2 ml. Then, the cells were cultured for an additional 24 h or 48 h at 37°C before analysis.
Animal IPF models
Balb/C mice (Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China) were maintained in a controlled environment and provided with water and standard rodent food. Twenty mice were randomly divided into the following 2 groups (n=10/group): vehicle group (saline-water) and treated group (BLM-water). Mice in the saline group were injected intratracheally with 2 ml/kg saline; the others were injected intratracheally with BLM (5 mg/kg, 2 ml/kg in saline). On day 14 following bleomycin or saline treatment, mice were sacrificed and lungs were collected for histology, immuno-histochemistry, real-time PCR, and western blot analysis. All procedures involving animals were approved by the Ethics Committee for Animal Research of Medical School of Nanjing University.
Immunofluorescence and histological study
In vitro, the treated NuLi-1 cells were pre-fixed with 4% paraformaldehyde for 30 min at room temperature. After washing with PBS, samples were incubated with PBS containing 0.1% Triton X-100 for 15 min. Following blocking with 3% bovine serum albumin (BSA) for 1 h, samples were stained with primary antibodies (anti-E-cadherin polyclonal antibody; anti-vimentin polyclonal antibody) overnight at 4°C. Then, cells were incubated with corresponding secondary antibodies (goat anti-rabbit IgG-FITC; goat anti-mouse IgG-PE) for 1 h at 25°C. Eventually, samples were stained with DAPI for 5 min. Images were captured using an Olympus FluoView 1000 confocal microscope (Olympus America Inc., Melville, NY, USA). For histological study, after euthanasia, mice lungs were perfused with normal saline and inflated with 1 ml of 10% neutral buffered formalin. Lungs were then removed en bloc after tracheal ligation and preserved in 10% neutral buffered formalin for 24 h at room temperature, and subsequently embedded in paraffin. Hematoxylin and eosin (H&E) stains were performed using a standard protocol.
Western-blot
Western blot was performed as following. Briefly, after separation on SDS-PAGE, proteins were transferred to a PVDF membrane. Protein bands were detected after incubation with primary antibody overnight at 4°C and HRP-goat anti-rabbit IgG for 2 h using the ECL plus western blotting detection system. GAPDH was used as an internal control. Blots were scanned using a FluorChemFC2 system (Alpha Innotech, San Leandro, CA, USA). Western blots were quantified using the Image 1.42 software. Relative protein expression of SMO, E-cadherin, and vimentin was calculated relative to GAPDH, separately.
Dual-luciferase reporter assay
The 3’-UTR sequence of SMO predicted to interact with miR-338* or a mutated sequence with the predicted target sites (Figure 1) were synthesized and inserted into a pGL3 vector (Promega, Madison, WI, USA). These constructs were named pGL3-SMO-3’UTR-wt and pGL3-SMO-3’UTR-mutant and used to transfect NuLi-1 cells. The cells were cultured in 12-well plates and each transfected with 50 ng of pGL3-SMO-3’UTR-wt or pGL3-SMO-3’UTR-mutant together with 50 ng of pGL3 (Promega, Madison, WI, USA) containing firefly and Renilla luciferase and 50 nM of miR-338* or control using Lipofectamine 3000. Forty-eight hours after transfection, cells were harvested and assayed with the Dual-Luciferase Reporter Assay (Promega), according to the manufacturer’s instructions.
Figure 1.
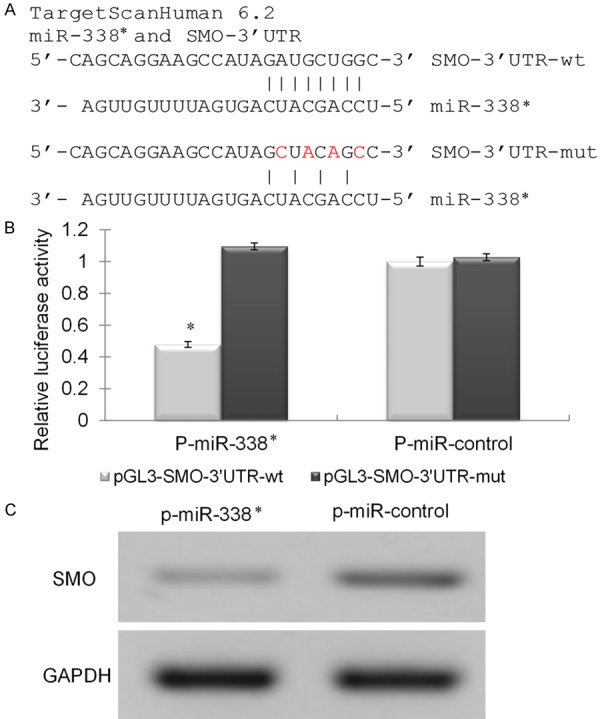
SMO was identified as the functional downstream targets of miR-338. A. Bioinformatics prediction the binding sites between miR-338* and SMO-3’UTR-wt and mutation. B. The relative luciferase activity of cells co-transfection with p-miR-338* or p-miR-control vector and pGL3-SMO-3’UTR-wt or mutation vector. C. Western blot analysis of total cell lysates extracted from p-miR-338* or p-miR-control vector transfected cells.
Statistical analysis
Data are presented as mean ± standard deviation (SD). The data were analyzed using the SPSS Windows software, version 12.0. Statistical analyses were performed using analysis of variance (ANOVA) or Student’s t test. P values <0.05 were considered to be statistically significant.
Results
SMO is identified as the functional downstream targets of miR-338* and being proved in cells
Since miR-338* has been shown to be associated with pulmonary fibrosis models in rats, we wondered whether the expression of miR-338* might be related to SMO, the downstream molecule of the Hh pathway. To confirm that SMO is a direct functional target of miR-338*, we investigated whether miR-338* targets the 3’-UTR of SMO mRNA using the dual-luciferase reporter assay. According to the predicted target sites from Microcosm (Figure 1A), we cloned the wild-type 3’-UTR fragment containing these predicted sites into the pGL3 luciferase reporter vector (pGL3-SMO-3’UTR-wt). Another 3’-UTR fragment with a mutation within each seed region was also cloned as a control (pGL3-SMO-3’UTR-mut). We observed that only the co-transfection of P-miR-338* and pGL3-SMO-3’UTR-wt significantly suppressed the luciferase activity by about 60% (Figure 1B). However, co-transfection of P-miR-control did not affect the luciferase activity of either the pGL3-SMO-3’UTR-wt or pGL3-SMO-3’UTR-mut expressed cell lines. These data show that SMO was a direct downstream target of miR-338*. We further measured the protein expression of SMO in Nuli-1 cells transfected with P-miR-338* or P-miR-control. The western blotting result also showed that over-expression of miR-338* reduced the endogenous protein expressions of SMO in NuLi-1 (Figure 1C).
TGF-beta-induced pulmonary fibrosis in vitro and silencing of SMO morphologically reverse epithelial to mesenchymal transition
To further investigate whether the suppression of SMO can reverse the process of fibrosis in cell lines, si-SMO was stably transfected into TGF-β-induced fibrotic Nuli-1 cells. As shown in Figure 2A, the expression of SMO was successfully knocked-down by siRNA in TGF-β-induced fibrotic cells. Then, we used transmission electron microscopy to observe the morphology of these cells. We found that, compared with wild-type Nuli-1 cells, TGF-β-induced fibrosis cells showed a fibroblast-like cell phenotype, such as loss of cell-to-cell contact and the capacity to form viable spheroids. While stably transfected with siSMO, fibroblast-like cells lose this phenotype, reversing to the epithelial phenotype and resembling the morphology of Nuli-1 cells (Figure 2B). These results suggested that the forced suppression of SMO may partially prevent the cellular fibrosis induced by TGF-β.
Figure 2.
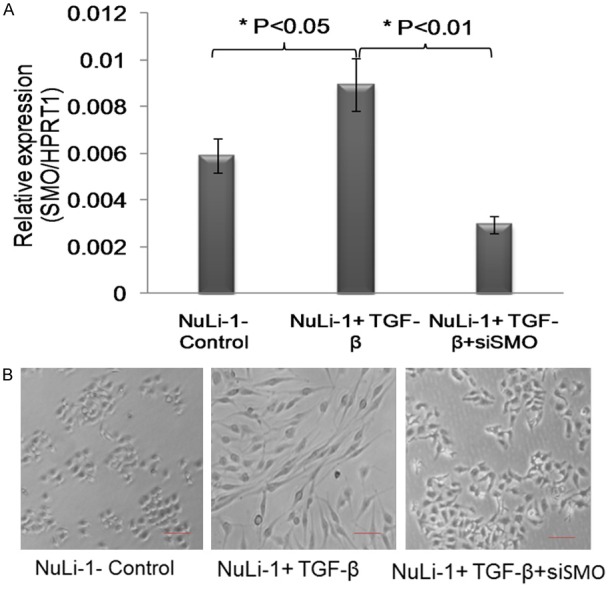
Silencing SMO protected TGF-beta induced pulmonary fibrosis. A. The relative expression of SMO in NuLi-1 cells treated with TGF-beta with SMO siRNA or siControl. The results showed that TGF-beta induced over-expression of SMO, while co-transfection with SMO siRNA decreased the expression of SMO. B. The morphology change of NuLi-1 cells treated with TGF-beta with SMO siRNA or siControl. The results showed that TGF-beta induced pulmonary fibrosis morphology of NuLi-1 cells, while co-transfection with SMO siRNA reversed the pulmonary fibrosis morphology of NuLi-1 cells.
SMO promotes epithelial-mesenchymal transition (EMT) in lung epithelial cells
Previously, it has been suggested that EMT was involved in repair and scar formation after epithelial injury during pulmonary fibrosis. Hence, we aimed to confirm whether SMO regulated by miR-338* participated in lung fibrosis by regulating the EMT procedure. Firstly, SMO was over-expressed in NuLi-1 cells through the transport plasmid pcDNA3.1 (Figure 3A). Secondly, we used western blotting to determine the expression of the EMT-induced markers E-cadherin, and vimentin in cells over-expressing SMO. Increased SMO expression levels were found to induce E-cadherin expression and decrease that of vimentin, while decreased SMO expression inhibited E-cadherin expression and promoted vimentin expression (Figure 3B). Similar to immunoblotting results, immunofluorescence analysis revealed the same changes in NuLi-1 cells (Figure 3C).
Figure 3.
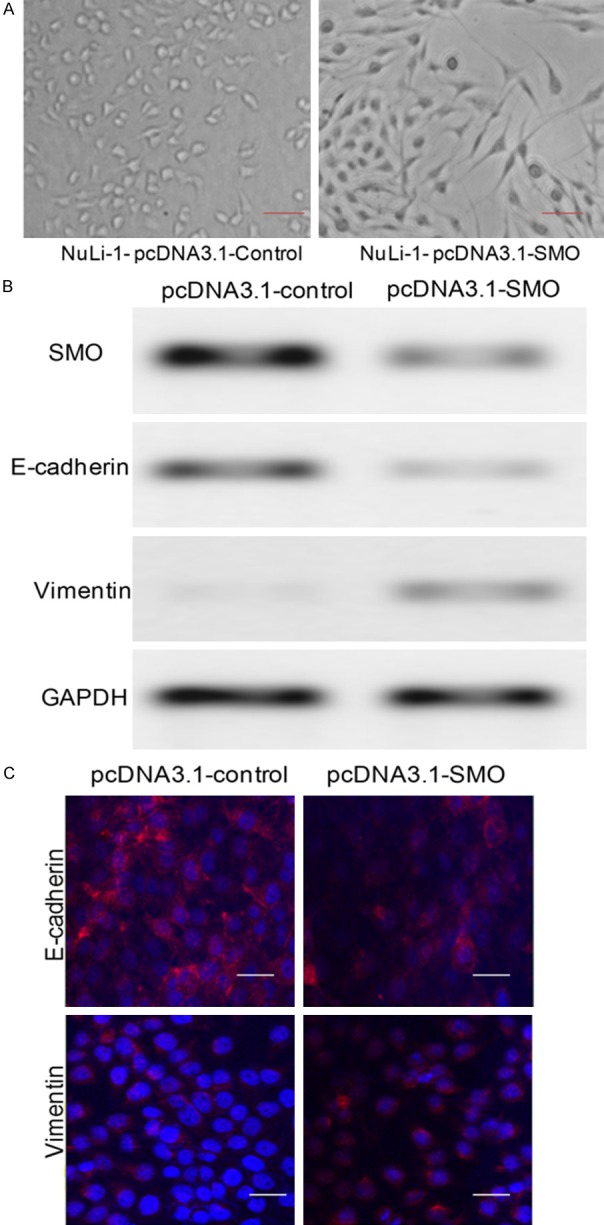
SMO promotes epithelial-mesenchymal transition in lung epithelial cells. (A) Stable over-expression of SMO induces fibrosis or mesenchymal morphology change. (B and C) The expression of SMO, epithelial marker Ecadherin, and mesenchymal marker vimentin in cells transfected with pcDNA3.1-control or pcDNA3-SMO vector by Western blotting (B) and Immunofluorescence (C) analysis.
Lentivirus-mediated miR-338* inhibits SMO expression to prevent bleomycin-induced pulmonary fibrosis in vivo
Using a well-established bleomycin (BLM)-induced pulmonary fibrosis animal model, we investigated whether the inhibited SMO expression regulated by miR-338* could prevent or reverse BLM-induced lung fibrosis in vivo. The transduced vehicle, lentivirus, was used to transduce miR-338* or miR-control into the animal to set up the different groups of mice. Identical to previous reports, miR-338* expression was significantly decreased after BLM treatment compared to the sample extracted from saline-treated mice. However, the transduction of lenti-miR-338* into the BLM-treated mice reconstituted the expression of miR-338* as shown in Figure 4A. Similarly, compared to saline-treated mice, histological findings indicated that treatment with bleomycin resulted in enhanced pulmonary alveolus inflammation. Additionally, broadened alveolar septa and increased fibroblasts were observed in multi-focus fibrosis areas that differed in size (data not shown). Transduction with lenti-MiR-control did not change the fibrotic phenotype of the histological findings of BLM-treated mice, while lenti-miR-338* partially reversed the fibrotic phenotype induced by BLM (Figure 4B). Then, we aimed to determine whether SMO expression could be affected by miR-338* in vivo and if this change could be associated with the phenotype of pulmonary fibrosis. By using immuno-histochemistry, we investigated SMO expression in the lung slide. As expected, SMO expression was significantly increased in BLM-treated mice and did not change following the transduction of lenti-miR-control. However, its expression was greatly reduced in BLM-treated mice transduced with lenti-miR-338*. Taken together, over-expression of miR-338* reversed the fibrosis procedure of the lung through the down-regulation of SMO.
Figure 4.
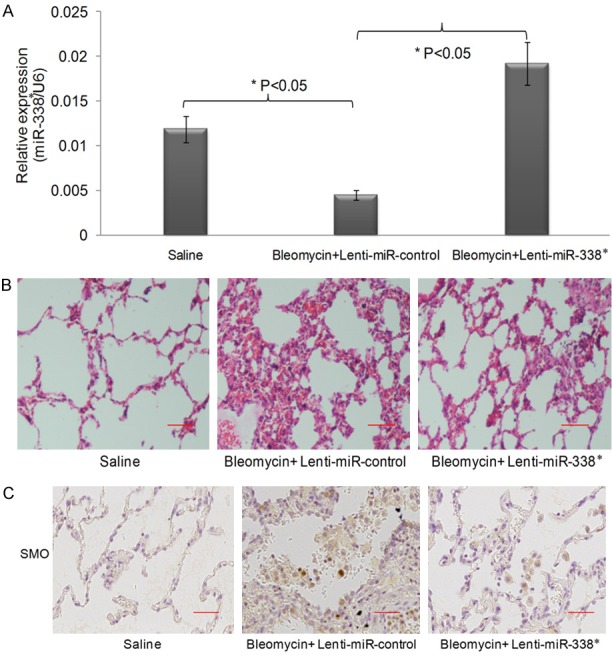
MiR-338 inhibits SMO expression to prevent bleomycin induced pulmonary fibrosis in vivo. A. The relative expression of miR-338* in mice treated with bleomycin to induce pulmonary fibrosis in vivo. The results showed that bleomycin decreased the expression of miR-338* in the lung of mice, while lentivirus mediated miR-338* infection increased the expression of miR-338*. B. Hematoxylin and eosin (H&E) staining for lung samples. C. The Immunofluorescence chemistry staining for the lung samples. The staining of lung samples showed that bleomycin induced pulmonary fibrosis in mice, while lentivirus mediated miR-338* infection reversed the pulmonary fibrosis morphology in mice.
Discussion
In this study, we found that miR-338* was down-regulated in bleomycin-induced pulmonary fibrosis. Moreover, SMO was shown to be the downstream target of miR-338* and silencing of SMO protected cell from TGF-β induced fibrosis in vitro by partially terminating the EMT procedure. Addi-tionally, over-expression of miR-338* overcame BLM-induced pulmonary fibrosis in vivo by down-regulating the expression of SMO. Taken together, our study demonstrated that miR-338* may target SMO to regulate the pathological process of pulmonary fibrosis by interfering with EMT, suggesting that miR-338* and SMO may serve as therapeutic targets for treating pulmonary fibrosis in the future.
Alveolar epithelial cell injury and subsequent uncontrolled repair are the major pathological observations in patients with IPF. Of these, EMT, which is thought to participate in the process of pulmonary fibrosis, is involved in the mechanism of injury and repair of lung epithelial cells. MiRs are small RNA molecules that play critical roles in various physiological and pathological processes such as cellular proliferation, tissue differentiation and repair [7]. Recently, miRs were demonstrated to be regulators of the EMT procedure and thus participate in the process of IPF [21,22]. Similar to a previous report, we also found that the expression of miR-338* was reduced in the BLM-treated pulmonary fibrosis mice in this study [18]. However, it does not fully explain the mechanism responsible for miR-338* regulating the fibrotic process.
The Hh pathway is confirmed to account for tumorigenesis in different sites [13,17], as well as lung fibrosis [18]. As a downstream molecule of the Hh pathway, the expression of SMO is manipulated by miR338-3p in tumor tissue [16]. Hence, it is reasonable to question whether the expression of SMO might be a direct target and tightly regulated by miR-338*, which has been shown to have reduced expression during pulmonary fibrosis in our study. As expected, SMO expression was elevated in both pulmonary fibrosis in vivo and TGFβ-treated cell lines in vitro in the current study. This finding is identical to previous reports of experiments that were conducted in vitro and/or in vivo [18,23,24].
Since the EMT procedure is regulated by miRs during IPF [21,22] and SMO is a direct target of miR-338*, confirmed by this study, we demonstrated that EMT was controlled by miR-338* by tightly regulating SMO expression. By knocking-down the expression of SMO in TGF-β-treated NuLi-1 cells in vitro, fibrotic-like changes of NuLi-1 were reversed. However, the over-expression of SMO induced fibrotic changes in the morphology of NuLi-1 whether treated with TGF-β or not. These fibrotic phenotypes were closely related to changes in the expression of EMT indicators, E-cadherin and vimentin, suggesting that SMO controls the process of EMT and thus contributes to the pathological changes of IPF. To the best of our knowledge, this is the first study to confirm that SMO participates in the patho-physiology process of IPF by regulating EMT.
In conclusion, we found that miR-338* was down-regulated in lung tissues from mice with BLM-induced pulmonary fibrosis. SMO, working as a downstream target of miR-338*, regulates the EMT procedure, thus contributing to alleviating cellular fibrosis in vitro. Our results indicate that miR-338* may be a potential therapeutic target for the treatment of pulmonary fibrosis in the future.
Acknowledgements
This study was supported by Grants from the National Natural Science Foundation of China (Grant No. 81370161, 81270152, 81470253), and the Fundamental Research Funds for the Central Universities (Grant No. 20620140732).
Disclosure of conflict of interest
None.
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, Bar-Joseph Z, Bitterman P, Blackbum MR, Bradford W, Brown KK, Chapman HA, Collard HR, Cosgrove GP, Deterding R, Doyle R, Flaherty KR, Garcia CK, Hagood JS, Henke CA, Herzog E, Hogaboam CM, Horowitz JC, King TE, Loyd JE, Lawson WE, Marsh CB, Noble PW, Noth I, Sheppard D, Olsson J, Ortiz LA, O’Riordan TG, Oury TD, Raghu G, Roman J, Sime PJ, Sisson TH, Tschumperlin D, Violette SM, Weaver TE, Wells RG, White ES, Kaminski N, Martinez FJ, Wynn TA, Thannickal VJ, Eu JP. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med. 2014;189:214–222. doi: 10.1164/rccm.201306-1141WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, Mikhail A, Hitchcock CL, Wright VP, Nana-Sinkam SP, Piper MG, Marsh CB. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187:397–405. doi: 10.1164/rccm.201205-0888OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milosevic J, Pandit K, Magister M, Rabinovich E, Ellwanger DC, Yu G, Vuga LJ, Weksler B, Benos PV, Gibson KF, McMillan M, Kahn M, Kaminski N. Profibrotic role of miR-154 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47:879–887. doi: 10.1165/rcmb.2011-0377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang S, Banerjee S, de Freitas A, Sanders YY, Ding Q, Matalon S, Thannickal VJ, Abraham E, Liu G. Participation of miR-200 in pulmonary fibrosis. Am J Pathol. 2012;180:484–493. doi: 10.1016/j.ajpath.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2007;96(Suppl):R40–44. [PubMed] [Google Scholar]
- 8.Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA? RNA. 2005;11:1753–1761. doi: 10.1261/rna.2248605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 10.Huang XH, Wang Q, Chen JS, Fu XH, Chen XL, Chen LZ, Li W, Bi J, Zhang LJ, Fu Q, Zeng WT, Cao LQ, Tan HX, Su Q. Bead-based microarray analysis of microRNA expression in hepatocellular carcinoma: miR-338 is downregulated. Hepatol Res. 2009;39:786–794. doi: 10.1111/j.1872-034X.2009.00502.x. [DOI] [PubMed] [Google Scholar]
- 11.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, Harris CC. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Liu X, Chen S, Wu J, Ye X, Xu L, Chen H, Zhang D, Tan R, Wang Y. Tectorigenin inhibits the in vitro proliferation and enhances miR-338* expression of pulmonary fibroblasts in rats with idiopathic pulmonary fibrosis. J Ethnopharmacol. 2010;131:165–173. doi: 10.1016/j.jep.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Coon V, Laukert T, Pedone CA, Laterra J, Kim KJ, Fults DW. Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol Cancer Ther. 2010;9:2627–2636. doi: 10.1158/1535-7163.MCT-10-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, Bernard K, Conklin JF, Szczepny A. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17:1504–1508. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 16.Huang XH, Chen JS, Wang Q, Chen XL, Wen L, Chen LZ, Bi J, Zhang LJ, Su Q, Zeng WT. miR-338-3p suppresses invasion of liver cancer cell by targeting smoothened. J Pathol. 2011;225:463–472. doi: 10.1002/path.2877. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad A, Maitah MY, Ginnebaugh KR, Li Y, Bao B, Gadgeel SM, Sarkar FH. Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J Hematol Oncol. 2013;6:77. doi: 10.1186/1756-8722-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolanos AL, Milla CM, Lira JC, Ramirez R, Checa M, Barrera L, Garcia-Alvarez J, Carbajal V, Becerril C, Gaxiola M, Pardo A, Selman M. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303:L978–990. doi: 10.1152/ajplung.00184.2012. [DOI] [PubMed] [Google Scholar]
- 19.Liang H, Gu Y, Li T, Zhang Y, Huangfu L, Hu M, Zhao D, Chen Y, Liu S, Dong Y, Li X, Lu Y, Yang B, Shan H. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014;5:e1238. doi: 10.1038/cddis.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Song X, Li Y, Han F, Gao S, Wang X, Xie S, Lv C. Low-dose paclitaxel ameliorates pulmonary fibrosis by suppressing TGF-beta1/Smad3 pathway via miR-140 upregulation. PLoS One. 2013;8:e70725. doi: 10.1371/journal.pone.0070725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oak SR, Murray L, Herath A, Sleeman M, Anderson I, Joshi AD, Coelho AL, Flaherty KR, Toews GB, Knight D, Martinez FJ, Hogaboam CM. A micro RNA processing defect in rapidly progressing idiopathic pulmonary fibrosis. PLoS One. 2011;6:e21253. doi: 10.1371/journal.pone.0021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huleihel L, Ben-Yehudah A, Milosevic J, Yu G, Pandit K, Sakamoto K, Yousef H, LeJeune M, Coon TA, Redinger CJ, Chensny L, Manor E, Schatten G, Schatten G, Kaminski N. Let-7d microRNA affects mesenchymal phenotypic properties of lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2014;306:L534–42. doi: 10.1152/ajplung.00149.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moshai EF, Wemeau-Stervinou L, Cigna N, Brayer S, Somme JM, Crestani B, Mailleux AA. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am J Respir Cell Mol Biol. 2014;51:11–25. doi: 10.1165/rcmb.2013-0154OC. [DOI] [PubMed] [Google Scholar]
- 24.Cigna N, Farrokhi Moshai E, Brayer S, Marchal-Somme J, Wemeau-Stervinou L, Fabre A, Mal H, Leseche G, Dehoux M, Soler P, Crestani B, Mailleux AA. The hedgehog system machinery controls transforming growth factor-beta-dependent myofibroblastic differentiation in humans: involvement in idiopathic pulmonary fibrosis. Am J Pathol. 2012;181:2126–2137. doi: 10.1016/j.ajpath.2012.08.019. [DOI] [PubMed] [Google Scholar]