

Abstract
Background
The incidence of lung adenocarcinoma (LUAD) is increasing worldwide with different prognosis even in early-stage patients. We aimed to identify a prognostic panel with multiple DNA methylation biomarkers to predict survival in early-stage LUAD patients of different racial groups.
Methods
The methylation array, pyrosequencing methylation assay, Cox regression and Kaplan-Meier analyses were conducted to build the risk score equations of selected probes in a training cohort of 69 Asian LUAD patients. The risk score model was verified in another cohort of 299 Caucasian LUAD patients in The Cancer Genome Atlas (TCGA) database.
Results
We performed a Cox regression analysis, in which the regression coefficients were obtained for eight probes corresponding to eight genes (AGTRL1, ALDH1A3, BDKRB1, CTSE, EFNA2, NFAM1, SEMA4A and TMEM129). The risk score was derived from sum of each methylated probes multiplied by its corresponding coefficient. Patients with the risk score greater than the median value showed poorer overall survival compared with other patients (p = 0.007). Such a risk score significantly predicted patients showing poor survival in TCGA cohort (p = 0.036). A multivariate analysis was further performed to demonstrate that the eight-probe panel association with poor outcome in early-stage LUAD patients remained significant even after adjusting for different clinical variables including staging parameters (hazard ratio, 2.03; p = 0.039).
Conclusions
We established a proof-of-concept prognostic panel consisting of eight-probe signature to predict survival of early-stage LUAD patients of Asian and Caucasian populations.
Electronic supplementary material
The online version of this article (doi:10.1186/s12929-016-0276-x) contains supplementary material, which is available to authorized users.
Keywords: Lung adenocarcinoma, DNA methylation array, Pyrosequencing, Risk score, Prognosis
Background
Lung cancer is the leading cause of cancer-related deaths with an increasing incidence of lung adenocarcinoma (LUAD) subtype worldwide [1]. Prognosis may vary in patients with the same stage tumor because cancer is characterized by genetic, epigenetic, and phenotypic changes that result in a tremendous variability in clinical behavior [2, 3]. Therefore, the development of additional molecular markers for survival prediction of LUAD is required.
DNA methylation, which usually occurs in CpG dinucleotides, is a major epigenetic modification in mammalian genome [4–6]. High-throughput methylation arrays are now available to determine DNA methylation levels of thousands of CpG sites, simultaneously [7–9]. This technology enables large-scale DNA methylation analysis to identify informative DNA methylation biomarkers in lung cancer [7, 10–16]. Many reports have demonstrated that each cancer subtypes such as lung adenocarcinoma and squamous cell carcinoma has its own methylation signature [12, 13, 15, 16].
Therefore, in the current study we focus on the development of survival predictors in early-stage LUAD patients by performing genome-wide methylation analysis and pyrosequencing quantitative methylation assay to select eight DNA methylation probes in a training cohort of 69 patients recruited in Taipei Veterans General Hospital (TVGH). We also included certain clinical parameters that are known to affect prognosis [2, 3, 17–19] along with the selected eight-probe panel to the Cox regression analysis. The relevance of our finding has been validated in a cohort of 299 patients as part of The Cancer Genome Atlas (TCGA) project.
Methods
Patients and tissue samples
A total of 69 surgically resected LUAD patients in early stage (stages I and II) were recruited from Taipei Veterans General Hospital (TVGH), after obtaining appropriate institutional review board permission (#98-03-18A) and informed consent from the patients. These LUAD patients with checked clinical data and sufficient amount of DNA available for successful genome-wide methylation and pyrosequencing quantitative methylation assays were defined as a training cohort. A validation cohort of 299 LUAD patients with clinical follow-up data and methylation microarray data available from TCGA were collected. The mean follow-up period for training cohort was 82 months (range 9–157 months) and for validation cohort was 37 months (range 12–242 months). The end of the follow-up in TVGH was defined as January 2016 and TCGA as April 2015. Patients with clinicopathological characteristics are shown in Table 1.
Table 1.
Characteristics of the lung adenocarcinoma patients included in the current study
| Cohort | TVGHa (%) | TCGAa (%) |
|---|---|---|
| N = 69 (100 %) | N = 299 (100 %) | |
| Age | ||
| < 65 year-old | 25 (36.2 %) | 124 (41.5 %) |
| ≥ 65 year-old | 44 (63.8 %) | 166 (55.5 %) |
| Stage | ||
| Stage IA | 13 (18.8 %) | 113 (37.8 %) |
| Stage IB | 42 (60.9 %) | 104 (34.8 %) |
| Stage IIA | 4 (5.8 %) | 32 (10.7 %) |
| Stage IIB | 10 (14.5 %) | 50 (16.7 %) |
| T stage | ||
| Stage 1 | 16 (23.2 %) | 122 (40.8 %) |
| Stage 2 | 51 (73.9 %) | 159 (53.2 %) |
| Stage 3 | 2 (2.9 %) | 18 (6.0 %) |
| N stage | ||
| N0 | 56 (81.2 %) | 236 (78.9 %) |
| ≥ N1 | 12 (17.4 %) | 57 (19.1 %) |
| M stage | ||
| M0 | 69 (100 %) | 299 (100 %) |
| ≥ M1 | 0 (0.0 %) | 0 (0.0 %) |
| Surgery | ||
| Lobectomy | 60 (87.0 %) | -b |
| Wedge resection | 8 (11.6 %) | -b |
| Segmentectomy | 1 (1.4 %) | -b |
| Chemotherapy | ||
| No | 51 (73.9 %) | -b |
| Yes | 15 (21.7 %) | -b |
| TKI treatment | ||
| No | 62 (89.9 %) | -b |
| Yes | 6 (8.7 %) | -b |
aTVGH: Taipei Veterans General Hospital; TCGA: The Cancer Genome Atlas
bInformation was not available for patients from TCGA
Genomic DNA extraction and sodium bisulfite conversion
Genomic DNA from primary tumor tissue samples of 69 patients from TVGH were extracted using proteinase K digestion and phenol-chloroform extraction. A total of 1 μg genomic DNA was used for bisulfite conversion using the EpiTect Bisulfite kit (Qiagen, Duesseldorf, Germany) according to the manufacturer’s protocols.
The genome-wide methylation analysis platform
The Illumina Infinium HumanMethylation27 BeadChip (27,578 CpG dinucleotides for 14,495 genes) was adapted for DNA methylation detection according to manufacturer’s manual. DNA methylation levels were reported as β-values by calculating the ratio of intensities between locus-specific methylated and unmethylated bead-bound probes. The β-value is a continuous variable, ranging from 0 (unmethylated) to 1 (fully methylated). The methylation array data can be viewed online under GEO accession number GSE83845.
Pyrosequencing assay
To quantify cytosine methylation in individual CpG sites of candidate methylation probes identified by methylation array, bisulfite-converted DNA was analyzed using a pyrosequencing system (PyroMark Q24, Qiagen, Hilden, Germany). Specific pyrosequencing primer and PCR primer were designed for “target” CpG sites in the probes to be analyzed. Pyrosequencing was carried out in accordance with the manufacturer’s protocol (Qiagen). The target CpG sites were evaluated by converting the resulting pyrograms to numerical values for peak heights. Primer sequences are listed in Additional file 1: Table S1, and the genomic map of the detected CpG sites are shown in Additional file 1: Figure S1.
Data processing and statistical analysis
Receiver operating characteristic (ROC) curve analysis was performed to determine the accuracy of the established CpG panel [area under the curve (AUC), sensitivity, and specificity]. The univariate and multivariate Cox regression analyses were conducted to explore the relationship between patient survival and several explanatory variables for defining the hazard ratio (HR) and confidence intervals (CI) of cancer death risk of variables using the Statistical Package for the Social Sciences version 17.0 (SPSS Inc., Headquarters Chicago, IL, USA). Overall survival curves were calculated according to the Kaplan-Meier method. p < 0.05 was considered statistically significant.
Results
Marker discovery in genome-scale DNA methylation dataset
In the marker selection phase of this study, we collected surgically dissected tumors of 69 early-stage LUAD patients from TVGH to form a training cohort for genome-wide methylation array analysis using Illumina Infinium HumanMethylation27 BeadChip. Procedures were performed as described below and shown in Fig. 1. First, we obtained 9384 qualified probes after removing potentially problematic probes, probes containing SNP, repeat sequencing, probes not in CpG island, probes in the X-chromosome and the non-differential probes with β-value greater than 0.9 or less than 0.1 in all samples after methylation analysis. Second, we selected 2815 informative probes with large variance of βtumor (top-30 % ranked) among the patients. Third, for each probe, a supervised principal components (Superpc) analysis [20] was applied to compare the survival distributions between patients with methylation levels above and below the mean level of all tumor tissues analyzed; 100 probes were chosen based on these tests. Fourth, since pyrosequencing assay is a highly sensitive method for detection of DNA methylation [12, 17, 21], we performed pyrosequencing methylation analyses. Of these probes, pyrosequencing was successfully designed and performed for 34 probes. A high concordance in quantifying the CpG methylation level was observed between DNA methylation array and pyrosequencing assay (Fig. 2). Fifth, eight specific candidate probes corresponding to eight genes showed significant correlation with survival by univariate Cox regression, including Angiotensin II receptor-like 1 (AGTRL1), Aldehyde dehydrogenase 1 family member A3 (ALDH1A3), Bradykinin receptor B1 (BDKRB1), Cathepsin E (CTSE), Ephrin A2 (EFNA2), NFAT activating protein with ITAM motif 1 (NFAM1), Semaphorin 4A (SEMA4A), and Transmembrane protein 129 (TMEM129) (Table 2). Sixth, we applied receiver operating characteristic (ROC) curve analysis to determine the diagnostic sensitivity and specificity of eight-probe panel in the validation cohort of TVGH patients. Finally, Kaplan-Meier method and multivariate Cox regression analyses were performed for the eight-probe panel in the TVGH patients and validated in 299 early-stage LUAD patients from TCGA datasets (as described below).
Fig. 1.
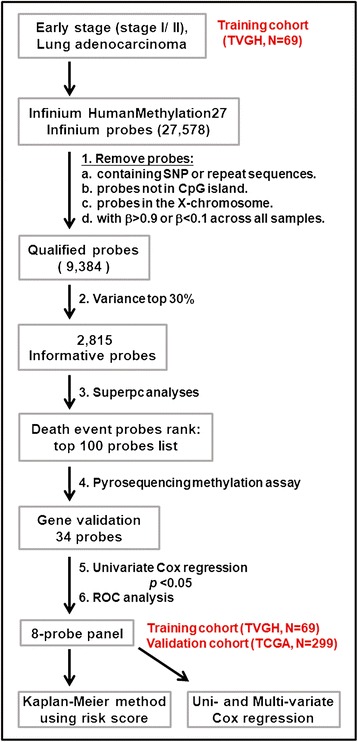
Flowchart of the probe selection and clinical validation procedures. Six steps were used to select the eight methylation gene probes from the methylation array in the training cohort of 69 LUAD patients from TVGH. The Kaplan-Meier survival analysis with the regression coefficients of eight probes was first performed to confirm the survival prediction of risk score calculation. The multivariate Cox regression was then performed to validate clinical performance of the eight-probe panel after adjusting for different clinical variables. The Kaplan-Meier survival analysis and multivariate Cox regression method were also performed in the validation cohort of 299 LUAD patients from TCGA database
Fig. 2.
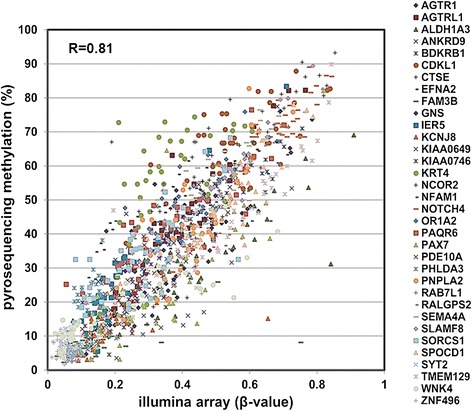
Correlation of methylation level between Illumina array and pyrosequencing method of top 34 methylated probes in early-stage LUAD patients. Dot-plot analyses show a high concordance of methylation level between pyrosequencing DNA methylation assay (Y-axis: %) and Illumina genome-wide methylation assay (X-axis: β value) and of 34 methylated probes. An average correlation coefficient was 0.81 (R = 0.81) among the probes
Table 2.
Univariate Cox model for 34 probes in the training cohort of LUAD by pyrosequencing methylation assay
| No. | probes IDa | gene symbol | p valueb | No. | probes IDa | gene symbol | p valueb |
|---|---|---|---|---|---|---|---|
| 1 | cg04878152 | AGTR1 | 0.680 | 18 | cg05973262 | NOTCH4 | 0.126 |
| 2 | cg25072179 | AGTRL1 c | 0.001 | 19 | cg16678925 | OR1A2 | 0.164 |
| 3 | cg27652350 | ALDH1A3 c | 0.007 | 20 | cg10046892 | PAQR6 | 0.386 |
| 4 | cg16787352 | ANKRD9 | 0.223 | 21 | cg11428724 | PAX7 | 0.444 |
| 5 | cg10528989 | BDKRB1 c | 0.021 | 22 | cg01431114 | PDE10A | 0.356 |
| 6 | cg16077929 | CDKL1 | 0.373 | 23 | cg13645078 | PHLDA3 | 0.877 |
| 7 | cg21478437 | CTSE c | 0.007 | 24 | cg24427660 | PNPLA2 | 0.443 |
| 8 | cg11885098 | EFNA2 c | 0.002 | 25 | cg09635067 | RAB7L1 | 0.679 |
| 9 | cg03158400 | FAM3B | 0.219 | 26 | cg10559803 | RALGPS2 | 0.175 |
| 10 | cg00626466 | GNS | 0.384 | 27 | cg15983538 | SEMA4A c | 0.030 |
| 11 | cg13228642 | IER5 | 0.275 | 28 | cg04275881 | SLAMF8 | 0.103 |
| 12 | cg01226811 | KCNJ8 | 0.564 | 29 | cg16415058 | SORCS1 | 0.265 |
| 13 | cg17536532 | KIAA0649 | 0.334 | 30 | cg15789095 | SPOCD1 | 0.811 |
| 14 | cg10150813 | KIAA0746 | 0.672 | 31 | cg22594309 | SYT2 | 0.944 |
| 15 | cg12610744 | KRT4 | 0.061 | 32 | cg21505886 | TMEM129 c | 0.004 |
| 16 | cg22820108 | NCOR2 | 0.546 | 33 | cg08108311 | WNK4 | 0.296 |
| 17 | cg17568996 | NFAM1 c | 0.002 | 34 | cg01184522 | ZNF496 | 0.560 |
aProbes ID is the CpG number of designated probe used in Illumina Human Methylation27 Bead Chip
bUnivariate Cox regression
cGenes in bold font indicated statistical significance (p < 0.05) thus were selected for further analyses
Sensitivity and specificity of the eight selected probes by ROC analysis
We examined the sensitivity and specificity of the eight selected probes by ROC curve analysis in the training cohort of 69 early-stage LUAD patients from TVGH. The area under the curve (AUC) of eight-probe together was 0.802 (Fig. 3a), indicating that the eight-probe signature showed good sensitivity and specificity in the ROC analysis. To assess the accuracy of the prognostic predictor panel, ROC curve analysis was performed on another randomly selected eight probes from the top 34 candidate probes. The AUC was 0.602 (Fig. 3b), suggesting a stronger prediction power of the specifically selected eight probes than the randomly selected probes. Thus, we defined this eight-probe signature as the prognostic predictor panel of early-stage LUAD.
Fig. 3.
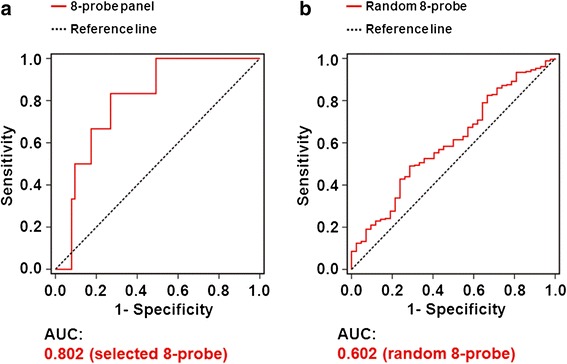
ROC curves of the prognostic predictor panel in the training cohort from TVGH. Sensitivity is indicated in the Y-axis, whereas 1 substrated by specificity (1-Specificity) is indicated in the X-axis. a The area under the curve (AUC) of ROC analysis for the eight selected probes panel. b The AUC of ROC analysis for the eight randomly selected genes
The risk score calculation and survival prediction of the eight-probe panel by Kaplan-Meier method
In the clinical validation phase, we first built the risk score for the eight selected methylation probes using the multivariate Cox regression analysis in the TVGH training cohort of 69 early-stage LUAD patients. These DNA methylation probe covariates were weighted by the regression coefficients to calculate the coefficient and hazard ratio for each patient. The risk score for each patient was derived from sum of methylation value of each probe multiplied by the corresponding coefficient, as following equation: risk score = AGTRL1 methylation value × (-0.015) + ALDH1A3 methylation value × (-0.023) + BDKRB1 methylation value × (-0.034) + CTSE methylation value × (0.022) + EFNA2 methylation value × (0.010) + NFAM1 methylation value × (-0.017) + SEMA4A methylation value × (-0.012) + TMEM129 methylation value × (-0.006). Example of risk score calculation for two patients is shown in Additional file 1: Figure S2.
Furthermore, we used the risk score calculation ranging from -1.03 to -4.95 to classify patients into two groups by median value of -2.63 in the TVGH training cohort of 69 early-stage LUAD patients (upper panel, Fig. 4a). The Kaplan-Meier overall survival analysis was performed to show the relative survival in each of the two groups identified by the risk score calculation (middle panel, Fig. 4a). Patients with high risk score indeed had a short median survival time (MST) of 58.9 months compared with other patients. The difference in the MST and 95 % confidence interval (CI) between the two groups was highly significant (lower panel, Fig. 4a). Therefore, the median risk score (as -2.63) was chosen as the cutoff value for survival prediction in the TVGH cohort.
Fig. 4.
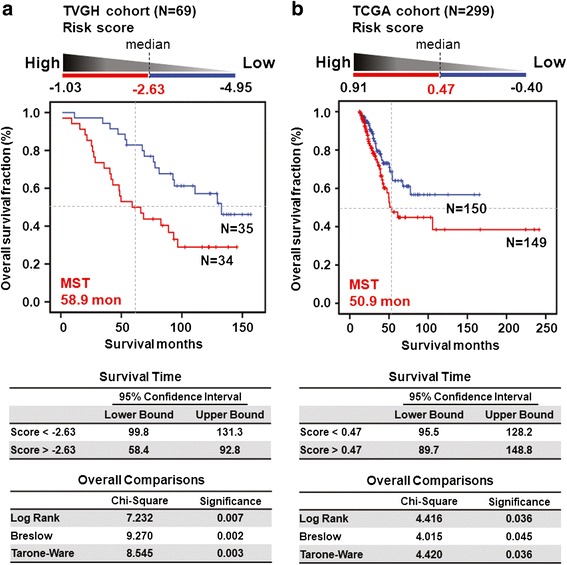
Survival risk score prediction based on the selected eight-probe in LUAD patients. a The risk score was used to classify 69 TVGH patients in the training cohort into two groups by median (as -2.63) (upper). The Kaplan-Meier overall survival analysis was performed to show the relative median survival time (MST) in two groups identified by the risk score calculation (middle). The 95 % confidence interval of survival time and p values of various methods are shown as indicated (lower). b The risk score, MST, and p values were analyzed in the validation cohort of 299 LUAD patients from TCGA database
We further applied our risk score model to determine whether our finding could be validated in another cohort of 299 early-stage LUAD patients whose follow-up data were available in TCGA project, and methylation level was also determined by the Infinium Methylation array. The risk score calculated with the median value (as 0.47) classified the 299 TCGA patients into two groups (upper panel, Fig. 4b). Such a calculation predicted a subset of patient with a high risk score showing poorer survival with MST of 50.9 months (middle panel, Fig. 4b) with statistical significance (lower panel, Fig. 4b). These results indicated that the prognostic predictor panel consisting of the selected eight-probe showed a strong prediction value in the TCGA validation cohort.
Univariate and multivariate Cox regression analysis of the eight-probe panel
To determine whether the eight-probe panel is an independent variable associated with poor survival of early-stage LUAD patients, we performed the univariate and multivariate Cox regression model in both TVGH and TCGA cohorts. The univariate Cox regression analysis revealed that patients with risk score > median of the eight-probe panel, stage IIA, stage IIB, or lymph node metastases had poor outcome (p = 0.009, HR = 2.37, 95 % CI = 1.24–4.53 for risk score > median of the eight-probe panel; Table 3). Notably, multivariate Cox regression analysis showed that the eight-probe panel correlated with a relative risk of death of 2.03 (p = 0.039), even after adjusting for the tumor staging and metastasis status (Table 3), suggesting that the eight-probe panel was an independent risk factor of poor outcome.
Table 3.
Univariate and multivariate Cox regression analyses of risk factors for cancer-related death in early-stage LUAD patients
| TVGH (N = 69)a | TCGA (N = 299)a | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristics | Univariate analysis | Multivariate analysisc | Univariate analysis | Multivariate analysisc | ||||
| HR (95 % CI)b | p-valueb | HR (95 % CI)b | p-valueb | HR (95 % CI)b | p-valueb | HR (95 % CI)b | p-valueb | |
| Eight-probe panel | ||||||||
| Risk < Median | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| Risk > Median | 2.37 (1.24–4.53) | 0.009 | 2.03 (1.04–3.98) | 0.039 | 1.66 (1.03–2.66) | 0.038 | 1.57 (0.96–2.57) | 0.073 |
| Gender | ||||||||
| Male | 1.00 | - | 1.00 | - | ||||
| Female | 1.43 (0.71–2.88) | 0.321 | - | - | 0.78 (0.49–1.26) | 0.315 | - | - |
| Stage | ||||||||
| Stage IA | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| Stage IB | 2.13 (0.73–6.16) | 0.164 | 2.01 (0.69–5.86) | 0.199 | 1.15 (0.63–2.11) | 0.642 | 1.17 (0.63–2.16) | 0.616 |
| Stage IIA | 8.76 (2.10–36.53) | 0.003 | 5.79 (0.72–46.58) | 0.099 | 2.17 (0.98–4.81) | 0.057 | 0.85 (0.21–3.38) | 0.817 |
| Stage IIB | 6.11 (1.81–20.64) | 0.003 | 3.65 (0.70–18.92) | 0.124 | 2.04 (1.07–3.91) | 0.031 | 0.91 (0.26–3.15) | 0.885 |
| T stage | ||||||||
| Stage 1–2 | 1.00 | - | 1.00 | - | ||||
| Stage 3–4 | 2.22 (0.53–9.28) | 0.277 | - | - | 0.62 (0.15–2.53) | 0.502 | - | - |
| T stage | ||||||||
| Stage 1 | 1.00 | - | 1.00 | - | ||||
| Stage 2 | 1.23 (0.56–2.68) | 0.613 | - | 1.28 (0.77–2.12) | 0.338 | - | ||
| Stage 3 | 2.59 (0.54–12.32) | 0.232 | - | - | 0.72 (0.17–3.07) | 0.657 | - | - |
| N stage | ||||||||
| N0 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| ≥ N1 | 3.95 (1.88–8.30) | 0.001 | 1.54 (0.35–6.91) | 0.571 | 2.27 (1.38–3.73) | 0.001 | 2.51 (0.74–8.43) | 0.138 |
| Chemotherapy | ||||||||
| No | 1.00 | - | ||||||
| Yes | 1.84 (0.92–3.69) | 0.084 | - | - | ||||
| TKI treatment | ||||||||
| No | 1.00 | - | ||||||
| Yes | 2.03 (0.79–5.26) | 0.144 | - | - | ||||
| Surgery | ||||||||
| Lobectomy | 1.00 | - | ||||||
| Wedge resection | 1.35 (0.53–3.45) | 0.536 | - | |||||
| Segmentectomy | 2.98 (0.40–22.31) | 0.288 | - | - | ||||
aTVGH: Taipei Veterans General Hospital; TCGA: The Cancer Genome Atlas
bCI, confidence interval; HR, hazard ratio. Bold values indicate statistical significance (p < 0.05)
cThe variables without significant HR in the univariate analysis were not included in the multivariate analysis
To further define the prognostic effects of the eight-probe panel in early-stage LUAD patients, univariate and multivariate Cox regression analyses were performed in the TCGA validation cohort of 299 early-stage LUAD patients. Univariate Cox regression analysis revealed that patients with the risk score > median of the eight-probe panel had poor outcome, with a relative risk of death of 1.66 (p = 0.038) (Table 3). However, the eight-probe panel showed a borderline significance by the multivariate analysis in the TCGA cohort.
Discussion
The incidence of LUAD is increasing worldwide [1]. Patients with the same stage of lung cancer may have different prognosis [22]. Development of prognostic markers is especially important in the patients with early-stage lung cancer, in whom clinical oncologists need selection factors to decide whether adjuvant therapy is necessary. In the present study, we develop a prognostic predictor panel for early-stage LUAD patients. This panel consists of eight DNA methylation probes corresponding to eight specific genes, including AGTRL1, ALDH1A3, BDKRB1, CTSE, EFNA2, NFAM1, SEMA4A, and TMEM129. The risk score calculated using the eight-probe panel served as an independent prognosis biomarker by Cox regression model and the multivariate analysis in our recruited patients. Therefore, the risk scores calculated from this eight-probe panel are valuable biomarkers for prognostic evaluation for early-stage LUAD patients to be tested in other cohorts.
Recently, Heller et al. identified a total of 12 genes that were differentially methylated in tumors compared with surrounding tissues in stage I, II or III Caucasian non-small cell lung cancer patients. Among the 12 genes, only the methylation patterns of HOXA2 and HOXA10 were independent prognostic factors in lung squamous cell carcinoma patients [15]. In addition, Esteller and the associates used methylation array to establish methylation profiles of stage I Caucasian non-small cell lung cancer and identified that methylation of two or more genes in HIST1H4F, PCDHGB6, NPBWR1, ALX1, and HOXA9 correlated with an increased risk of cancer recurrence [16]. Interestingly, HOXA9 promoter methylation was associated with high risk in stage I LUAD patients of two independently cohorts by another study [23]. To date, all studies that have been executed in an attempt to find markers for clinical use do not include patients from different racial groups. In our study, the prognostic predictor panel comprising eight DNA methylation biomarkers was an independent risk factor of poor outcome in Asian LUAD patients. We further applied our risk score model to determine whether our finding could be validated in another cohort of 299 early-stage LUAD patients whose follow-up data were available in TCGA project. The new coefficient and hazard ratio were defined according to the methylation value of the eight probes given in TCGA database of these patients. The Kaplan-Meier overall survival analysis showed that TCGA patients with risk score greater than median value had a shorter MST compared with other patients (Fig. 4b). However, the result of multivariate Cox regression was only close to significance in the Caucasian LUAD patients (Table 3). One of the limitations of the current TCGA study is that we are unable to acquire the data on treatment or surgery performed on the TCGA patients (Table 1). We believe that these results could be improved after including data from more patients when they are available in TCGA dataset or by validating in other cohorts of Caucasian LUAD patients.
The identification of the eight probes that can predict the clinical outcome in patients may reveal causes of the cancer development and tumorigenesis. For example, Angiotensin II receptor-like 1 (AGTRL1) and Bradykinin receptor B1 (BDKRB1) are G-protein-coupled receptors (GPCRs). GPCRs, which represent by far the largest family of cell-surface molecules involved in signal transduction, have recently emerged as crucial players in tumor growth and metastasis [24]. AGTRL1 is Apelin receptor. Apelin is an angiogenic factor secreted by tumor cells in order to promote the formation of new vessels necessary for tumor growth [25]. In addition, crosstalk between BDKRB1 and EGFR has been shown to maintain tumor growth in the breast cancer [26]. Aldehyde dehydrogenase 1 family, member A3 (ALDH1A3) is the retinoic acid biosynthesis enzyme, and plays a major role in the detoxification of aldehydes generated by alcohol metabolism and lipid peroxidation. Promoter hypermethylation of ALDH1A3 has been reported to be a prognostic marker for lung cancer, gastric cancer, and invasive bladder cancer [27–30]. Cathepsin E (CTSE) prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble trail from tumor cell surface [31]. Ephrin A2 (EFNA2), which belongs to ephrins family, regulates cell adhesion, motility, survival, proliferation, and differentiation. Semaphorins 4A (SEMA4A) suppresses endothelial cell migration and proliferation in vitro and angiogenesis in vivo mediated by vascular endothelial growth factor [32]. Further characterization of the probes validated in our panel could help to dissect the mechanism of LUAD tumorigenesis and progression.
The advantages of our prognostic predictor panel are as follows. First, the methylation level of the eight probes could be analyzed by DNA methylation array or pyrosequencing in patients. Second, the stepwise multivariate Cox regression analysis, in which the coefficients were obtained for the selected eight probes, could generate the risk score equations specifically for the cohort of patients to be tested. Third, any newly recruited patients could be assigned into risk groups once the risk score equations are determined. Therefore, the prognostic predictor panel could calculate the risk score not only in the Asian but also in the Caucasian LUAD patients. However, some technical limitations such as sample collection and preprocessing as well as experimental procedures of DNA methylation array or pyrosequencing assay need to be controlled to avoid batch effects. In addition, clinical variables such as adjuvant therapy and surgical methods may affect outcome prediction. Large-scale, multicenter and prospective studies are necessary to validate our risk score model in early-stage LUAD patients.
Conclusions
Our study provides a proof-of-concept prognostic prediction panel consisting of eight methylated probes that are closely associated with survival in the early-stage LUAD patients. This prediction panel could be useful in stratifying patients according to the Cox-model and risk score before further treatment for early-stage LUAD patients who in dire need of intensive care.
Abbreviations
AGTRL1, Angiotensin II receptor-like 1; ALDH1A3, Aldehyde dehydrogenase 1 family member A3; BDKRB1, Bradykinin receptor B1; CI, confidence intervals; CTSE, Cathepsin E; EFNA2, Ephrin A2; HR, hazard ratio; LUAD, lung adenocarcinoma; MST, median survival time; NFAM1, NFAT activating protein with ITAM motif 1; SEMA4A, Semaphorin 4A; Superpc, supervised principal components; TCGA, the cancer genome atlas; TMEM129, Transmembrane protein 129; TVGH, Taipei Veterans General Hospital
Acknowledgements
The authors thank Ms. Ching-Hsi Lin, Mr. Chi-Huei Hsiung, and Mr. Chien-Hsun Lin for technical support.
Funding
This study was supported by Taiwan National Science Council (98-3112-B-006-014-CC1, 99-3112-B-006-013-CC1), Taiwan Ministry of Science and Technology (103-2627-B-006-007), and Taiwan Ministry of Health and Welfare (105-TDU-B-211-124-003).
Availability of data and materials
Data and materials related to this work are available upon request.
Authors’ contributions
IYK performed the experiments. IYK and JJ did the data analysis in this study. LHH, HSH, WWL provided clinical samples. IYK, JJ and YCW wrote the paper. All authors read and approved the manuscript. YCW and WWL obtained funding.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors approve the manuscript for publication.
Ethics approval and consent to participate
Surgically resected LUAD patients were recruited from TVGH, after obtaining appropriate institutional review board permission (#98-03-18A) and informed consent from the patients.
Additional file
The primers used for pyrosequencing analysisa. Figure S1. The genomic maps of the selected genes and CpG sites in DNA methylation biomarker studies. cg_number is the CpG number of selected probes from methylation array. TSS: transcription start site. The black arrows (▼) indicate the detected CpG sites in Infinium array and the white arrows (△) indicate the sites in pyrosequencing. The nucleotides relative to TSS are shown. Figure S2. Risk score calculation and risk group assignment of two example patients. A Coefficient (coef) of genes and clinical variables were established by multivariate Cox regression model. A patient’s risk score was derived from sum of each probe methylation level multiplied by its corresponding coefficient. The equations used are as follows: Risk score = AGTRL1 methylation value × (-0.015) + ALDH1A3 methylation value × (-0.023) + BDKRB1 methylation value × (-0.034) + CTSE methylation value × (0.022) + EFNA2 methylation value × (0.010) + NFAM1 methylation value × (-0.017) + SEMA4A methylation value × (-0.012) + TMEM129 methylation value × (-0.006). B The risk score ranging from -1.03 to 4.95 was used to classify patients into two groups by the median value (as -2.63). Patient A with risk score of -1.0318 was assigned to the high risk group and patient B with -4.4262 was assigned to the low risk group. (DOCX 360 kb)
Contributor Information
I-Ying Kuo, Email: yihyng@msn.com.
Jayu Jen, Email: jayujen1021@gmail.com.
Lien-Huei Hsu, Email: polohsu33@gmail.com.
Han-Shui Hsu, Email: hsuhs@vghtpe.gov.tw.
Wu-Wei Lai, Phone: +886-6-2353535, Email: wwlai@mail.ncku.edu.tw.
Yi-Ching Wang, Phone: +886-6-2353535, Email: ycw5798@mail.ncku.edu.tw.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 4.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 5.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 6.Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–8. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 7.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–93. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones PA. At the tipping point for epigenetic therapies in cancer. J Clin Invest. 2014;124:14–6. doi: 10.1172/JCI74145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma X, Wang YW, Zhang MQ, Gazdar AF. DNA methylation data analysis and its application to cancer research. Epigenomics. 2013;5:301–16. doi: 10.2217/epi.13.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai Z, Lakshmanan RR, Zhu WG, Smiraglia DJ, Rush LJ, Frühwald MC, et al. Global methylation profiling of lung cancer identifies novel methylated genes. Neoplasia. 2001;3:314–23. doi: 10.1038/sj.neo.7900162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S, et al. Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res. 2009;69:6315–21. doi: 10.1158/0008-5472.CAN-09-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Son JW, Jeong KJ, Jean WS, Park SY, Jheon S, Cho HM, et al. Genome-wide combination profiling of DNA copy number and methylation for deciphering biomarkers in non-small cell lung cancer patients. Cancer Lett. 2011;311:29–37. doi: 10.1016/j.canlet.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 13.Kwon YJ, Lee SJ, Koh JS, Kim SH, Lee HW, Kang MC, et al. Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. J Thorac Oncol. 2012;7:20–33. doi: 10.1097/JTO.0b013e3182307f62. [DOI] [PubMed] [Google Scholar]
- 14.Park JY, Kim D, Yang M, Park HY, Lee SH, Rincon M, et al. Gene silencing of SLC5A8 identified by genome-wide methylation profiling in lung cancer. Lung Cancer. 2013;79:198–204. doi: 10.1016/j.lungcan.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heller G, Babinsky VN, Ziegler B, Weinzierl M, Noll C, Altenberger C, et al. Genome-wide CpG island methylation analyses in non-small cell lung cancer patients. Carcinogenesis. 2013;34:513–21. doi: 10.1093/carcin/bgs363. [DOI] [PubMed] [Google Scholar]
- 16.Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013;31:4140–7. doi: 10.1200/JCO.2012.48.5516. [DOI] [PubMed] [Google Scholar]
- 17.Vaissiere T, Hung RJ, Zaridze D, Moukeria A, Cuenin C, Fasolo V, et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009;69:243–52. doi: 10.1158/0008-5472.CAN-08-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang R, Chu M, Zhao Y, Wu C, Guo H, Shi Y, et al. A genome-wide gene-environment interaction analysis for tobacco smoke and lung cancer susceptibility. Carcinogenesis. 2014;35:1528–35. doi: 10.1093/carcin/bgu076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forrest LF, Adams J, White M, Rubin G. Factors associated with timeliness of post-primary care referral, diagnosis and treatment for lung cancer: population-based, data-linkage study. Br J Cancer. 2014;111:1843–51. doi: 10.1038/bjc.2014.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:E108. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vasiljević N, Wu K, Brentnall AR, Kim DC, Thorat MA, Kudahetti SC, et al. Absolute quantitation of DNA methylation of 28 candidate genes in prostate cancer using pyrosequencing. Dis Markers. 2011;30:151–61. doi: 10.1155/2011/157829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deppermann KM. Lung cancer screening--where we are in 2004 (take home messages) Lung Cancer. 2004;45:S39–42. doi: 10.1016/j.lungcan.2004.07.994. [DOI] [PubMed] [Google Scholar]
- 23.Robles AI, Arai E, Mathé EA, Okayama H, Schetter AJ, Brown D, et al. An integrated prognostic classifier for stage I lung adenocarcinoma based on mRNA, microRNA, and DNA methylation biomarkers. J Thorac Oncol. 2015;10:1037–48. doi: 10.1097/JTO.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 25.Sorli SC, Le Gonidec S, Knibiehler B, Audigier Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene. 2007;26:7692–9. doi: 10.1038/sj.onc.1210573. [DOI] [PubMed] [Google Scholar]
- 26.Molina L, Matus CE, Astroza A, Pavicic F, Tapia E, Toledo C, et al. Stimulation of the bradykinin B(1) receptor induces the proliferation of estrogen-sensitive breast cancer cells and activates the ERK1/2 signaling pathway. Breast Cancer Res Treat. 2009;118:499–510. doi: 10.1007/s10549-009-0314-4. [DOI] [PubMed] [Google Scholar]
- 27.Kim YJ, Yoon HY, Kim JS, Kang HW, Min BD, Kim SK, et al. HOXA9, ISL1 and ALDH1A3 methylation patterns as prognostic markers for nonmuscle invasive bladder cancer: Array-based DNA methylation and expression profiling. Int J Cancer. 2013;133:1135–42. doi: 10.1002/ijc.28121. [DOI] [PubMed] [Google Scholar]
- 28.Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 29.Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N, et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006;3:e486. doi: 10.1371/journal.pmed.0030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita S, Tsujino Y, Moriguchi K, Tatematsu M, Ushijima T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2'-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci. 2006;97:64–71. doi: 10.1111/j.1349-7006.2006.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawakubo T, Okamoto K, Iwata J, Shin M, Okamoto Y, Yasukochi A, et al. Cathepsin E prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble TRAIL from tumor cell surface. Cancer Res. 2007;67:10869–78. doi: 10.1158/0008-5472.CAN-07-2048. [DOI] [PubMed] [Google Scholar]
- 32.Toyofuku T, Yabuki M, Kamei J, Kamei M, Makino N, Kumanogoh A, et al. Semaphorin-4A, an activator for T-cell-mediated immunity, suppresses angiogenesis via Plexin-D1. EMBO J. 2007;26:1373–84. doi: 10.1038/sj.emboj.7601589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and materials related to this work are available upon request.