

Abstract
Pulmonary alveolar proteinosis (PAP) is a rare diffuse lung disease characterized by accumulation of lipoproteinacious material in alveoli, with distinct features on high resolution computed tomography and biopsy. Its association with pulmonary fibrosis is infrequently encountered, and a clear understanding of the underlying pathogenesis is yet to be established. We report the case of a 48‐year‐old woman with known autoimmune PAP (aPAP) first diagnosed 20 years ago, who presented with worsening hypoxemia and radiological features consistent with pulmonary fibrosis, after many years of stable disease. We present a review of previously considered mechanisms of causation behind such changes, and in particular, postulate the role of granulocyte‐macrophage colony‐stimulating factor deficiency in pulmonary fibrosis seen in aPAP.
Keywords: Autoimmune disease, granulocyte‐macrophage colony stimulating factor, pulmonary alveolar proteinosis, pulmonary fibrosis, rare lung diseases
Introduction
Pulmonary alveolar proteinosis (PAP) is a rare diffuse lung disease, characterized by intra‐alveolar accumulation of surfactant. We report an unusual case, where over the course of two decades, imaging and pathology transition from a pattern consistent with PAP to that of pulmonary fibrosis.
Case Report
The patient initially presented aged 29 with an eight‐year history of breathlessness and cough. Risk factors for lung disease included cigarette smoking, and previous employment in a steel factory, although any significant dust exposure is uncertain. Clinical examination revealed pulse oximetry of 87% on room air, peripheral cyanosis, and right basal crackles on chest auscultation. Chest radiograph demonstrated extensive bilateral alveolar opacities. The baseline computed tomography scan is shown in Figure 1. Lung function tests revealed restrictive disease with vital capacity 47% predicted, and diffusing capacity of the lungs for carbon monoxide (DLCO) 20% predicted. Diagnosis was confirmed by video‐assisted thoracoscopic biopsy. The histopathology was typical of PAP with well‐preserved pulmonary parenchymal architecture, and confluent filling of alveoli by eosinophilic and amorphous material, with areas of cholesterol cleft formation (Fig. 2). Anti granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) antibodies were elevated at diagnosis, confirming autoimmune PAP (aPAP). Hematological malignancy and immunodeficiency were excluded with normal serum immunoglobulins, serum electrophoresis, and peripheral flow cytometry.
Figure 1.
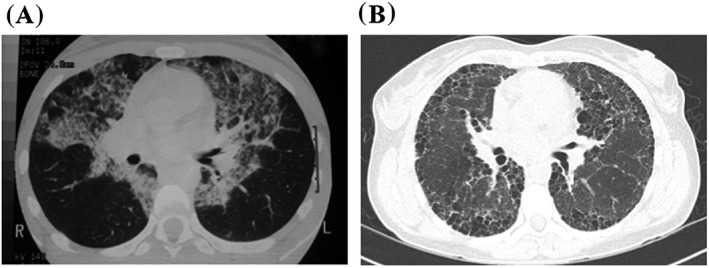
A: Baseline high‐resolution computed tomography from 1996, showing bilateral alveolar opacities and thickened inter‐lobular septa. B: High‐resolution computed tomography from 2015, with subpleural honeycombing and bullae.
Figure 2.
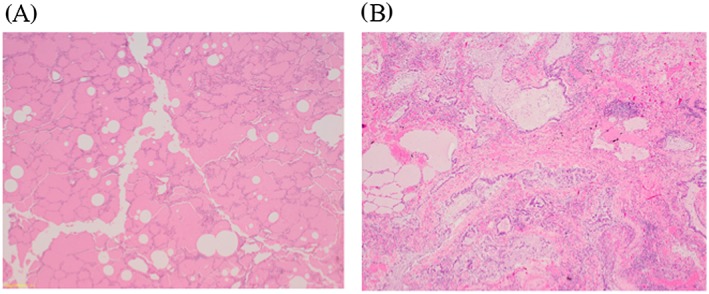
A: Photomicrograph at magnification ×4 of right lower lobe lung biopsy from 1996. There is confluent filling of alveoli by eosinophilic, finely granular, and amorphous material with areas of cholesterol cleft formation. B: Photomicrograph at magnification ×40 of post‐mortem lung biopsy from 2015, demonstrating end stage fibrotic lung disease. There is no evidence of residual pulmonary alveolar proteinosis. Images courtesy of Dorevitch Pathology Frankston, Victoria.
Over the subsequent 12 months, the patient became increasingly hypoxemic requiring domiciliary oxygen. She was treated with whole lung lavage on four occasions, with only transient improvements in symptoms and lung function (Table 1). The final lavage was complicated by right cerebellum infarction, likely secondary to hypoxia, although complete recovery was observed. She was finally enrolled in a trial using subcutaneous GM‐CSF, which resulted in a sustained improvement over the next decade, with symptom resolution, normalization of oxygen saturation, and improved lung function.
Table 1.
Trend of lung function test results.
| Baseline |
WLL 1 +1 month |
WLL 2 +3 months |
WLL 3 +7 months |
WLL 4 +10 months |
GM‐CSF +3 years |
+10 years | +18 years | |
|---|---|---|---|---|---|---|---|---|
| VC L (% predicted) | 1.67 (47) | 1.95 (54) | 2.27 (64) | 2.04 (57) | 1.88 (53) | 2.33 (66) | 2.85 (83.9) | 1.75 (54.8) |
| FVC L (% predicted) | 1.57 (44) | 1.79 (51) | 2.22 (63) | 1.89 (54) | 1.85 (52) | 2.17 (62.6) | 2.85 (85.6) | 1.62 (52.2) |
| FEV1 L (% predicted) | 1.56 (51) | 1.54 (50) | 1.90 (62) | 1.66 (54) | 1.64 (54) | 1.95 (64.6) | 2.31 (80.2) | 1.27 (47.8) |
| FEV1/FVC % | 99.6 | 85.6 | 85.7 | 87.8 | 88.8 | 89.6 | 80.9 | 78.4 |
| DLCO % predicted | 20 | 22 | 29 | 22 | 22 | 44 | 37 | 20 |
|
DLCO/VA % predicted |
Not measured | 43 | 54 | 42 | 50 | 63 | 50 | 43 |
DLCO, diffusing capacity of the lungs for carbon monoxide; DLCO/VA, DLCO corrected for alveolar volume; FEV1, forced expiratory volume in one second; FVC, forced vital capacity; GM‐CSF, granulocyte macrophage‐colony stimulating factor; VC, vital capacity; WLL, whole lung lavage.
In 2015, she represented with worsening dyspnea, having been lost to follow‐up for many years, and had continued to smoke. Notable examination findings included pulse oximetry of 83% on room air, finger clubbing, and bilateral crepitations on chest auscultation. Lung function tests showed marked deterioration compared with results post GM‐CSF treatment, and DLCO had returned to levels of initial presentation. Arterial blood gases breathing room air at sea level showed PaO2 45 mmHg, PaCO2 43 mmHg. Inflammatory markers were not elevated. Transthoracic echocardiogram demonstrated mild pulmonary hypertension.
Repeat high‐resolution computed tomography depicted a very different appearance (Fig. 1). There were extensive areas of subpleural honeycombing consistent with fibrosis, in addition to large bullae and minor traction bronchiectasis. The classic alveolar pattern of PAP was absent. Anti‐GM‐CSF antibody however was again elevated at 0.43 OD units [<0.23]. Undertaking another lung biopsy was considered to pose an unacceptably high risk of death given the patient's poor clinical state. She was discharged on domiciliary oxygen after pledging smoking cessation. Application for repeat GM‐CSF access was submitted. Referral was also made for lung transplant assessment.
Subsequently, deterioration was progressive and rapid. During the final hospitalization, she was severely hypoxic, requiring continuous high‐flow oxygen using 0.95 fraction of inspired oxygen to maintain pulse oximetry of 70%. There were no identifiable reversible causes. She declined non‐invasive ventilation, and was unsuitable for invasive ventilation because of poor underlying reserve. The patient died with type 1 respiratory failure, 8 months after re‐presentation. Inhaled GM‐CSF was approved and made available one day prior, and two doses were administered. We do not believe this precipitated her death, as the declining trajectory well preceded GM‐CSF. Post‐mortem lung biopsy was obtained, and confirmed end‐stage fibrotic lung disease (Fig. 2).
Discussion
Pulmonary fibrosis is infrequently seen in aPAP. There have been few reports on this association, and little is known regarding the underlying mechanism or progression. From the small pool of known cases, the predominant hypotheses proposed include fibrosis as an end‐stage phase of PAP, that chronic infections may predispose to fibrosis, or that fibrogenic reactions may occur against the accumulated lipoproteinacious material 1. A case series of patients with PAP and hypersensitivity pneumonitis also observed fibrosis in two patients, attributing this to a chronic phase of hypersensitivity pneumonitis 2. In this case, tobacco smoke is also undoubtedly a risk factor for both PAP and fibrosis, as well as contributing to any underlying emphysema.
The pathogenesis of aPAP is understood to be secondary to autoantibodies against GM‐CSF, leading to impaired GM‐CSF function, and consequently its role in effective surfactant homeostasis by alveolar macrophages 3. It is noteworthy that in experimental models of pulmonary fibrosis in mice, GM‐CSF deficiency is associated with worse fibrosis after bleomycin administration. This has been demonstrated both by neutralization with anti‐GM‐CSF‐IgG 4, and in genetically altered mice through GM‐CSF gene deletion 5. In the latter study, absence of GM‐CSF led to reduced synthesis by alveolar macrophages of the anti‐fibrotic prostaglandin E2 (PGE2). These studies demonstrate that there is an important role for GM‐CSF in minimizing lung injury after insult.
Long‐term results on GM‐CSF therapy efficacy are lacking, with an absence of consistent data on duration of benefit after treatment. In the longest prospective study available, serum anti‐GM‐CSF‐antibody levels remained unchanged 30 months following inhaled GM‐CSF 3. Any correlation with disease activity or treatment response is however uncertain. We postulate that in this case, relapse and progression of PAP had occurred, associated with new pulmonary fibrosis, both of which driven by ongoing functional GM‐CSF deficiency as a possible unifying mechanism.
Whilst rare, it is important to recognize that fibrosis may develop in PAP. Further insights are required into the pathogenesis, as well as the clinical course of those following GM‐CSF therapy.
Disclosure Statement
No conflict of interest declared.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
Sha, J. , and Langton, D. (2016) Role of granulocyte‐macrophage colony‐stimulating factor in pulmonary fibrosis following pulmonary alveolar proteinosis. Respirology Case Reports, 4 (4), e00159. doi: 10.1002/rcr2.159.
References
- 1. Agarwal PP, Seely JM, Perkins DG, et al 2005. Pulmonary alveolar proteinosis and end‐stage pulmonary fibrosis: a rare association. J. Thorac. Imaging 20:242–244. [DOI] [PubMed] [Google Scholar]
- 2. Verma H, Nicholson AG, Kerr KM, et al. 2010. Alveolar proteinosis with hypersensitivity pneumonitis: a new clinical phenotype. Respirology 15:1197–1202. [DOI] [PubMed] [Google Scholar]
- 3. Tazawa R, Inoue Y, Arai T, et al. 2014. Duration of benefit in patients with autoimmune pulmonary alveolar proteinosis after inhaled granulocyte‐macrophage colony‐stimulating factor therapy. Chest 145:729–737. [DOI] [PubMed] [Google Scholar]
- 4. Christensen PJ, Bailie MB, Goodman RE, et al. 2000. Role of diminished epithelial GM‐CSF in the pathogenesis of bleomycin‐induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L487–495. [DOI] [PubMed] [Google Scholar]
- 5. Moore BB, Coffey MJ, Christensen P, et al. 2000. GM‐CSF regulates bleomycin‐induced pulmonary fibrosis via a prostaglandin‐dependent mechanism. J. Immunol. 165:4032–4039. [DOI] [PubMed] [Google Scholar]