

Abstract
Background
Disease progression in multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), as one of its animal models, is characterized by demyelination and neuronal damage in white and gray matter structures, including the hippocampus. It is thought that dysfunction of the hippocampus, a primary locus of learning and memory consolidation, may contribute to cognitive impairment in MS patients. Previously, we reported an increased generation of hippocampal neuronal progenitors in the acute stage of EAE, whereas the microenvironmental signals triggering this process remained uninvestigated.
Results
In the present study, we used the Wnt signaling reporter mouse Axin2LacZ, to elucidate the molecular mechanisms underlying the activation of the hippocampal neurogenic niche upon autoimmune neuroinflammation. Histological and enzymatic examinations of β-gal during the disease course of EAE, allowed us to survey hippocampal Wnt/β-catenin activity, one of the key signaling pathways of adult neurogenesis. We found that Wnt signaling is transiently upregulated in the acute stage of disease, consistent with a timely induction of canonical Wnt ligands. The enhancement of signaling coincided with hippocampal neuronal damage and local expression of immune cytokines such as TNFα and IFNγ, implicating the role of the inflammatory milieu in activation of the Wnt/β-catenin pathway. Supporting this finding, we show that transient exposure to pro-inflammatory cytokine TNFα triggers Wnt signaling in hippocampal organotypic slice cultures. Importantly, inflammation-mediated activation of the Wnt/β-catenin pathway was associated with enhanced neurogenesis in vitro and in vivo, indicating its potential role in hippocampal tissue regeneration and repair.
Conclusions
This study raises the possibility that enhancement of Wnt signaling may support neurogenic processes to cope with neuronal deficits upon immune-mediated neuroinflammation.
Electronic supplementary material
The online version of this article (doi:10.1186/s13024-016-0117-0) contains supplementary material, which is available to authorized users.
Keywords: Wnt, Hippocampus, Neurogenesis, EAE, Multiple sclerosis
Background
A large body of research highlights the involvement of hippocampal pathology in cognitive disability that is commonly observed in patients with multiple sclerosis (MS), an inflammatory demyelinating and neurodegenerative disease of the central nervous system (CNS) [1, 2]. Structural changes in the hippocampus were shown to be associated with MS-related memory impairment [3] and may occur prior to cognitive dysfunction [4, 5], suggesting a therapeutic opportunity for reversing memory decline in MS patients (reviewed in [5–7]). Examinations of neurobiological alterations in post-mortem brain tissue indicate frequent and extensive hippocampal demyelination [8] resulting in alterations of synaptic function, axonal transport [9] and neuronal loss in the Ammon’s horn 1–3 (CA1-3) regions [9, 10]. Furthermore, axonal demyelination was shown to be accompanied by drastic changes in the expression of specific neuronal genes [9] and neuronal miRNA [11] suggesting disturbance of molecular pathways involved in physiological homeostasis of hippocampal tissue. Experimental autoimmune encephalomyelitis (EAE), an animal model of MS, mimics many aspects of hippocampal pathology and its clinical expression in the human disease such as demyelination, alteration in synaptic plasticity and transmission [12, 13], neuronal loss [12], cognitive deficits [14] and thus, becomes an important model for the characterization of mechanisms involved in hippocampal neuropathology and MS-related memory deficits. Our previous data identified adult hippocampal neurogenesis, an important player in cognition [15], as a primary target of autoimmune-mediated neuroinflammation [16]. The analysis of hippocampal gene expression in acute and chronic stage of EAE revealed alterations in the transcriptional profile of genes relevant for key neurogenic pathways in the adult brain, including those of Wnt signaling [16]. Considering the potential role of the Wnt pathway in tissue renewal and regeneration [17, 18], we here examined the impact of autoimmune neuroinflammation on hippocampal Wnt/β-catenin activity by using Axin2lacZ/+ reporter mice. We found that T cell-mediated autoimmune neuroinflammation leads to hippocampal neuronal damage occurring early in disease progression and may potentially trigger the activation of Wnt signaling via upregulation of a specific set of Wnt ligands. Transient increase in the Wnt activity in the acute phase was associated with increased proliferation and generation of doublecortin-positive (Dcx+) neuronal precursor cells. Further, we identified the inflammatory mediator tumor necrosis factor alpha (TNFα) as a potent enhancer of Wnt signaling activity and subsequently, the proliferation of progenitor cells in the hippocampal neurogenic niche.
Results
Increased Wnt activity in the acute phase of EAE
In the adult brain, Wnt signaling pathway is persistently active in the hippocampus and is of high importance for homeostatic neurogenesis [19]. To evaluate the impact of autoimmune-mediated neuroinflammation on hippocampal Wnt/β-catenin signaling we induced EAE in Axin2lacZ/+ reporter mice by immunization with an encephalitogenic CNS self-antigen, myelin oligodendrocyte glycoprotein (MOG35–55) peptide. Like other feedback inhibitors in various pathways, Axin2 is induced in response to the Wnt signaling cascade and is widely used as readout of Wnt/β-catenin activity. Mutant mice, lacking one functional Axin2 allele, are born normal and upon immunization developed typical non-remitting chronic EAE with a disease onset at 11–12 days post immunization (d.p.i.). (Fig. 1a). Histological examinations revealed the occurrence of typical perivascular infiltrates, predominantly in the white matter of the spinal cord (Additional file 1: Figure S1a-b). These infiltrates were characterized by increased Iba1 immunoreactivity (the marker for microglia and macrophages). Interestingly, microglia activation was evident in the hippocampus and particularly in the hippocampal subgranular zone (SGZ). In this region, microglia were localized in close proximity to SOX2/GFAP-double positive cells with prototypical morphology of radial glia-like cells (RGCs or type-1 cell) and horizontal progenitors, both the primary stem cells of the adult dentate gyrus (DG) (Fig. 1b and Additional file 1: Figure S1d) [20, 21]. Despite the rare occurrence, we also found perivascular lesions enriched with Iba1 immunoreactive cells located in close proximity to the SGZ, the germinative zone of the hippocampal DG (Additional file 1: Figure S1c). Analysis of β-galactosidase (β-gal) activity in Axin2lacZ/+ mice showed the enhancement of Wnt signaling in the hippocampal tissue at days 20 and 30 after disease induction (1.31–1.78 fold), while at day 50 its activity resumed to the levels observed in control CFA/PTX-immunized mice (Fig. 1c). In the spinal cord, increased Wnt signaling was observed in all disease stages examined (Fig. 1c), whereas no significant changes were detected in the frontal/motor cortex (Additional file 1: Figure S1e), the area that was virtually not affected by inflammation. Only modest increase in β-gal activity was observed in the cerebellum (Additional file 1: Figure S1f). Of note, only trace amounts of β-gal activity were detected in naive or EAE tissue from wt (Axin2+/+) mice (data not shown), proving the accuracy of the β-gal measurements and the minor impact of senescence-associated processes. Additionally, we confirmed the results of the β-gal assay in the spinal cord and different parts of the brain by qPCR analysis of the Axin2 gene expression in the acute stage of EAE (Additional file 1: Figure S1g). The distribution pattern of LacZ expression in the DG examined by X-gal staining revealed increased signal intensity mostly in cells of the granular layer including the SGZ and in some cells of the hilus (Fig. 1d). β-gal activity in the hippocampal and spinal cord tissues positively correlated with clinical disease score in individual mice (Additional file 1: Figure S1.h, Pearson correlation, r = 0.58; p = 0.018 and r = 0.52; p = 0.036, respectively). Taken together, our results revealed a brain area-specific and disease severity-dependent upregulation of Wnt signaling in EAE mice.
Fig. 1.
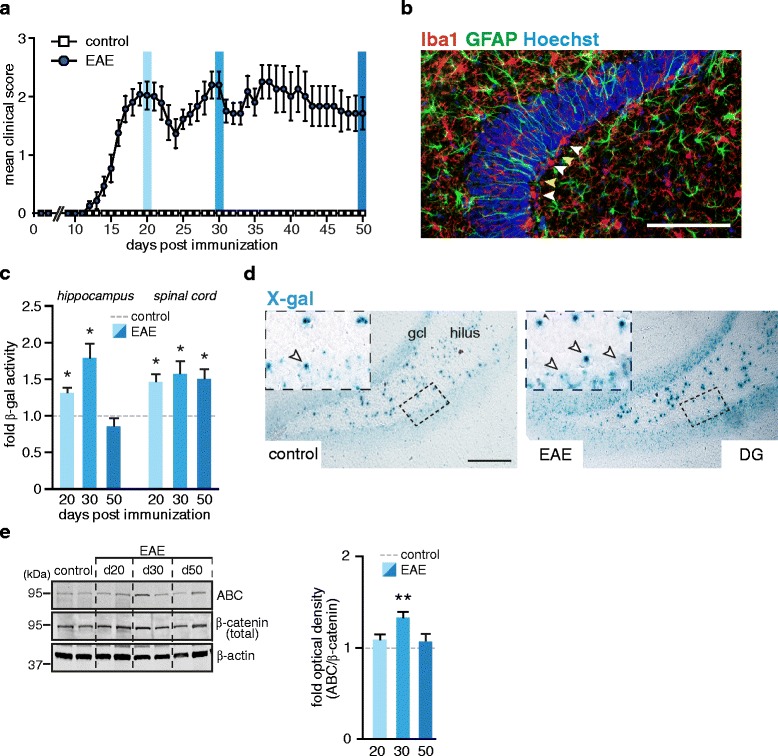
Analysis of Wnt activity in EAE. a Disease course in Axin2lacZ/+ mice with chronic EAE. EAE was induced by immunization with MOG35–55. Control group (CFA) were injected with an adjuvant cocktail without antigen. Data are shown as a mean clinical score ± SEM. Blue columns indicate the time points when mice were sacrificed for analysis: days 20, 30 and 50 post immunization. b Representative image of Iba1, GFAP co-immunostaining in hippocampal section of an EAE mouse at day 30. White arrow heads indicate reactive microglia Iba1+ (red) located in close proximity to radially oriented GFAP+ cells (green; yellow arrow heads) resembling radial glia progenitor cells of the SGZ. Nuclei were counterstained with Hoechst (blue). Scale bar: 100 μm. c β-gal assay of hippocampal and spinal cord tissue. Histogram represents mean + SEM of fold-changes of β-gal activity in EAE mice relative to respective controls (CFA) set as 1. Day 20 (EAE, n = 6; CFA, n = 3); day 30 (EAE, n = 4; CFA, n = 3); day 50 (EAE, n = 8; CFA, n = 4). Two-tailed, unpaired Student’s t-test, *p < 0.05. d Upregulation of Axin2 expression in the DG of EAE mice. Expression of lacZ and thus activation of Axin2 promoter were detected by X-gal staining on hippocampal sections from EAE mice (day 20) and respective controls (CFA). Representative images are shown. Top insets show magnified cells of the outlined SGZ region. Arrow heads mark X-gal positive cells. Gcl, granular cell layer. Scale bar, 100 μm. e Immunoblot analysis of β-catenin in the hippocampus of EAE and control (CFA) animals. Hippocampal proteins were extracted and processed for Western blotting with antibodies against active β-catenin (ABC) and total β-catenin. β-actin was probed for analysis of protein loading. Right histogram represents mean + SEM of fold-changes of normalized optical densities in EAE mice relative to controls (CFA) set as 1. Day 20 (n = 6); day 30 (n = 4); day 50 (n = 6) and control CFA mice (n = 6). Two-tailed, unpaired Student’s t-test, *p < 0.05 and ** p < 0.01
Next, we examined the activation state of Wnt signaling mediators in naive and EAE mice. The ligation of Wnts inducing the canonical pathway leads to stabilization (de-phosphorylation) of β-catenin via inactivation (phosphorylation at Ser9) of GSK-3β, a member of the β-catenin destructive complex. Supporting the results of the enzymatic assay, we found a significant increase in active β-catenin (ABC)/total β-catenin ratio at day 30 of EAE mice in comparison to their respective controls (Fig. 1e). Taken together, our results indicate a transient upregulation of hippocampal Wnt signaling in acute stages of EAE (20–30 d.p.i.). In contrast to this, sustained increase in Wnt activity was observed in the spinal cord through all EAE stages examined (Fig. 1c), which was correlated with a large number of inflammatory foci (Additional file 1: Figure S1a-b).
Induction of Wnt ligands in passive EAE
The Wnt gene family encodes for 19 cysteine-rich secreted signaling molecules. To determine the particular Wnt ligands activated in the hippocampi of EAE mice, we performed Wnt gene expression profiling analysis. To exclude possible confounders associated with an active immunization process, this examination was performed in the passive model of EAE induced by adoptive transfer of myelin specific T cells (Fig. 2a). As a first result, we noticed a transient upregulation of hippocampal Axin2 on the transcriptional level (Fig. 2b). This observation extends our findings from actively immunized Axin2lacZ/+ mice and is indicative of enhanced Wnt signaling in the acute stage of disease independent from immunization process. Notably, induction of Axin2 messenger RNA (mRNA) expression has occurred neither prior to the onset of disease (day six after adoptive transfer) nor in the chronic stage (day 50) (Fig. 2b). At day 20, EAE mice exhibited higher mRNA levels of a number of Wnt genes relevant for the canonical β-catenin/TCF pathway (Wnt2, Wnt3, Wnt3a, Wnt8b, Wnt9a and Wnt16; fold inductions ranging from 2 to 6) and those signaling through β-catenin-independent mechanisms (Wnt4, Wnt5a, Wnt5b; fold inductions ranging from 1.5 to 3.5) (Fig. 2c). Other Wnt ligands found to be expressed in the hippocampus (Wnt7a, Wnt7b, Wnt10a, Wnt11) exhibited no significant differences compared to controls (Additional file 2: Figure S2a). We also elucidated the gene expression profile of up-regulated Wnt ligands in other CNS regions of EAE mice. In the acute stage, we found an induction of Wnt2, Wnt3, Wnt5a, Wnt5b in the spinal cord tissue and Wnt5a in the cerebellum (Additional file 2: Figure S2b). As it was observed in actively-immunized mice (Fig. 1c), the transcriptional levels of Wnts in the chronic stage of passive EAE returned to the levels detected in control animals (injected with PBS) confirming the transient profile of enhanced Wnt signaling (Fig. 2b, c). Consistent with previous reports in other CNS regions [22, 23], our data revealed that the acute phase of EAE is associated with the induction of several Wnt ligands in the hippocampus.
Fig. 2.
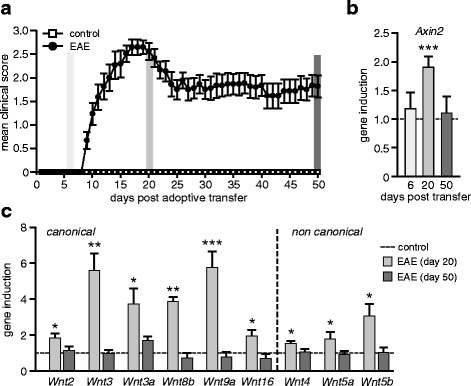
Gene expression analysis of Wnts in the hippocampus of EAE mice. a Disease course in passively transferred EAE. Female SJL/J mice received activated encephalitogenic T lymphocytes derived from immunized donors and re-stimulated with encephalitogenic PLP139–151 peptide. Control mice were injected with PBS. Data are shown as mean clinical score ± SEM. Grey columns indicate the time points when mice were sacrificed for analysis: days 6, 20 and 50 after adoptive transfer. b qPCR analysis of Axin2 gene expression in the hippocampus of mice with passively transferred EAE. Data represent mean of fold-changes + SEM of gene expression in EAE mice relative to respective controls set as 1. Expression of Axin2 was normalized to Gapdh. Day 6 (EAE, n = 5; control, n = 4); day 20 (EAE, n = 6; control, n = 8); day 50 (EAE, n = 12; control, n = 11). Two-tailed, unpaired Student’s t-test, *** p < 0.001. c qPCR analysis of selected Wnts genes in the hippocampus at early (day 20) and chronic (day 50) stages of passive EAE. Histogram represents mean + SEM of fold-changes relative to control group set as 1. Gene expression was normalized to Gapdh. Day 20 (EAE, n = 4–6; control, n = 4); day 50 (EAE, n = 8–13; control, n = 7–11). Two-tailed, unpaired Student’s t-test, * p < 0.05, ** p < 0.01 and *** p < 0.001
Upregulation of Wnt activity correlated with neuronal injury and inflammatory processes
Upregulation of Wnt signaling pathway members was observed in early stages of various pathologies associated with inflammation-mediated tissue injury [17, 18]. To take a closer look at hippocampal neuronal injury in early and late stages of EAE, we analyzed the expression of the axonal marker neurofilament-H (NF-H) and the postsynaptic density protein-95 (PSD-95). The protein levels of PSD-95 were diminished throughout all stages of disease, reflecting the reduction in the number of hippocampal synapses (Fig. 3a). We also found a persistently decreased protein level of NF-H (Fig. 3a), indicating that early neuronal injury is not compensated for in the chronic stage of non-remitting EAE (Fig. 1a). Early neuronal damage was associated with active inflammatory processes, as suggested by an elevated number of Iba1-positive cells (Fig. 3b-c), immunoreactivity for activation markers MHC class I (Fig. 3b), CD68 (Fig. 3e) and significant upregulation of gene transcription of inflammatory mediators (TNFα, IFNγ and interleukin-1 beta (IL-1β)) (Fig. 3f). Of note, strong positive correlation was observed between the transcriptional level of Axin2 and TNFα mRNA in individual mice (Additional file 2: Figure S2.c, Pearson correlation, r = 0.845; p < 0.001).We also found a significant induction of mRNA levels of transforming growth factor 1 beta (TGFβ1) (Fig. 3f) and Smad7 (Additional file 2: Figure S2d), the negative feedback regulator of TGFβ signaling. Upregulation of hippocampal TGFβ signaling in early stages of EAE is consistent with its CNS specific role in disease initiation [24]. In contrast to the significant increase in microglia number in the acute stage, the number of astrocytes remained unaffected by neuronal injury; however, it was significantly increased in the chronic stage (Fig. 3d). Together these data indicate that the upregulation of Wnt activity described above coincides with occurrence of neuronal damage and inflammatory processes at the early stage of disease, whereas in the chronic stage it returned to the basal level.
Fig. 3.
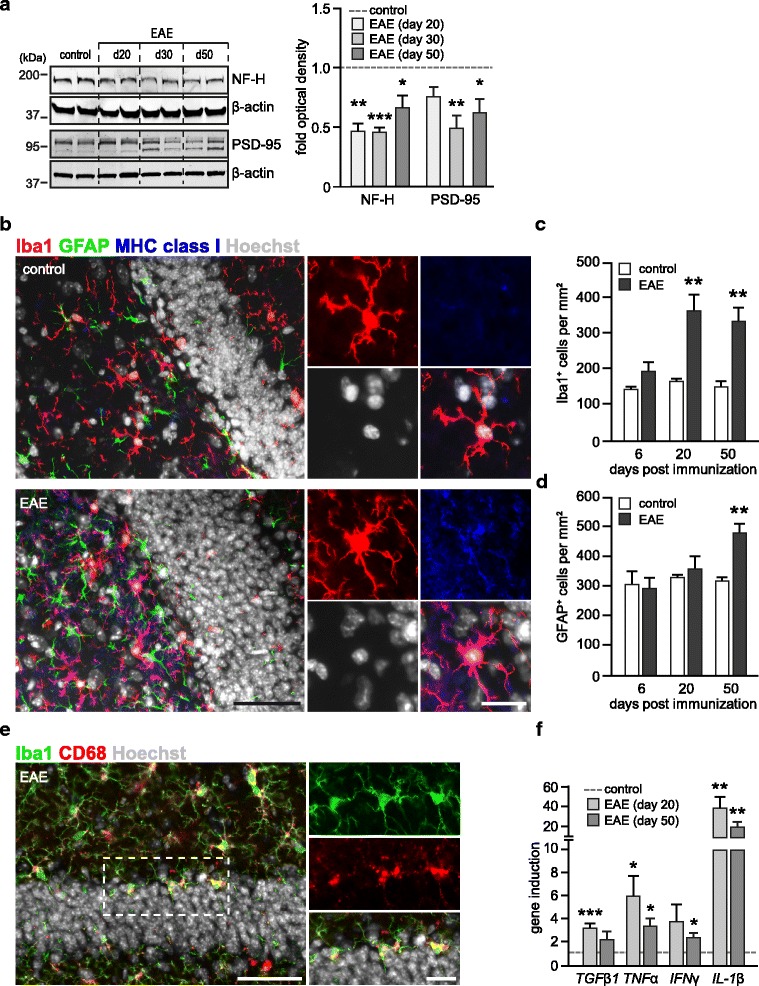
Neuronal damage and neuroinflammation in the hippocampus of EAE mice. a Immunoblot analysis of neuronal proteins in the hippocampus of Axin2lacZ/+ EAE and control (CFA) animals. Hippocampal proteins were extracted and processed for Western blotting with antibodies against neurofilament-H (NF-H) and postsynaptic protein 95 (PSD-95). β-actin was probed for analysis of protein loading. Right histogram represents mean + SEM of fold-changes of normalized optical densities in EAE mice relative to controls (CFA) set as 1. Day 20 (n = 6); day 30 (n = 4); day 50 (n = 6) and control CFA mice (n = 6). Two-tailed, unpaired Student’s t-test, *p < 0.05 and ** p < 0.01 b Representative image of Iba1, GFAP and MHC class I co-immunostaining in a hippocampal section of an EAE mouse and a healthy control at day 20. The insets on the right show magnified Iba1+ macrophage/microglia cells (red) co-labelled with MHC class I (blue). Nuclei were counterstained with Hoechst (grey). Scale bar: 50 μm and 10 μm. c-d Quantification of Iba1+ (c) and GFAP+ (d) cells in the DG of EAE (n = 4) and control (n = 4) mice. Data represents mean + SEM. Two-tailed, unpaired Student’s t-test, * p < 0.05. e Representative image of Iba1, CD68 co-immunostaining in the DG of EAE mouse at day 20. The insets on the right show magnified microglia cells (green) co-labelled with the activation marker CD68 (red). Nuclei were counterstained with Hoechst (grey). Scale bar: 50 μm and 10 μm. f qPCR analysis of gene expression of inflammatory cytokines in the hippocampus at early (day 20) and chronic (day 50) stages of EAE. Histogram represents mean + SEM of fold-changes relative to control (CFA) group set as 1. Expression was normalized to Gapdh. Day 20 (EAE, n = 3–6; control, n = 3–8); day 50 (EAE, n = 9–10; control, n = 6–7). Two-tailed, unpaired Student’s t-test, * p < 0.05, ** p < 0.01 and *** p < 0.001
Wnt signaling promotes neurogenic processes in the DG
In the adult brain, the Wnt/β-catenin pathway regulates proliferation and self-renewal of hippocampal stem/progenitor cells [19]. Therefore, we next asked if the increased Wnt activity in EAE may trigger proliferation and generation of hippocampal neuronal precursor cells. First, in mice with passively induced EAE, we examined the gene expression of pro-neurogenic transcription factors, known to be regulated by β-catenin/TCF transcriptional activity. We found that the increase of Wnt activity during the early stage of disease (day 20) correlates with an upregulation of Neurogenic differentiation 1 (NeuroD1), prospero homeobox 1 (Prox1), distal-less homeobox 2 (Dlx2) and lymphoid enhancer-binding factor 1 (Lef1) mRNA levels (Fig. 4a). Next, we used the BrdU labeling approach to mark dividing progenitors in the hippocampus and to trace the cell fate of newborn cells. Considering the early induction of Wnt signaling (Fig. 1c), we administered BrdU at 15 d.p.i. for five consecutive days, followed by a period of 2-weeks without BrdU treatment (Fig. 4b). We found a significant increase in the total number of label-retaining cells in the DG of EAE mice (14 ± 2 cells/mm) in comparison to control mice (10 ± 1 cells/mm). Similarly, the number of newly generated neuroblasts, measured by co-labeling of BrdU+-retaining cells with the neuronal progenitor marker Dcx, was increased twofold in EAE mice (8 ± 2 cells/mm in EAE mice; 4 ± 1 cells/mm in control mice) (Fig. 4c-d). Taken together, these results support our previous findings indicating an enhanced de novo generation of hippocampal neuronal progenitors in EAE mice [16] and are consistent with the notion that augmented Wnt activity in the hippocampus is relevant for activation of neurogenesis in acute disease stages.
Fig. 4.
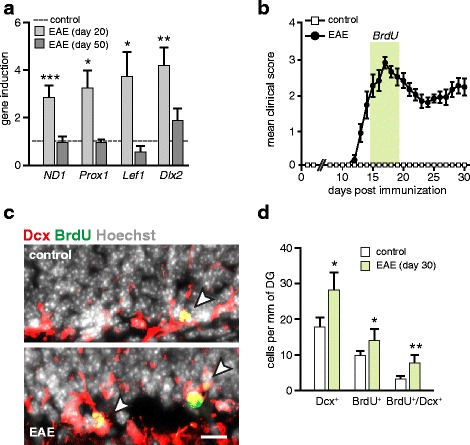
Hippocampal neurogenesis is increased in the acute phase of EAE. a Induction of pro-neuronal transcription factors in the hippocampus of mice with passive EAE. qPCR analysis of the Wnt-dependent genes (NeuroD1 (ND1), Prox1, Lef1) and Dlx2, a marker of transient amplifying neuroblasts. Histogram represents mean + SEM of fold-changes in gene expression of EAE mice relative to control (PBS) group set as 1. Gapdh was used as an endogenous reference. Day 20 (EAE, n = 4–8; control, n = 4–8); day 50 (EAE, n = 4–10; control, n = 4–11). Two-tailed, unpaired Student’s t-test, * p < 0.05, ** p < 0.01 and *** p < 0.001. b Disease course of C57/B6 EAE mice immunized with encephalitogenic MOG35–55. Control group (CFA) were injected with an adjuvant cocktail without antigen. Data are shown as a mean clinical score ± SEM. In acute phase of disease (days 15–20; highlighted by green line) EAE and respective control mice received daily i.p. injections of BrdU (50 mg/kg body weight). After a 2-weeks chase period without administration of BrdU mice were sacrificed for histological assessment (day 30). c Histological analysis of proliferating cells in the DG of EAE mice. Immunostaining for BrdU (green) and Dcx (red) in hippocampal sections. Arrow heads indicate BrdU+/Dcx+ co-labeled neuronal progenitors. Nuclei were counterstained with Hoechst (grey). Scale bar, 50 μm. d The frequency of BrdU label-retaining cells in the DG is increased in mice with EAE (day 30; n = 3 mice; 18–28 sections per mouse) as compared to control mice (n = 3 mice; 17–28 sections per mouse). Data is shown as mean + SEM of positive cells per mm of the DG. Two-tailed, unpaired Student’s t-test,* p < 0.05 and ** p < 0.01
Effects of TNFα on the Wnt signaling and hippocampal neurogenesis
Though the induction of Wnt signaling was previously reported in the context of various tissue damage paradigms [17, 18], the mechanisms triggering the Wnt pathway remain unknown. Therefore, we used the Axin2lacZ/+ reporter mice to characterize the possible contribution of particular cytokines, known to be secreted by activated microglia, to promote Wnt activity in neural tissue.
We switched our experimental setup to hippocampal slice cultures, which represent a useful model to study neurogenic processes ex vivo [25]. We focused on the role of TNFα and IFNγ, because their expression was significantly induced in the hippocampus of EAE mice (Fig. 3c) and because of independent findings on their potential contribution in tissue regeneration during demyelinating pathology [26–28]. Similar to freshly isolated hippocampal tissue (Fig. 1d), the pattern of Wnt activity in cultured slices examined by X-gal staining was mostly confined to the neuronal compartment of the granular layer (Additional file 3: Figure S3a). Treatment with TNFα or Wnt3a (used as a positive control [29, 30]) for 6 h and subsequent incubation of Axin2lacZ/+ slices in cytokine-free media for 24 h led to a significant upregulation of Wnt activity (1.56 ± 0.20 fold and 2.39 ± 0.18 fold, respectively) (Fig. 5a). The increase of Wnt signaling was limited to this particular immune cytokine, as the treatment with IFNγ did not have a similar effect. We then investigated whether TNFα may have a direct effect on the induction of Axin2 in cultured hippocampal progenitor cells. However, the treatment with TNFα was not associated with upregulation of Axin2 transcription (Additional file 3: Figure S3b). In contrast, Wnt3a potently induced Axin2 gene expression in these cells (Additional file 3: Figure S3b). This result led us to the hypothesis that enhancement of Wnt signaling in slice cultures is mediated via an indirect effect of the cytokine on other cell types. To test this, we separated hippocampal astrocytes (GLAST+) and microglia (CD11b+) from mixed glial cultures using microbeads-based sorting procedure and exposed them to TNFα. Analysis of gene expression of Wnt ligands found to be upregulated in acute stage (Fig. 2c) revealed that TNFα significantly induced the expression of Wnt2, Wnt3 and Wnt5a in astrocyte cultures and Wnt3 and Wnt5a in microglia cultures (Additional file 3: Figure S3c). We further investigated the effect of inhibition of endogenous Wnt ligand secretion on the induction of Axin2 in hippocampal slice cultures. Treatment with TNFα enhanced Axin2 expression (Fig. 5b), supporting our results of the β-gal assay (Fig. 5a). Co-treatment with IWP-2 inhibitor, that inactivates porcupine and suppresses the secretion of Wnt ligands [31], potently abrogated the positive effect of TNFα (Fig. 5b). Taken together, these data suggest a role of Wnt ligands in TNFα-mediated upregulation of hippocampal Wnt signaling.
Fig. 5.
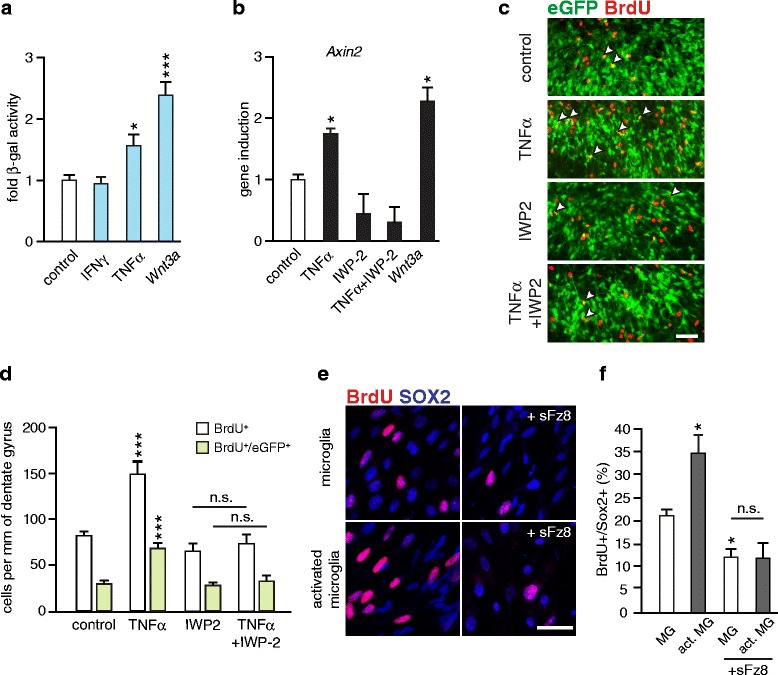
Analysis of Wnt activity in organotypic slice cultures (OSCs). a β-gal assay. Hippocampal Axin2lacZ/+ OSCs were treated for 6 h with Wnt3a (20 ng/ml), TNFα (1 ng/ml) or IFNγ (100 U/ml) and the medium was replaced with fresh cytokine-free medium for additional 24 h. Histogram represents mean + SEM of fold-changes of β-gal activity after treatment with Wnt3a (n = 15), TNFα (n = 14) or IFNγ (n = 14) relative to controls (PBS, n = 20) set as 1. Two-tailed, unpaired Student’s t-test, * p < 0.05 and *** p < 0.001. b qPCR analysis of the Axin2 gene expression. OSCs were treated as described above. IWP-2 (4 μM) was present in the media during the whole experiment. Histogram represents mean + SEM of fold-changes in Axin2 gene expression after treatment with Wnt3a (n = 3), TNFα (n = 4) or TNFα with IWP-2 (n = 3) relative to control (PBS, n = 4) OSCs set as 1. Gapdh was used as an endogenous reference. Two-tailed, unpaired Student’s t-test, * p < 0.05. c-d Histological analysis of proliferating cells in the DG of hippocampal NestineGFP OSCs. BrdU was administered to slice culture for 24 h prior to treatment with cytokines (performed as described for Fig. 5a-b). c Representative images show the immunostaining for BrdU (red) and eGFP (green). Arrow heads indicate BrdU+/eGFP+ co-labeled hippocampal progenitors. Scale bar, 50 μm. d Inhibition of Wnts secretion with IWP-2 (4 μm) abrogates the proliferative effect of TNFα on mitotic activity of hippocampal eGFP+ progenitors. Data are shown as mean + SEM of BrdU+ or BrdU+/eGFP+ co-labeled cells per mm of the DG. Control (PBS, n = 20 slices); TNFα (n = 9 slices), IFNγ (n = 11 slices) and Wnt3a (n = 9 slices); Two-tailed, unpaired Student’s t-test, * p < 0.05, ** p < 0.01 and *** p < 0.001. e-f TNFα - activated hippocampal microglia promotes the proliferation of hippocampal progenitors in vitro. Microglia cultured on transwell inserts were primed by TNFα (1 ng/ml) for 4 h. Transwells were set onto the hippocampal progenitor cultures in proliferating medium containing 5 ng/ml of bFGF in the presence or absence of soluble Frizzled-8 (Fz8; 1 μg/ml). After 24 h dividing cells were labelled with BrdU (2 h) and cells were fixed for immunocytochemistry. e Representative images of SOX2+ (blue) hippocampal progenitors stained for BrdU (red). Scale bar, 25 μm. f Quantification of BrdU-retaining SOX2+ cells. Data are shown as mean of four experiments + SEM. Two-tailed, unpaired Student’s t-test,* p < 0.05
We next asked whether TNFα affects hippocampal neurogenesis and whether Wnt ligands contribute to this effect. To assess the proliferation rate of hippocampal progenitors in NestineGFP hippocampal slice cultures, we labeled mitotically active cells with BrdU prior to 6 h treatment with the individual cytokines. After a 24-h chase in cytokine-free media the quantification of BrdU-retaining cells was examined. Treatment with TNFα (Fig. 5c-d) or Wnt3a, but not IFNγ (Additional file 3: Figure S3d-e), led to a significant increase in the number of BrdU+/eGFP+ neural progenitor cells. Inhibition of Wnt ligand secretion by IWP-2 suppressed the proliferative effect of TNFα (Fig. 5c-d). We further confirmed these results using co-culture experiments. For this approach we used hippocampal microglia because of its prominent activation pattern during the EAE progression (Fig. 3b-c). CD11b-sorted hippocampal microglia cultured on transwell inserts were activated with TNFα for 6 h. After several washing steps, transwells were set onto hippocampal progenitor cultures in proliferating conditions in the presence or absence of soluble Frizzled-8-Fc protein (sFz8). This recombinant protein contains a conservative cysteine-rich region (CRD) and was efficiently used to block Wnt ligand binding in hippocampal progenitor cultures [30]. After 24 h, mitotic cells were labeled with BrdU (2 h treatment). TNFα -primed microglia significantly enhanced mitotic activity of SOX2+ progenitor cells (Fig. 5e-f). Blockade of Wnt signaling with sFz8 decreased the proliferating rate to the level observed in progenitors co-cultured with non-activated microglia in the presence of sFz8. Taken together, these results are consistent with the observation that TNFα promotes Wnt signaling to enhance the initial steps of the hippocampal neurogenic program. In part, this effect is mediated through microglia-dependent Wnt ligand production, supporting the pro-neurogenic role of activated microglia in the hippocampus [32–34].
Discussion
The discovery of Wnt pathway activity in the adult CNS in physiological and pathological conditions has raised new questions about the mechanism of its activation. Moreover, it is of therapeutic interest whether the unique involvement of the Wnt pathway in developmental processes (such as neurogenesis, oligodendrogenesis, synaptogenesis and axonal guidance) may be useful for facilitation of tissue renewal and repair.
In the present study we made use of Axin2lacZ/+ reporter mice to monitor the dynamic changes in hippocampal Wnt activity upon acute and chronic stages of EAE, an animal model of MS. Our major finding is that CNS pathology, relevant for acute tissue injury and inflammation, is associated with a transient increase of hippocampal Wnt signaling. Enhanced Wnt/β-catenin activity was correlated with transient upregulation of canonical Wnt ligand (Wnt2, Wnt3, Wnt3a, Wnt8b and Wnt9a) mRNA levels. We also show that TNFα, an inflammatory cytokine upregulated in the hippocampus of EAE mice (Fig. 3d), can be considered as a potent factor contributing to Wnt signaling activation. Furthermore, the induction of Wnt activity led to increased proliferation of neuronal progenitors and generation of Dcx+ neuronal precursor cells (also known as Type2b). Taken together, our study revealed a link between neuroinflammation and Wnt-dependent activation of hippocampal neurogenesis regarding the tissue repair program.
Our finding that activation of Wnt signaling is mediated by active inflammatory processes is in line with previous reports on MS pathology. Using different biochemical techniques, the upregulation of Wnt ligands (Wnt2, Wnt7a: microarray data [35]); Wnt3a: proteomic analysis [36]) and Wnt pathway members such as Axin2 (histological examination [37]), GSK-3β, β-catenin and Tcf4 (proteomic analysis [36]) was demonstrated in active plaques in the white matter and was absent in chronic silent plaques or normal-appearing white matter. In the EAE model, transient induction of Wnt1 was demonstrated in the subventricular zone (SVZ) of the lateral ventricle [23].
We found that the upregulation of Wnt/β-catenin activity in the hippocampus is associated with an induction of several canonical Wnts involved in self-renewal and cell fate determination of stem/progenitor cells, synaptic plasticity, axon guidance and tissue homeostasis (reviewed in [38, 39]). Thus, Wnt3 and Wnt3a are potent activators of self-renewal and neuronal fate in the adult hippocampus [19, 29] and cultured neural progenitor cells [30]. Wnt2 enhances neurogenesis in the developing brain [40] and its induction in the dentate gyrus after electroconvulsive seizures (ECS) [41] was reported to be associated with increased hippocampal neurogenesis [42]. Wnt8b regulates dorsal thalamic neurogenesis during zebrafish development [43] and was also shown to enhance the proliferation of cultured rat adult hippocampal progenitors [30]. Expression of Wnt16a (human analogue of mouse Wnt16) was found in the brain [44]. However, its role in the CNS remains to be examined. Considering the transient induction of Wnt16 and activation of Wnt/β-catenin pathway at the early stage of osteoarthritis [45], it is plausible to assume that Wnt16 mediates the response to neural tissue injury via promotion of β-catenin signaling. Remarkably, in addition to their role in neural plasticity, Wnts likely participate in neuroimmune interactions (reviewed in [46]).
Consistent with previous reports demonstrating the upregulation of different components of canonical Wnt pathway in various models of acute spinal cord pathology (such as EAE [22], spinal cord injury [47] and lysolecithin-induced demyelination [37]), we found the enhancement of Wnt/β-catenin signaling in the spinal cord of Axin2lacZ/+ EAE mice. Notably, in this part of the CNS Wnt activity was increased throughout the course of disease and correlated with a large number of inflammatory foci (Additional file 1: Figure S1b), indicating ongoing active inflammatory processes. Thus, it is conceivable that the new waves of inflammation-mediated damage trigger the persistent activation of Wnt signaling in the spinal cord.
In line with the idea that acute inflammation enhances Wnt signaling, we showed that TNFα is a potent inducer of Wnt activity in hippocampal slice cultures. This finding is consistent with previous reports demonstrating the interplay between those two signaling pathways in other cellular contexts [48–51]. Notably, increased proliferation and induction of Wnt genes (such as of Wnt3, Wnt5a, β-catenin) was observed upon TNFα-induced trans-differentiation of human mesenchymal stem cells towards the neural lineage [52]. Taking advantage of hippocampal slice cultures as a useful model to study neurogenic processes ex vivo [25], we also demonstrated that TNFα-dependent activation of the Wnt pathway induces the proliferation of Nestin+ progenitor cells in the DG,and this effect is mediated via production of Wnt ligands. We further consider hippocampal microglia as the responder cells to TNFα-dependent enhancement of Wnt signaling, although other targets of TNFα may also mediate its effect on adult neurogenesis. While the mechanism remains to be elucidated, our data is in line with the role of TNFα-TNFR2 signaling in hippocampal neurogenesis in healthy mice [53] and upon various CNS pathologies. Thus, in the model of status epilepticus, deficiency for TNFR2 drastically abrogates the number of newly generated neurons in the hippocampus [54], though not in the SVZ [55]. Complementary to these findings, infusion of neutralizing antibodies against TNFα had an anti-proliferative effect on hippocampal progenitors in a rat model of stroke [56]. In cultured progenitor cells, TNFα potentially triggers the proliferation of neural stem/progenitor cells [57–59].
Understanding the role of TNFα in MS is complex and has been re-considered in the last years [60, 61]. While anti-TNFα therapy has been used successfully to treat various autoimmune pathologies, it was proven to be deleterious in MS patients [62]. The aggravation of EAE in TNFR2-deficient mice, particularly in the chronic phase [63], may indicate a principal involvement of the TNFα-TNFR2 axis in processes of regeneration and tissue repair. Supporting this idea, recent findings demonstrated the beneficial effect of TNFα/TNFR2 signaling on expansion of the endogenous pool of oligodendrocyte progenitors and subsequent remyelination in a toxin-induced demyelinating model [26].
Activation of neurogenesis was reported in various models of CNS injuries and diseases [64, 65]. In MS brains, immature neurons were found in a subpopulation of subcortical white matter lesions, which may indicate an activation of neurogenic processes serving to compensate neuronal deficits [66]. However, the origin of these cells is unknown. In contrast to this, specification of stem/progenitor cells seems to be shifted towards the glial lineage in the SVZ of MS patients compared to the healthy brain, leading to a diminished generation of Dcx+ neuroblasts [67]. Increased proliferation and enhanced generation of immature neurons in the acute stage of EAE was previously reported by our [16] and other groups [68, 69]. Enhancement of neurogenesis in EAE is transient [16, 68] and seems to be specific to the hippocampal neurogenic niche, whereas in the SVZ, the generation of neuroblasts is reduced [23].
Our data raised the possibility that enhancement of Wnt activity may trigger hippocampal neurogenesis in order to replenish damaged neurons. Regarding this, it was shown that lentiviral-based overexpression of Wnt3a in the striatum enhances neurogenesis and neuronal differentiation, leading to an improved neuronal function in the model of focal ischemic injury [70]. In addition to its neurogenic role, Wnts may promote axonal regeneration [71], synaptic transmission [72] and neuronal differentiation [73] in the injured CNS. Prolonged activation of Wnt signaling by Wnt-activating small molecule potentiator-1 (WASP-1) infusion into the hippocampus improves cognitive function in healthy adult mice and rescues memory loss in a mouse model of Alzheimer’s disease [72]. Our data support these findings, which already raise a considerable interest in Wnts regarding the development of novel therapeutic approaches to treat neuronal injury.
Conclusion
Taken together, our data argue for a role of inflammation in transient activation of hippocampal Wnt signaling upon autoimmune mediated injury. Induction of Wnts can be accounted as a key factor triggering endogenous repair by engaging the activation of neuronal progenitors in the hippocampal neurogenic niche. Further experiments are needed to test whether therapeutic boosting of Wnt activity leads to tissue regeneration and thus provides a strategy to improve hippocampal function.
Methods
Mice
The following mice were used for experiments: 6 weeks old female SJL/J mice were purchased from Janvier (Le Genest-St-Isle, France). NestineGFP+ mice [74] were provided by the animal facility of the University of Düsseldorf. In these transgenic mice the second intron enhancer of the rat nestin gene was placed upstream of the minimum promoter of heat shock protein 68 (HSP68) fused to eGFP cDNA and a polyadenylation signal. Axin2lacZ/+ [75] were received from the European Mouse Mutant Archive (EMMA). In Axin2lacZ/+ mice the lacZ gene, containing a nuclear localization signal, was introduced in frame to the endogenous Axin2 (also known as conductin or Axil) by replacing exon 2 [75]. Axin2 is a direct target of TCF/LEF1-mediated transcriptional activation that encodes an inhibitor of the Wnt signaling pathway. All mice were housed in the animal research facility of the University of Düsseldorf under specific pathogen free conditions, a dark/light cycle of 12 h, a stable temperature of 22–24 °C and unlimited access to food and water.
EAE induction
All experimental procedures were conducted following the guidelines and protocols approved by the local animal welfare committee (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV); under protocol numbers G388-11 (active EAE in Axin2lacZ/+mice), G197-09 (BrdU injections in EAE mice), G363-09 (passive EAE in SJL/J)) and follow the animal research: reporting of in vivo experiments (ARRIVE) criteria and EAE induction guideline [76]. For passive EAE, female SJL/J mice were subcutaneously immunized with 200 μg of recombinant proteolipid protein peptide (PLP139–151) (Pepceuticals) supplemented with 800 μg heat-inactivated Mycobacterium tuberculosis (strain H37RA, Difco) emulsified in 200 μl Complete Freund’s Adjuvant (CFA, Sigma-Aldrich) per mouse. On day 10 post immunization (p.i.), spleen and lymph node cells were isolated, and the resulting single-cell suspension was re-stimulated with PLP139–151 (10 μg/mL) in RPMI 1640 medium supplemented with 10 % fetal calf serum (FCS), penicillin/streptomycin, glutamate, and 2-mercaptoethanol (all from Invitrogen Life Technologies). After four days in culture, cells were harvested and injected intraperitoneal (i.p.) into naive female SJL/J mice (3 × 107 cells per mouse). Control mice received a single i.p. injection of PBS. Animals were weighed and scored daily, according to following clinical scale: 0 (no clinical signs), 1 (tail plegia), 2 (abnormal gait), 3 (hind limb paralysis), 4 (complete paralysis) to 5 (death or euthanasia) with intervals of 0.5 [16]. Mice were euthanized either on day 6, 20 or 50 post-transfer. Hippocampi were dissected and stored at −80 °C for total RNA and protein isolation.
To induce active EAE in the Axin2lacZ/+ mouse strain, animals were immunized by subcutaneous injection with 200 μg of recombinant myelin oligodendrocyte glycoprotein (MOG35–55) peptide (Pepceuticals) in CFA (Sigma-Aldrich), supplemented with 800 μg heat-inactivated Mycobacterium tuberculosis (strain H37RA, Difco). Intraperitoneal injections of 200 ng pertussis toxin (PTX, Sigma-Aldrich) were performed on the day of immunization and on day 2 [16]. The control group was treated with CFA only and with 200 ng pertussis toxin on day 0 and day 2. Mice were euthanized either on day 20, 30 or 50 post immunization.
β-Galactosidase assay
Tissue or organotypic slices isolated from Axin2lacZ/+ mice were lysed on ice for 15 min in triton-containing lysis buffer (Applied Biosystems) and cleared by centrifugation at 10,000 g for 10 min at 4 °C. Then protein concentration was determined by BCA protein assay kit (Interchim). 50 μg of protein were applied for measuring β-galactosidase (β-gal) activity in triplicates using the “Galactostar Chemiluminescence Kit” (Applied Biosystems) in a white 96-well NUNC plate (Thermo Scientific) at room temperature (RT) for 2 h using a microplate reader (TECAN). The maximum of luminescence intensity for each individual sample was used to calculate mean of triplicate measurements. Lysates of corresponding β-gal-negative brain tissues were used to calculate background intensity.
Western blotting
Hippocampal tissue isolated from EAE mice was lysed on ice for 15 min in RIPA buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1 % (v/v) NP40, 0.5 % (v/v) sodium deoxycholate, 0.1 % (v/v) SDS) with protease and phosphatase inhibitors (Roche). Lysates were cleared by centrifugation at 14,000 g for 20 min at 4 °C and protein concentration was determined using a BCA protein assay Kit (Interchim). Protein samples were loaded on 8–16 % gradient “Mini-Protean TGX” gel (Biorad). Blotting onto a nitrocellulose membrane was performed using the “Trans-Blot Turbo System” (Biorad) for 7 min at 25 Volts. Afterwards, membranes were incubated with blocking buffer (5 % (v/v) skimmed milk in 0.05 % (v/v) PBS/Tween) for 1 h at RT and incubated overnight at 4 °C with following antibodies: mouse anti-neurofilament 160/200 (Sigma-Aldrich), mouse anti-PSD-95 (Abcam), rabbit anti-β-catenin (Cell Signalling), mouse anti-active-β-catenin (ABC, Cell Signalling), mouse anti-β-actin (Sigma-Aldrich). Primary antibodies were detected by incubation with IR-Dye secondary antibodies (Li-COR) for 1 h at RT and subsequently quantified using the Odyssey infrared imaging system (LI-COR). Optical density analysis was performed with ImageJ software (NIH).
RNA isolation, cDNA synthesis and qPCR
RNA was isolated with a RNA isolation Kit (Macherey-Nagel) or Trizol (Invitrogen Life Technologies). 1 μg of purified RNA was used for first-strand cDNA synthesis using “Superscript III Reverse Transcriptase” and oligo(dT) in a final volume of 20 μl according to manufacturer’s instruction (Invitrogen Life Technologies). Real-time quantification of gene expression was performed using a SYBR Green qPCR assay (Applied Biosystems) or TaqMan 5′FAM- and 3′TAMRA-labeled fluorescent probes (Eurofins MWG). PCR was performed using an “ABI 7500” real-time PCR system (Applied Biosystems) with standard cycling conditions: 40 cycles of 10 s at 95 °C, 60 s at 60 °C. Specificity of the PCR product was confirmed by examination of the dissociation reaction plot and PCR product detection on 2 % agarose gel. The samples were run in duplicates and the level of expression of each gene was normalized to the expression of the housekeeping gene glycerinaldehyd-3-phosphat-dehydrogenase (GAPDH). The sequences of all PCR primers used are available on request.
Organotypic slice cultures
Organotypic slice cultures (OSCs) were generated from 10 days old Axin2lacZ/+/nestineGFP+ pups as previously described [77]. Briefly, the hippocampus was cut into 350 μm thick slices using a McIllwain tissue chopper (GaLa Instrumente). OSCs were dissociated in ice-cold dissecting medium (Hank’s Balanced Salt Solution (HBSS), Invitrogen Life Technologies) complemented with penicillin/streptomycin (100 U/ml, Invitrogen Life Technologies), 2.5 mg/ml glucose (Sigma Aldrich) and 10 mM kynurenic acid (Sigma Aldrich). OSCs were cultured on Millicell-CM culture plate inserts (Millipore) in culture medium (50 % (v/v) MEM, 25 % (v/v) HBSS, 25 % (v/v) heat-inactivated horse serum, 2 mM glutamine, penicillin/streptomycin (100 U/ml) (all from Invitrogen Life Technologies) and 2.5 mg/ml glucose (Sigma Aldrich) for 1 week at 37 °C in a humidified atmosphere with 5 % CO2. At this point OSCs were used for further experiments. For analysis of β-gal activity and Axin2 gene expression hippocampal Axin2lacZ/+ OSCs were cultured for seven days followed by treatment with Wnt3a (20 ng/ml; R&D Systems), interferon gamma (IFNγ, 100 U/ml; Immunotools), TNFα (1 ng/ml; Immunotools) or TNFα (1 ng/ml) with the inhibitor of Porcn function, IWP-2 (4 μM; R&D Systems). After 6 h of treatment, the medium was replaced with fresh cytokine-free medium and OSCs were cultured for additional 24 h. Afterwards, tissue was either fixed (β-gal assay; discussed below) or collected for RNA isolation. For the double treatment with IWP-2, the inhibitor was all the time present in the media.
For analysis of neurogenesis in hippocampal OSCs, cultures were first pretreated for 24 h with BrdU (0.5 μM, Sigma) to allow detection of proliferating cells [25]. Next, recombinant cytokines: TNFα (1 ng/ml), interferon gamma (IFNγ, 100 U/ml) (both from ImmunoTools) or Wnt3a (20 ng/ml, R&D Systems) were added to the culture medium for 6 h, followed by washing and replacement with cytokine-free media for additional 24 h. IWP-2 (4 μM, R&D Systems) was added at the same time as the cytokines and was present until the end of the experiment. After this period, slice cultures were directly used to analyse β-gal activity (see above), RNA isolation or fixed with aldehydes for histological examination.
Histology
Mice were anaesthetized using isoflurane (Piramal Healthcare) and perfused with PBS. Brain and spinal cord were dissected and either directly frozen for X-gal staining or postfixed with 4 % (v/v) paraformaldehyde (PFA, Roth) for 16 h, followed by dehydration in a 25–30 % (v/v) sucrose solution. Tissue samples were cryopreserved in “TissueTek” (Sakura Fintek Europe) at −80 °C. For histological examination brains and spinal cords were cut on a cryostat (Leica). 20 μm slices were permeabilized with 0.5 % (v/v) Triton X-100 and blocked in 5 % (v/v) horse serum (Invitrogen Life Technologies) and 1 % (v/v) bovine serum albumin in PBS for 2 h. OSCs were washed two times in warm PBS, fixed for 1 h in 4 % PFA and permeabilized for 1 h with 1 % (v/v) Triton X-100 (Sigma Aldrich) in PBS. Primary antibodies were diluted in PBS containing 2.5 % (v/v) horse serum with 0.25 % (v/v) Triton X-100 and incubated with tissue samples overnight (or 48 h for OSCs) at 4 °C. Following primary antibodies were used: guinea pig anti-GFAP (1:1000; Synaptic Systems), rat anti-CD68 (1:350; Biolegend), goat anti-SOX2 (1:250; Santa Cruz); mouse anti-iNOS (1:100; BD Transduction Laboratories), rabbit anti-Iba1 (1:500, Wako Pure Chemical Industries), rabbit anti-GFP (1:1000, Abcam) goat anti-Doublecortin (Dcx, 1:250, Santa Cruz Biotech) and Hoechst 33342 dye (Molecular Probes). Visualization was performed by incubation with fluorescent Cy2-, Cy3- and Cy5-conjugated secondary antibodies (Millipore) for 1–2 h at RT. Hoechst dye 33258 (Molecular Probes) was used to counterstain nuclei followed by mounting on glass slides with “Immuno Mount” (Thermo Scientific). Immunostainings were analyzed on an Olympus BX51 microscope and overlaid using Photoshop 13.0 software (Adobe).
Quantification of Iba1+ and GFAP+ cells in the dentate gyrus has been performed in four animals per group. The number of positive cells per animal was calculated as a mean of labeled cells in mm2 of the DG area in 3–4 sections (200 μm distance between sections). The ratio of micron per pixel (1 pixel = 0.16125 μm at magnification x 40) was used to calculate the area analyzed.
For X-gal staining, 20 μm thick cryoslices or OSCs, prepared from Axin2lacZ/+ mice, were fixed with PBS containing 2 % (v/v) formaldehyde, 0.2 % (v/v) glutaraldehyde and 0.02 % (v/v) NP40 (all from Sigma Aldrich) for 5 min. After washing, slices were incubated for 2 h at 37 °C in X-gal staining solution, containing 10 mM K3Fe(CN)6, 10 mM K4Fe(CN)6, 0.02 % (v/v) NP40, 2 mM MgCl2 and X-gal (0.5 mg/ml) (all from Sigma Aldrich) dissolved in PBS. Samples were fixed with 4 % (v/v) PFA for 15 min and mounted on glass slides with “Immuno Mount” (Thermo Scientific).
BrdU treatment and immunostaining
For labeling of proliferating cells, 50 mg/kg body weight of 5-bromo-20-desoxyuridine (BrdU; Sigma Aldrich) was injected i.p. daily for five following days starting at the peak of disease (14 d.p.i.). EAE animals and healthy controls were euthanized and perfused 2 weeks after the first BrdU-injection. Mouse hippocampi were collected and cryopreserved (see histology part) and afterwards processed as previously described [16]. Briefly, sections were incubated in 2 N HCl for 30 min at 37 °C and rinsed with 100 mM tetraborate buffer. Sections were incubated with primary rat anti-BrdU antibody (1:400; Upstate) and goat anti-Dcx antibody (1:500; Santa Cruz Biotechnology) overnight at 4 °C, followed by incubation with appropriate Cy2/Cy3-conjugated secondary antibodies (Millipore) for 1 h at RT.
Primary hippocampal cell cultures
Tissue processing and preparation of mixed glial cell population
Whole hippocampus from 2 to 5-day-old C57BL/6 mice were dissected and washed with ice cold HBSS supplemented with 0.2 % glucose. Tissue was finely minced with a razor blade and digested by StemPro Accutase (Gibco) for 20 min at 37 °C followed by 10 min digestion with 0.05 % Trypsin-EDTA (Gibco), DNase (250 U/ml, Worthington). Tissue was rinsed with DMEM supplemented with 10 % heat-inactivated fetal calve serum (FCS; Gibco), gently triturated by passing through a 1-ml serological pipette until a homogenous suspension appeared and filtered through a 40 μm cell strainer (BD Biosciences). Cells were suspended in Neurobasal medium supplemented with B27, glutamax, 1 % penicillin/streptomycin (all from Gibco) (hereinafter referred to as NB media).
Microglia and astrocyte cell cultures
Isolated hippocampal cells were plated on poly-ornithine-coated petri dishes and cultured in 10 ml of NB media (see above) supplemented with 10 % (v/v) FCS and 20 ng/ml of macrophage colony stimulating factor (M-CSF; Immunotools). 50 % of culture media was replaced every 4 days with fresh media. After 2–3 weeks incubation in a 5 % CO2 incubator at 37 °C, cells were collected by dissociation with StemPro Accutase (Gibco) for positive selection of CD11b+ cells or GLAST+ cells using magnetic microbeads (Miltenyi Biotech), according to the manufacturer’s instructions. This approach results in about 90 % purity of primary microglia cells and 95 % purity of primary astrocytes determined by immunostaining for Iba-1 and GFAP, respectively. Microglia and astrocytes were harvested, washed and plated on poly-L-ornitine coated plates or coverslips at densities appropriate for each assay.
Hippocampal progenitor cultures
Isolated hippocampal cells were cultured in serum-free proliferating conditions on uncoated Petri plates in NB supplemented with 20 ng/ml bFGF (Immunotools). After 3–4 days divided cells formed free-floating neurospheres. bFGF was added every second day. 30 % of culture media was replaced every 4 days with fresh media. After 1–2 weeks in culture neurospheres were collected and cells were dissociated by trypsin-EDTA solution. For further analysis, cells were cultured as adherent cells in proliferating medium on poly-ornithine-coated petri dishes or glasses. For gene expression analysis cells were treated for 6 h with mouse recombinant TNFα (0.2 ng/ml or 1 ng/ml; R&D) or Wnt3a (20 ng/ml; R&D).
Coculture of hippocampal progenitors with microglia
2 × 105 CD11b+ cells in 200 μl NB media were added to the upper well of each Transwelltm inserts (0.2 μm diameter holes, Ibidi) for 24 h. In parallel, 4 × 104 hippocampal progenitor cells dissociated from neurospheres were cultured separately on poly-L-ornithine coated coverslips in proliferating NB media containing 20 ng/ml bFGF. 24 h after culturing of microglia on transwells, cells were activated with TNFα (1 ng/ml) for 4 h and washed carefully with NB media. After this, the transwells containing microglia were set on hippocampal progenitor cultures and incubated for 24 h in proliferating NB media (containing 5 ng/ml bFGF) in the presence or absence of soluble Frizzled-8 (sFz8; 1 μg/ml; R&D). 10 μg/ml of BrdU was added for last 2 h of experiment to allow detection of proliferating cells. Goat anti-SOX2 (1:250; Santa Cruz) and rat anti-BrdU (1:500; Upstate) primary antibody were used to detect mitotically active cells in hippocampal progenitor cultures.
Statistical Analysis
All values in the figures are shown as mean + SEM. Statistical analysis was performed with GraphPad Prism 5 (GraphPad Software). The p values of *p < 0.05; **p < 0.01; ***p < 0.001 were determined to be statistically significant by using Student’s t-test.
Abbreviations
ABC, active-β-catenin; ARRIVE, Animal Research: Reporting of In Vivo Experiments; BrdU, 5-Bromo-20-desoxyuridine; CFA, Complete Freund’s Adjuvant; CNS, central nervous system; d.p.i., days post immunization; Dcx, doublecortin; DG, dentate gyrus; EAE, experimental autoimmune encephalomyelitis; ECS, electroconvulsive seizures; FCS, fetal calf serum; GAPDH, glycerinaldehyd-3-phosphat dehydrogenase; GSK-3β, glycogensynthase-kinase-3 beta; HBSS, hank’s balanced salt solution; HSP68, heat shock protein 68; i.p., intraperitoneal; IFNγ, interferon gamma; IL-1β, interleukin-1 beta; LANUV, Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen; Lef1, lymphoid enhancer-binding factor 1; LPC, lysolecithin; MOG, myelin oligodendrocytes glycoprotein; MS, multiple sclerosis; NeuroD1, neurogenic differentiation 1; NF-H, neurofilament-H; OSC, organotypic slice culture; PFA, paraformaldehyde; PLP, proteolipid protein peptide; Prox1, prospero homeobox 1; PSD-95, postsynaptic protein 95; PTX, pertussis toxin; RT, room temperature; SEM, standard error of mean; SGZ, subgranular zone; SVZ, subventricular zone; TCF, transcription factor; TGFβ1, transforming growth factor beta 1; TNFα, Tumor necrosis factor alpha; WASP-1, Wnt-activating small molecule potentiator-1.
Acknowledgements
We would like to thank EMMA (European Mouse Mutant Archive) for providing the Axin2lacZ/+ mice.
Funding
Our research is supported by grants from the “Forschungskommission” of the Medical Faculty (Heinrich-Heine-University Düsseldorf) to T.P. and O.A. (06/2012), the Deutsche Forschungsgemeinschaft (SPP1710, BE3529/5-1 to CB) and the Walter and Ilse Rose Foundation (to O.A.).
Availability of data and materials
All requested data are available via T.P.
Authors’ contributions
RS and TP designed the experiments and performed the biochemical analysis of tissue. TP, RS, CB, JC and OA wrote the manuscript. J.I. and R.S. have performed the EAE experiments, including immunization, treatment, scoring, isolation and processing of animal tissue. F.S. and B.K. contributed to treatment of animals and tissue collection. HPH and OA provided critical input. All authors read, revised and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All experimental procedures were conducted following the guidelines and protocols approved by the local animal welfare committee (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV); under protocol numbers G388-11 (active EAE in Axin2lacZ/+mice), G197-09 (BrdU injections in EAE mice), G363-09 (passive EAE in SJL/J)).
Additional files
Upregulation of Wnt activity in the hippocampus and the spinal cord correlates with disease severity. (a) Iba1 immunostaining (red) of spinal cord sections from Axin2lacZ/+ EAE mice at day 20. The white dashed line highlights lesion areas; white matter tissue is outlined by yellow dashed line. Nuclei were counterstained with Hoechst (blue). Scale bar, 100 μm. (b) Histogram represents the average number of lesions per section of each EAE time point as mean + SEM. Spinal cord: day 20 (n = 4; 15–35 sections/mouse), day 30 (n = 3; 10–23 sections/mouse), day 50 (n = 3; 10–17 sections/mouse). (c) Representative image of lesion site in the DG. Iba1 (red) immunostaining in hippocampal section from EAE mouse at day 30. Nuclei (grey). Scale bar, 50 μm. (d) Representative image of neural stem cells located in the SGZ. SOX2 (green) and GFAP (red). Arrowheads indicate SOX2+/GFAP+ radial glia-like cells and arrows indicate SOX2+/GFAP+ horizontal progenitors. Nuclei (grey). Scale bar, 25 μm. (e+f) β-gal assay of cortical and cerebellar EAE tissue revealed no changes in Wnt activity. Histograms represent mean + SEM of fold-changes of β-gal activity in EAE mice relative to controls (CFA) set as 1. Day 20 (EAE, n = 6; CFA, n = 3); day 30 (EAE, n = 4; CFA, n = 3); day 50 (EAE, n = 8; CFA, n = 4). (g) qPCR analysis of Axin2 in different CNS parts in acute EAE (day 20). Data represent mean of fold-changes + SEM of gene expression in EAE mice (n = 6) relative to controls (CFA; n = 6) set as 1. Expression of Axin2 was normalized to Gapdh. (d) Regression analysis shows correlation between β-gal activity in inflamed CNS tissues and clinical disease score in EAE animals. Pearson correlation, r = 0.58; p = 0.018 (hippocampus) and r = 0.52, p = 0.036 (spinal cord). Statistics: Two-tailed, unpaired Student’s t-test,* * p < 0.05 and ** p < 0.01. (PDF 6160 kb)
Gene expression analysis in the hippocampal tissue of EAE mice. (a) qPCR analysis of selected Wnt ligands in the hippocampus at early (day 20) and chronic (day 50) stages of passive EAE. Histogram represents mean + SEM of fold-changes relative to control group (PBS) set as 1. Gene expression was normalized to Gapdh. Day 20 (EAE, n = 4–6; control, n = 4); day 50 (EAE, n = 8–13; control, n = 7–11). (b) qPCR analysis of selected Wnt ligands in the cortex, cerebellum and spinal cord at early stages of passive EAE (day 20). Histogram represents mean + SEM of fold-changes relative to control group (PBS) set as 1. Gene expression was normalized to Gapdh. Six control and six EAE mice were analysed for each part of the CNS. n.d. not detectable. (c) A regression analysis shows correlation between the TNFα and the Axin2 gene expression levels examined in hippocampal tissue of individual EAE animals. Pearson correlation, r = 0.8458; p < 0. 001. (d) qPCR analysis shows transient upregulation of Smad7 expression in the hippocampus of mice with passively transferred EAE. Data represent mean + SEM of fold-changes of gene expression in EAE mice relative to respective controls (PBS) set as 1. Expression was normalized to Gapdh. Day 20 (EAE, n = 4; control, n = 4); day 50 (EAE, n = 4; control, n = 4). Two-tailed, unpaired Student’s t-test, * p < 0.05 and ** p < 0.01. (PDF 430 kb)
Effect of TNFα on gene expression of Axin2 and Wnt ligands in hippocampal cells. (a) The distribution pattern of LacZ expression in Axin2lacZ/+ hippocampal slice cultures. X-gal staining of hippocampal slices (postnatal day 10) shows LacZ expression predominantly in the granular cell layer (gcl). Scale bar, 50 μm. (b) qPCR analysis of Axin2, Wnt3, Wnt8b and Wnt5a gene expression in hippocampal progenitors after 6 h treatment with TNFα (0.2 ng/ml or 1.0 ng/ml; n = 3) or Wnt3a (20 ng/ml; n = 3). Histogram represents mean + SEM of fold-changes of gene expression relative to controls (PBS, n = 3) set as 1. (c) qPCR analysis of gene expression of selected Wnt ligands in hippocampal astrocytes (GLAST+) and microglia (CD11b+) cells treated with TNFα (0.2 ng/ml or 1.0 ng/ml; n = 3) for 6 h. Histogram represents mean + SEM of fold-changes of gene expression relative to controls (PBS, n = 3) set as 1. (c) Histological analysis of proliferating cells in the DG of hippocampal NestineGFP OSCs. Hippocampal slices were cultured for seven days followed by 6 h treatment with Wnt3a (20 ng/ml) or IFNγ (100 U/ml) and additional incubation in cytokine-free medium for 24 h. BrdU was administered for 24 h prior to treatment with cytokines. Representative images show BrdU (red) and eGFP (green). Arrow heads indicate BrdU+/eGFP+ co-labeled hippocampal progenitors. Scale bar, 50 μm. (d) The frequency of BrdU label-retaining cells in the DG of hippocampal NestineGFP OSCs is increased after treatment with Wnt3a. Data are shown as mean + SEM of cells per mm of the DG. Control (PBS, n = 15–20 slices); IFNγ (n = 4–11 slices) and Wnt3a (n = 8–14 slices). Statistics: Two-tailed, unpaired Student’s t-test,* * p < 0.05, ** p < 0.01 and *** p < 0.001. (PDF 1448 kb)
Contributor Information
Reiner Schneider, Email: reiner.schneider@hhu.de.
Barbara Koop, Email: b_koop@gmx.de.
Friederike Schröter, Email: friederike.schroeter@med.uni-duesseldorf.de.
Jason Cline, Email: jasoncline1279@gmail.com.
Jens Ingwersen, Email: jens.ingwersen@hhu.de.
Carsten Berndt, Email: carsten.berndt@med.uni-duesseldorf.de.
Hans-Peter Hartung, Email: hans-peter.hartung@uni-duesseldorf.de.
Orhan Aktas, Phone: +49-211-81 04170, Email: orhan.aktas@uni-duesseldorf.de.
Tim Prozorovski, Phone: +49 211 302039220, Email: Timur.Prozorovskiy@med.uni-duesseldorf.de.
References
- 1.Aktas O, Ullrich O, Infante-Duarte C, Nitsch R, Zipp F. Neuronal damage in brain inflammation. Arch. Neurol. 2007;64:185–9. doi: 10.1001/archneur.64.2.185. [DOI] [PubMed] [Google Scholar]
- 2.Trapp BD, Nave K. Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008;31:247–69. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 3.Sicotte NL, Kern KC, Giesser BS, Arshanapalli A, Schultz A, Montag M, et al. Regional hippocampal atrophy in multiple sclerosis. Brain. 2008;131:1134–41. doi: 10.1093/brain/awn030. [DOI] [PubMed] [Google Scholar]
- 4.Roosendaal SD, Hulst HE, Vrenken H, Feenstra HEM, Castelijns JA, Pouwels PJW, et al. Structural and functional hippocampal changes in multiple sclerosis patients with intact memory function. Radiology. 2010;255:595–604. doi: 10.1148/radiol.10091433. [DOI] [PubMed] [Google Scholar]
- 5.Rao SM, Leo GJ, Bernardin L, Unverzagt F. Cognitive dysfunction in multiple sclerosis. I. Frequency, patterns, and prediction. Neurology. 1991;41:685–91. doi: 10.1212/WNL.41.5.685. [DOI] [PubMed] [Google Scholar]
- 6.Chiaravalloti ND, DeLuca J. Cognitive impairment in multiple sclerosis. Lancet Neurol. 2008;7:1139–51. doi: 10.1016/S1474-4422(08)70259-X. [DOI] [PubMed] [Google Scholar]
- 7.Aktas O, Hartung H. Beyond axonal transection: hippocampal damage in multiple sclerosis. Ann. Neurol. 2011;69:433–6. doi: 10.1002/ana.22409. [DOI] [PubMed] [Google Scholar]
- 8.Geurts JJG, Bö L, Roosendaal SD, Hazes T, Daniëls R, Barkhof F, et al. Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2007;66:819–27. doi: 10.1097/nen.0b013e3181461f54. [DOI] [PubMed] [Google Scholar]
- 9.Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA, et al. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011;69:445–54. doi: 10.1002/ana.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papadopoulos D, Dukes S, Patel R, Nicholas R, Vora A, Reynolds R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. 2009;19:238–53. doi: 10.1111/j.1750-3639.2008.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutta R. Gene expression changes underlying cortical pathology: clues to understanding neurological disability in multiple sclerosis. Mult. Scler. 2013;19:1249–54. doi: 10.1177/1352458513500554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziehn MO, Avedisian AA, Tiwari-Woodruff S, Voskuhl RR. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis. EAE. Lab. Invest. 2010;90:774–86. doi: 10.1038/labinvest.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nisticò R, Mango D, Mandolesi G, Piccinin S, Berretta N, Pignatelli M, et al. Inflammation subverts hippocampal synaptic plasticity in experimental multiple sclerosis. PLoS ONE. 2013;8:e54666. doi: 10.1371/journal.pone.0054666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandolesi G, Grasselli G, Musumeci G, Centonze D. Cognitive deficits in experimental autoimmune encephalomyelitis: neuroinflammation and synaptic degeneration. Neurol. Sci. 2010;31:S255–9. doi: 10.1007/s10072-010-0369-3. [DOI] [PubMed] [Google Scholar]
- 15.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010;11:339–50. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huehnchen P, Prozorovski T, Klaissle P, Lesemann A, Ingwersen J, Wolf SA, et al. Modulation of adult hippocampal neurogenesis during myelin-directed autoimmune neuroinflammation. Glia. 2011;59:132–42. doi: 10.1002/glia.21082. [DOI] [PubMed] [Google Scholar]
- 17.Whyte JL, Smith AA, Helms JA. Wnt signaling and injury repair. Cold Spring Harb Perspect Biol. 2012;4:a008078. doi: 10.1101/cshperspect.a008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert C, Cisternas P, Inestrosa NC. Role of Wnt Signaling in Central Nervous System Injury. Mol. Neurobiol. 2015 doi: 10.1007/s12035-015-9138-x. [DOI] [PubMed] [Google Scholar]
- 19.Lie D, Colamarino SA, Song H, Désiré L, Mira H, Consiglio A, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–5. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 20.Gebara E, Bonaguidi MA, Beckervordersandforth R, Sultan S, Udry F, Gijs P, et al. Heterogeneity of Radial Glia-Like Cells in the Adult Hippocampus. Stem Cells. 2016;34:997–1010. doi: 10.1002/stem.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–4. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan S, Shi Y, Tang S. Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J Neuroimmune Pharmacol. 2012;7:904–13. doi: 10.1007/s11481-012-9370-3. [DOI] [PubMed] [Google Scholar]
- 23.Pluchino S, Muzio L, Imitola J, Deleidi M, Alfaro-Cervello C, Salani G, et al. Persistent inflammation alters the function of the endogenous brain stem cell compartment. Brain. 2008;131:2564–78. doi: 10.1093/brain/awn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo J, Ho PP, Buckwalter MS, Hsu T, Lee LY, Zhang H, et al. Glia-dependent TGF-beta signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J Clin Invest. 2007;117:3306–15. doi: 10.1172/JCI31763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raineteau O, Rietschin L, Gradwohl G, Guillemot F, Gähwiler BH. Neurogenesis in hippocampal slice cultures. Mol. Cell. Neurosci. 2004;26:241–50. doi: 10.1016/j.mcn.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–22. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 27.Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, et al. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:2013–24. doi: 10.1523/JNEUROSCI.4689-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Invest. 2007;117:448–56. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SM, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–67. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- 30.Wexler EM, Paucer A, Kornblum HI, Palmer TD, Plamer TD, Geschwind DH. Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells. 2009;27:1130–41. doi: 10.1002/stem.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan C, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5:100–7. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, Sekino Y, Sato K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci. 2014;34:2231–43. doi: 10.1523/JNEUROSCI.1619-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sierra A, Encinas JM, Deudero JJP, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–95. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–60. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 36.Han MH, Hwang S, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–81. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 37.Fancy SPJ, Baranzini SE, Zhao C, Yuk D, Irvine K, Kaing S, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–85. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- 40.Sousa KM, Villaescusa JC, Cajanek L, Ondr JK, Castelo-Branco G, Hofstra W, et al. Wnt2 regulates progenitor proliferation in the developing ventral midbrain. J Biol Chem. 2010;285:7246–53. doi: 10.1074/jbc.M109.079822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madsen TM, Newton SS, Eaton ME, Russell DS, Duman RS. Chronic electroconvulsive seizure up-regulates beta-catenin expression in rat hippocampus: role in adult neurogenesis. Biol Psychiatry. 2003;54:1006–14. doi: 10.1016/S0006-3223(03)00700-5. [DOI] [PubMed] [Google Scholar]
- 42.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–10. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JE, Wu S, Goering LM, Dorsky RI. Canonical Wnt signaling through Lef1 is required for hypothalamic neurogenesis. Development. 2006;133:4451–61. doi: 10.1242/dev.02613. [DOI] [PubMed] [Google Scholar]
- 44.Fear MW, Kelsell DP, Spurr NK, Barnes MR. Wnt-16a, a novel Wnt-16 isoform, which shows differential expression in adult human tissues. Biochem Biophys Res Commun. 2000;278:814–20. doi: 10.1006/bbrc.2000.3852. [DOI] [PubMed] [Google Scholar]
- 45.Dell’accio F, de Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. 2008;58:1410–21. doi: 10.1002/art.23444. [DOI] [PubMed] [Google Scholar]
- 46.Marchetti B, Pluchino S. Wnt your brain be inflamed? Yes, it Wnt! Trends Mol Med. 2013;19:144–56. doi: 10.1016/j.molmed.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.González-Fernández C, Fernández-Martos CM, Shields SD, Arenas E, Javier RF. Wnts are expressed in the spinal cord of adult mice and are differentially induced after injury. J. Neurotrauma. 2014;31:565–81. doi: 10.1089/neu.2013.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hiyama A, Yokoyama K, Nukaga T, Sakai D, Mochida J. A complex interaction between Wnt signaling and TNF-α in nucleus pulposus cells. Arthritis Res Ther. 2013;15:R189. doi: 10.1186/ar4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cawthorn WP, Heyd F, Hegyi K, Sethi JK. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 2007;14:1361–73. doi: 10.1038/sj.cdd.4402127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008;27:1671–81. doi: 10.1038/emboj.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Egea V, von Baumgarten L, Schichor C, Berninger B, Popp T, Neth P, et al. TNF-α respecifies human mesenchymal stem cells to a neural fate and promotes migration toward experimental glioma. Cell Death Differ. 2011;18:853–63. doi: 10.1038/cdd.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Z, Palmer TD. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav Immun. 2013;30:45–53. doi: 10.1016/j.bbi.2013.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJH, Bonde S, Kokaia Z, et al. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–12. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iosif RE, Ahlenius H, Ekdahl CT, Darsalia V, Thored P, Jovinge S, et al. Suppression of stroke-induced progenitor proliferation in adult subventricular zone by tumor necrosis factor receptor 1. J Cereb Blood Flow Metab. 2008;28:1574–87. doi: 10.1038/jcbfm.2008.47. [DOI] [PubMed] [Google Scholar]
- 56.Heldmann U, Thored P, Claasen J, Arvidsson A, Kokaia Z, Lindvall O. TNF-alpha antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp Neurol. 2005;196:204–8. doi: 10.1016/j.expneurol.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 57.Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;7:64. doi: 10.1186/1471-2202-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peng H, Whitney N, Wu Y, Tian C, Dou H, Zhou Y, et al. HIV-1-infected and/or immune-activated macrophage-secreted TNF-alpha affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia. 2008;56:903–16. doi: 10.1002/glia.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernardino L, Agasse F, Silva B, Ferreira R, Grade S, Malva JO. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells. 2008;26:2361–71. doi: 10.1634/stemcells.2007-0914. [DOI] [PubMed] [Google Scholar]
- 60.Finsen B, Antel J, Owens T. TNFalpha: kill or cure for demyelinating disease? Mol Psychiatry. 2002;7:820–1. doi: 10.1038/sj.mp.4001120. [DOI] [PubMed] [Google Scholar]
- 61.Wood H. Multiple sclerosis: TNF receptor 1 gene variant could explain failure of TNF-blocking drugs in multiple sclerosis. Nat Rev Neurol. 2012;8:476. doi: 10.1038/nrneurol.2012.149. [DOI] [PubMed] [Google Scholar]
- 62.van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47:1531–4. doi: 10.1212/WNL.47.6.1531. [DOI] [PubMed] [Google Scholar]
- 63.Eugster HP, Frei K, Bachmann R, Bluethmann H, Lassmann H, Fontana A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur J Immunol. 1999;29:626–32. doi: 10.1002/(SICI)1521-4141(199902)29:02<626::AID-IMMU626>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 64.Quadrato G, Elnaggar MY, Di Giovanni S. Adult neurogenesis in brain repair: cellular plasticity vs. cellular replacement. Front Neurosci. 2014;8:17. doi: 10.3389/fnins.2014.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur. J. Neurosci. 2011;33:1139–51. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- 66.Chang A, Smith MC, Yin X, Fox RJ, Staugaitis SM, Trapp BD. Neurogenesis in the chronic lesions of multiple sclerosis. Brain. 2008;131:2366–75. doi: 10.1093/brain/awn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tepavčević V, Lazarini F, Alfaro-Cervello C, Kerninon C, Yoshikawa K, Garcia-Verdugo JM, et al. Inflammation-induced subventricular zone dysfunction leads to olfactory deficits in a targeted mouse model of multiple sclerosis. J Clin Invest. 2011;121:4722–34. doi: 10.1172/JCI59145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aharoni R, Arnon R, Eilam R. Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J. Neurosci. 2005;25:8217–28. doi: 10.1523/JNEUROSCI.1859-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giannakopoulou A, Grigoriadis N, Bekiari C, Lourbopoulos A, Dori I, Tsingotjidou AS, et al. Acute inflammation alters adult hippocampal neurogenesis in a multiple sclerosis mouse model. J. Neurosci. Res. 2013;91:890–900. doi: 10.1002/jnr.23226. [DOI] [PubMed] [Google Scholar]
- 70.Shruster A, Ben-Zur T, Melamed E, Offen D. Wnt signaling enhances neurogenesis and improves neurological function after focal ischemic injury. PLoS One. 2012;7:e40843. doi: 10.1371/journal.pone.0040843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suh HI, Min J, Choi KH, Kim SW, Kim KS, Jeon SR. Axonal regeneration effects of Wnt3a-secreting fibroblast transplantation in spinal cord-injured rats. Acta Neurochir (Wien) 2011;153:1003–10. doi: 10.1007/s00701-011-0945-1. [DOI] [PubMed] [Google Scholar]
- 72.Vargas JY, Fuenzalida M, Inestrosa NC. In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J. Neurosci. 2014;34:2191–202. doi: 10.1523/JNEUROSCI.0862-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yin Z, Zu B, Chang J, Zhang H. Repair effect of Wnt3a protein on the contused adult rat spinal cord. Neurol Res. 2008;30:480–6. doi: 10.1179/174313208X284133. [DOI] [PubMed] [Google Scholar]
- 74.Kawaguchi A, Miyata T, Sawamoto K, Takashita N, Murayama A, Akamatsu W, et al. Nestin-EGFP transgenic mice: visualization of the self-renewal and multipotency of CNS stem cells. Mol. Cell. Neurosci. 2001;17:259–73. doi: 10.1006/mcne.2000.0925. [DOI] [PubMed] [Google Scholar]
- 75.Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 2002;22:1184–93. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baker D, Amor S. Publication guidelines for refereeing and reporting on animal use in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012;242:78–83. doi: 10.1016/j.jneuroim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 77.Aktas O, Schulze-Topphoff U, Zipp F. The role of TRAIL/TRAIL receptors in central nervous system pathology. Front. Biosci. 2007;12:2912–21. doi: 10.2741/2281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All requested data are available via T.P.