

Abstract
Organic anion transporting polypeptide (OATP) 1B1 mediates the hepatic uptake of many drugs including lipid-lowering statins. Decreased OATP1B1 transport activity is often associated with increased systemic exposure of statins and statin-induced myopathy. Antimalarial drug chloroquine (CQ) is also used for long-term treatment of rheumatoid arthritis and systemic lupus erythematosus. CQ is lysosomotropic and inhibits protein degradation in lysosomes. The current studies were designed to determine the effects of CQ on OATP1B1 protein degradation, OATP1B1-mediated transport in OATP1B1-overexpressing cell line, and statin uptake in human sandwich-cultured hepatocytes (SCH). Treatment with lysosome inhibitor CQ increased OATP1B1 total protein levels in HEK293-OATP1B1 cells and in human SCH as determined by OATP1B1 immunoblot. In HEK293-FLAG-tagged OATP1B1 stable cell line, co-immunofluorescence staining indicated that intracellular FLAG-OATP1B1 is colocalized with lysosomal associated membrane glycoprotein (LAMP)-2, a marker protein of late endosome/lysosome. Enlarged LAMP-2-positive vacuoles with FLAG-OATP1B1 protein retained inside were readily detected in CQ-treated cells, consistent with blocking lysosomal degradation of OATP1B1 by CQ. In HEK293-OATP1B1 cells, without pre-incubation, CQ concentrations up to 100 μM did not affect OATP1B1-mediated [3H]E217G accumulation. However, pre-incubation with CQ at clinically relevant concentration(s) significantly decreased [3H]E217G and [3H]pitavastatin accumulation in HEK293-OATP1B1 cells and [3H]pitavastatin accumulation in human SCH. CQ pretreatment (25 μM, 2 h) resulted in ∼1.9-fold decrease in Vmax without affecting Km of OATP1B1-mediated [3H]E217G transport in HEK293-OATP1B1 cells. Pretreatment with monensin and bafilomycin A1, which also have lysosome inhibition activity, significantly decreased OATP1B1-mediated transport in HEK293-OATP1B1 cells. Pharmacoepidemiologic studies using data from the U.S. Food and Drug Administration Adverse Event Reporting System indicated that CQ plus pitavastatin, rosuvastatin, and pravastatin, which are minimally metabolized by the cytochrome P450 enzymes, led to higher myopathy risk than these statins alone. In summary, the present studies report novel findings that lysosome is involved in degradation of OATP1B1 protein and that pre-incubation with lysosomotropic drug CQ downregulates OATP1B1 transport activity. Our in vitro data in combination with pharmacoepidemiologic studies support that CQ has potential to cause OATP-mediated drug–drug interactions.
Keywords: organic anion transporting polypeptide (OATP), chloroquine (CQ), human sandwich-cultured hepatocytes (SCH), lysosomotropic drug, drug–drug interactions (DDIs), FDA Adverse Event Reporting System (FAERS)
Graphical abstract
Introduction
Drug–drug interactions (DDIs), which often lead to adverse drug effects, are a major concern in patients receiving multidrug therapy.1 The organic anion-transporting polypeptides (OATPs) belong to the solute carrier organic anion (SLCO) transporter superfamily.2 OATP1B1 is predominantly expressed on the basolateral membranes of hepatocytes and mediates the hepatic uptake of many drugs such as HMG-CoA reductase inhibitor statins, rifampicin, and paclitaxel as well as endogenous compounds.3,4 Decreased transport activity of OATP1B1 due to genetic variation [e.g., the single nucleotide polymorphism (SNP) V174A of OATP1B1] and DDIs is often associated with increased systemic exposure of statins and statin-induced myopathy.5–7 OATP1B1 has been recognized as an important determinant of transporter-mediated DDIs.8
Chloroquine (CQ), a 4-aminoquinoline class of drug, is a lysosomotropic agent that accumulates preferentially within the lysosome. It increases the intralysosomal pH,9 and inhibits lysosomal degradation of proteins including many transport proteins such as glutamate transporter10 and glucose transporter (GLUT) 1 and 2.11,12 The effect of CQ on OATP1B1 protein degradation has not been reported. CQ interacts with several transporters as substrate and/or inhibitor. CQ is a substrate and inhibitor of multidrug and toxin extrusion (MATE) 1.13 It also inhibits substrates transport mediated by P-glycoprotein (P-gp)14 breast cancer resistance protein (BCRP)14 and OATP1A2.15 In rat sandwich-cultured hepatocytes (SCH), pretreatment with 10 μM CQ for 48 h significantly decreased total accumulation of rosuvastatin in cells and bile canaliculi.16 Since rosuvastatin is a substrate of multiple transporters including OATP1B1,17 we hypothesize that CQ pretreatment may modulate the transport function of OATP1B1.
In addition to being used for treatment of malaria and amebic infection,18 CQ is also used for long-term treatment of rheumatoid arthritis and systemic lupus erythematosus.19–22 It is currently in clinical trials for cancer therapy either alone23–25 or in combination with chemotherapy drugs.26,27 Considering the widespread use of statins in cardiovascular diseases, concurrent administration of statins and CQ is likely. To the best of our knowledge, the effects of CQ on systemic exposure of statins and statin-associated myopathy have not been reported in clinical studies. The FDA Adverse Event Reporting System (FAERS) is a database that contains information on adverse events and medication error reports submitted to the FDA.28 This database is designed to support the FDA's postmarketing safety surveillance program for drug and therapeutic biologic products. The FAERS has been widely used for pharmacoepidemiologic studies including assessing statin-related myopathy risk due to DDIs.29,30 The current studies were designed to determine the effects of CQ on OATP1B1 protein degradation, OATP1B1-mediated transport in OATP1B1-overexpressing cell line, and statin uptake in human SCH. The myopathy risk was also compared in patients concomitantly taking CQ and statins vs statins alone using data from the FAERS.
The effects of CQ on OATP1B1 protein degradation and transport activity were determined in HEK293 stable cell lines overexpressing OATP1B1 (HEK293-OATP1B1) or FLAG-tagged OATP1B1 (HEK293-FLAG-OATP1B1). Pitavastatin and estradiol 17β-d-glucuronide (E217G), which are sensitive OATP1B1 substrates for studying the inhibition of OATP1B1 transport activity in vitro,31 were used as OATP1B1 probe substrates in HEK293-OATP1B1 and -FLAG-OATP1B1 stable cell lines. The effect of CQ on hepatic uptake of therapeutic drug pitavastatin was also determined in the physiologically relevant human SCH model. Pitavastatin, pravastatin, and rosuvastatin are OATP1B1 substrates32 and are minimally metabolized by the cytochrome P450 enzymes.33–35 These three statins were selected in current pharmacoepidemiologic studies to determine whether CQ plus statins (pitavastatin, rosuvastatin, and pravastatin) leads to a higher risk for myopathy than these statins alone, using data from the FAERS.
Experimental Section
Materials
Estradiol 17β-d-glucuronide (E217G), chloroquine diphosphate, IGEPAL (NP-40), Hanks balanced salt solution (HBSS), dexamethasone, dimethyl sulfoxide (DMSO), Triton X-100, Dulbecco's modified Eagle medium (DMEM), fetal bovine serum (FBS), trypsin–EDTA solution, antibiotic antimycotic solution, Dulbecco's phosphate-buffered saline (DPBS), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Monensin sodium salt was purchased from Santa Cruz (Dallas, TX). Bafilomycin A1 was purchased from LC Laboratories (Woburn, MA). Insulin, minimum essential medium (MEM) nonessential amino acids (NEAA), l-glutamine, and penicillin–streptomycin were purchased from Life Technologies (Grand Island, NY). Matrigel and insulin/transferrin/selenium (ITS+ premix) were purchased from BD Biosciences (Bedford, MA). Cultrex poly-l-lysine was purchased from Trevigen, Inc. (Gaithersburg, MD). Geneticin was purchased from Life Technology (Grand Island, NY). Complete protease inhibitor cocktail tablets were purchased from Roche Diagnostics (Indianapolis, IN). [3H]Pitavastatin (specific activity 5 Ci/mmol) and unlabeled pitavastatin were purchased from American Radiolabeled Chemicals (St. Louis, MO). [3H]E217G (specific activity 41.4 Ci/mmol) was purchased from PerkinElmer Life Science (Waltham, MA). Bio-Safe II liquid scintillation mixture was obtained from Research Products International (Mt. Prospect, IL). All other reagents were purchased from VWR International (Radnor, PA).
Estimation of Maximum Unbound Concentration of CQ at the Inlet to Liver
Maximum concentration of CQ at the inlet of the liver (Iin,max) was estimated according to eq 1, described previously.36
| (1) |
The values for the unbound fraction of CQ in the plasma (fu), absorption rate constant (ka), and hepatic blood flow (Qh) used in eq 1 are 0.45,18 0.1 min−1,37 and 1500 mL/min,38 respectively. The maximum concentration of CQ in the systemic circulation (Cmax) and associated dose (D) were obtained from the literature and are summarized in Table 2. Since CQ has high bioavailability and almost complete absorption after oral administration,18 the fraction of CQ absorbed from the gastrointestinal tract intact (FaFg) was set at 1.18,39 Unbound concentration of Iin,max (Iu,in,max) was calculated as fuIin,max.
Table 2. Estimation of CQ Maximum Unbound Concentration at the Inlet to the Liver (Iu,in,max).
| variable | |||
|---|---|---|---|
| dose of CQ (mg)a | 19148 | 30039 | 60049 |
| Cmax (μM) | 0.72–12.1848 | 0.2439 | 1.249 |
| Iu,in,maxbb (μM) | 18.3–23.4 | 28.2 | 56.8 |
CQ doses were converted to CQ base.
Estimated on the basis of eq 1.
Cell Culture
The human embryonic kidney (HEK) 293 stable cell line overexpressing OATP1B1 (HEK293-OATP1B1) was provided by Dr. Dietrich Keppler.3 A HEK293 stable cell line overexpressing FLAG-tagged OATP1B1 (HEK293-FLAG-OATP1B1) was established by transfection of a mammalian plasmid expression vector encoding C-terminus FLAG-tagged OATP1B1 (pCMV6-FLAG-OATP1B1), which was custom constructed through Origene (Rockville, MD). Both cell lines were maintained in DMEM medium containing 10% FBS, 1% antibiotic and antimycotic solution, and 600 μg/mL Geneticin (Life Technologies, Grand Island, NY). Cells were cultured in a humidified atmosphere (95% O2, 5% CO2) at 37 °C.
Human Sandwich-Cultured Hepatocytes (SCH)
Human hepatocytes were purchased from Triangle Research Laboratories, LLC (Research Triangle Park, NC). Demographics of the donors are listed in Table 1. Human SCH were cultured as described previously.40 In brief, on day 0, cells were plated at 3.5 × 105 cells per well in 24-well Biocoat culture plates in phenol red free DMEM containing 5% (v/v) FBS, 1% (v/v) MEM NEAA, 2 mM l-glutamine, 100 units of penicillin G sodium/mL, 100 g/mL of streptomycin sulfate, 1 μM dexamethasone, and 4 μg/mL insulin. Cells were cultured at 37 °C in a humidified incubator (95% O2, 5% CO2). At 6 h after seeding, when the cells were attached to the plate, cells were overlaid with Matrigel at a final concentration of 0.25 mg/mL in phenol red free DMEM supplemented with 2 mM l-glutamine, 1% (v/v) MEM NEAA, 100 units/mL of penicillin G sodium, 100 g/mL of streptomycin sulfate, 0.1 μM dexamethasone, and 1% (v/v) ITS+ premix. Culture medium was replaced every 24 h. Uptake experiments were conducted on day 2 of culture.
Table 1. Demographics of Human Hepatocyte Donors.
| donors | age | gender | race | BMIa | smoking | alcohol use | experiments |
|---|---|---|---|---|---|---|---|
| GC4008 | 69 | M | Caucasian | 24.7 | no | no | Figures 2C, 7B |
| HUM 4059 | 17 | F | Caucasian | 25.5 | no | no | Figure 7B |
| HUM 4089 | 36 | M | Caucasian | 30 | no | no | Figure 7A |
| HUM 4110 | 56 | M | Caucasian | 26 | yes | 8–10 beers/week | Figure 7B |
| HUM 4130 | 64 | M | African American | 23.6 | quit 3 months ago | none reported | Figure 2C |
| HUM 4140 | 21 | M | Caucasian | 32 | yes | rare | Figure 2C |
Body mass index.
Immunoblotting
Immunoblotting was conducted similarly to those described previously.41 In brief, cells were seeded at a density of 1.1 × 105 cells per well in 24-well culture plates precoated with poly-l-lysine and cultured for 48 h. Two cell lysis methods (with or without trypsinization) were compared to optimize the OATP1B1 immunoblot. After aspirating culture media and rinsing once with DPBS, whole cell lysates (WCL) were prepared by adding ice cold lysis buffer, which contained 50 mM Tris/HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% (v/v) NP-40, 0.5% Na-deoxycholate, and Complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), either directly onto the cells in culture plate (without trypsinization) or to cell pellets collected after trypsinization with 1× Trypsin/EDTA (Sigma-Aldrich, St. Louis, MO) and subsequent washing with DPBS (with trypsinization). Whole cell lysates (WCL) (50 μg) were resolved on 10% SDS–PAGE (Bio-Rad, Hercules, CA). Proteins were then transferred to nitrocellulose membranes and subsequently probed with the following primary antibodies: rabbit polyclonal OATP1B1 antibody (Fisher Scientific Company, LLC, Denver, CO) custom-generated according to a previous publication (1:1000 dilution),43 rabbit polyclonal anti-FLAG antibody (1:4000 dilution) and mouse monoclonal anti-β-actin antibody (1:5000 dilution) (Sigma-Aldrich, St. Louis, MO), mouse monoclonal anti-GAPDH antibody (1:2000) (Santa Cruz Biotechnology, Inc., Dallas, TX), and anti-alpha 1 sodium potassium ATPase antibody (1:8000 dilution) (Abcam, Cambridge, MA). After incubation with HRP-conjugated secondary antibody (Santa Cruz Biotechnology, Inc., Dallas, TX), signals were detected by Supersignal West Duro (Pierce, Rockford, IL) using a Bio-Rad ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). Image Lab v4.1 software (Bio-Rad Laboratories, Hercules, CA) was used to perform the densitometry analysis.
Immunofluorescence Staining
Experiments were conducted similarly to those published previously, with slight modifications.44 In brief, HEK293-FLAG-OATP1B1 cells were grown to confluence on poly-l-lysine coated coverslips in 24-well plates. Cells were fixed for 20 min with methanol, permeabilized with Triton X-100 (0.25%) in DPBS for 10 min at room temperature, and blocked with 5% BSA in DPBS for 1 h at room temperature. For co-immunofluorescence staining of FLAG-OATP1B1 and LAMP-2, cells were co-incubated with the rabbit polyclonal anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO, 1:100 dilution) and mouse monoclonal antibody of LAMP-2 (Clone H4B4, Santa Cruz Biotechnology, Inc., Dallas, TX, 1:50 dilution) at 37 °C for 2 h. After washing 3 times with DPBS, cells were co-incubated with secondary antibodies Alexa Fluor 488-conjugated goat anti-mouse IgG (Life Technologies, Grand Island, NY, 1:200 dilution) and Alexa Fluor 594-conjugated goat anti-rabbit IgG (Life technologies, Grand Island, NY, 1:200 dilution) at 37 °C for 1 h. Both primary and secondary antibodies were diluted in DPBS containing 5% BSA. Diamidino-2-phenylindole (DAPI) (300 nM) was used for counterstaining nuclei. After washing, the coverslips were mounted on glass slides with ProLong gold antifade reagent (Life Technologies, Grand Island, NY). Images were taken using Leica SP2 MP (Leica Microsystems, Inc., Buffalo Grove, IL) or Olympus FV1000 (Olympus Scientific Solutions Americas Corp., Waltham, MA) confocal microscopes as indicated in the figure legends.
Transport Studies
Transport studies in HEK293 stable cell lines and in human SCH were conducted similarly to those described previously.41,42 HEK293-OATP1B1, -FLAG-OATP1B1, and -Mock cells were seeded at a density of 0.8–1.1 × 105 cells/well in 24-well culture plates precoated with poly-l-lysine and were cultured for 48–72 h. Cells from stable cell lines or human SCH were either pretreated with culture medium containing CQ, monensin, bafilomycin A1, or vehicle control for the designated times or not pretreated. 0.1% DMSO was used as vehicle control for monensin and bafilomycin A1 treatment. As CQ stock (50 mM) was resolved in water, CQ-free fresh medium was used as vehicle control for CQ pretreatment. To determine the effects of CQ on OATP1B1-mediated transport after prolonged treatment, HEK293-OATP1B1 cells were preincubated with CQ-free (CTL) or 25 μM CQ-containing medium for 2 h. At the end of pre-incubation, the culture medium was aspirated and CTL cells were cultured in CQ-free medium and cells pretreated with 25 μM CQ were cultured in medium containing 1.5 μM CQ, for the indicated time, up to 24 h.
At the time of uptake experiments, after rinsing with prewarmed (37 °C) HBSS buffer (pH 7.4) three times, cells were incubated with HBSS containing [3H]E217G (1 μM, 2 min) or [3H]pitavastatin (1 μM, 0.5 min) in the absence or presence of testing drugs (CQ or rifampicin). At the end of incubation, the buffer was aspirated rapidly, and the cells were rinsed with ice-cold HBSS three times and then lysed with Triton X-100 (0.5% v/v) in DPBS. An aliquot of the lysate was subjected to liquid scintillation counting (LS6500 scintillation counter, Beckman Coulter, Brea, CA). Substrate accumulation was normalized to protein concentration determined by BCA assay (Pierce Chemical, Rockford, IL) and corrected for nonspecific binding of the substrate by including a non-overlaid poly-l-lysine coated blank plate for uptake studies in stable cell lines and a Matrigel overlaid blank plate for human SCH, respectively.
To determine [3H]E217G transport kinetic parameters, the maximal transport velocity (Vmax) and the affinity constant (Km), [3H]E217G accumulation (0.1–40 μM, 2 min) was determined in HEK293-OATP1B1 and -Mock cells following pretreatment with CQ(25 μM) or CTL. Values of [3H]E217G accumulation in Mock cells were subtracted from those in HEK293-OATP1B1 cells. The Vmax and Km values of E217G transport were estimated by fitting the Michaelis–Menten equation (eq 2) to the data using Phoenix WinNonlin, v6.3 (Certara, St. Louis, MO), where ν is E217G transport velocity and S is E217G concentration.
| (2) |
Lactate Dehydrogenase (LDH) Cytotoxicity Assay
Experiments were conducted similarly to those published previously.45 After treatment with CQ, monensin, bafilomycin A1, or respective vehicle control, cell culture medium was assayed for LDH activity with a cytotoxicity detection kit (Roche Diagnostics GmbH, Mannheim, Germany), according to the manufacturer's instructions. Triton X (2%) treated and non-treated cells served as the 100% cytotoxicity positive control and negative control, respectively.
Pharmacoepidemiologic Studies Using the FDA Adverse Event Reporting System (FAERS)
Studies using the FAERS database were similar to those published previously.29 All of the adverse event case reports from quarter 1 of 2004 to quarter 3 of 2012 were used for data analysis (n = 6.47 millions). The myopathy adverse event was defined as published previously.29 It includes all the mild or severe myopathy symptoms. The drug names and their synonyms were mapped based on their DrugBank46 drug names. Our drug interaction hypothesis was whether CQ plus statins (pitavastatin, rosuvastatin, and pravastatin) leads to higher myopathy risk than these statins alone. A similar hypothesis was posed to test the interactions between sulfonylureas and statins in our previous work.47 The interaction between CQ and statins was further tested in different subpopulations defined by age and gender.
Data Analysis
For the statistical analysis in Figures 4, 5, 6 and 7B, fold changes and associated standard errors (SEs) were estimated by generalized linear mixed models with the log link function, a fixed effect (treatment time or group), and a random effect (experiment date or hepatocyte donor), adjusting for treatment time/group-specific overdispersion. In the case of multiple comparisons, p-values were adjusted based on Bonferroni's method. The Chi-square test and Student's t test were used for statistical analysis in Table 3 and the Figure 8 inset, respectively. A two-sided p-value of <0.05 defines statistical significance. SAS software (version 9.3, Cary, NC) was used for statistical analyses.
Figure 4.
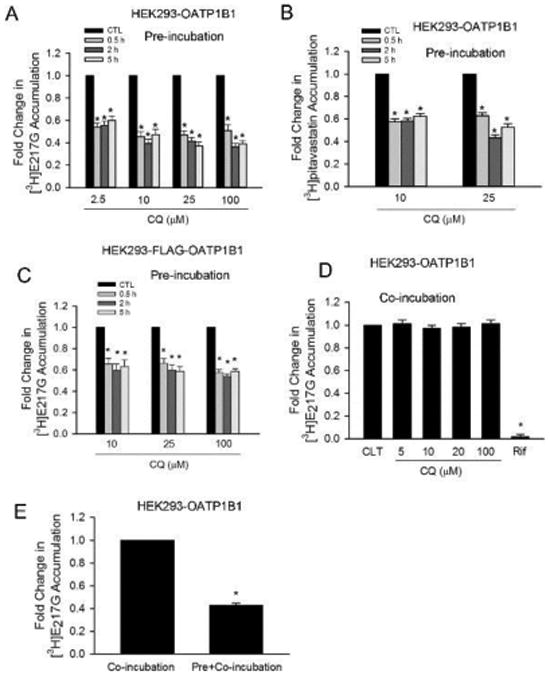
Pretreatment effect and direct inhibition of CQ on OATP1B1-mediated transport. Cells were seeded at 1.1 × 105 cells/well in 24-well plates and cultured for 48 h (A, B, C, and E) or 72 h. (D) Accumulation of [3H]E217G (1 μM, 2 min) or [3H]pitavastatin accumulation (1 μM, 0.5 min) was determined in HEK293-OATP1B1 or -FLAG-OATP1B1 cells, as indicated in the figures. (A) Model-estimated fold change and associated SE in [3H]E217G accumulation vs CTL treatment in HEK293-OATP1B1 cells at each indicated pretreatment concentration and time (pre-incubation). (B) Model-estimated fold change and associated SE in [3H]pitavastatin accumulation vs CTL treatment in HEK293-OATP1B1 cells at each indicated pretreatment concentration and time (pre-incubation). (C) Model-estimated fold change and associated SE in [3H]E217G accumulation vs CTL treatment in HEK293-FLAG-OATP1B1 cells at each indicated pretreatment concentration and time (pre-incubation). (D) Model-estimated fold change and associated SE in [3H]E217G accumulation in the presence of 5–100 μM CQ or 25 μM rifampicin (Rif) vs CTL in HEK293-OATP1B1 cells without CQ pretreatment (co-incubation). (E) Model-estimated fold change and associated SE in [3H]E217G accumulation vs co-incubation control. Following pretreatment in culture medium containing 100 μM CQ (pre+co-incubation) or vehicle control (co-incubation) for 5 h, HEK293-OATP1B1 cells were rinsed three times with HBSS, and the [3H]E217G accumulation was determined in the presence of 100 μM CQ. A generalized linear mixed model as described in the Experimental Section was fit to data in A–E (n = 3 in triplicate for all panels of A–E). To account for multiple comparisons, p-values were adjusted based on Bonferroni's method. * indicates a statistically significant difference (adjusted p < 0.05) vs CTL.
Figure 5.
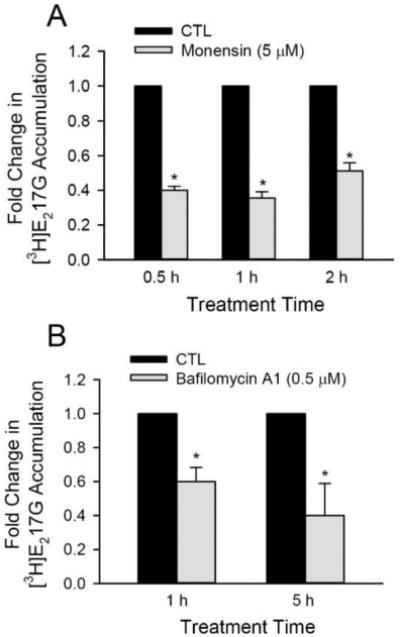
Effects of monensin and bafilomycin A1 on OATP1B1-mediated [3H]E217G transport. HEK293-OATP1B1 were seeded at 1.1 × 105 cells/well in 24-well plates for up to 72 h. (A) Model-estimated fold change and associated SE in [3H]E217G accumulation vs CTL at each indicated monensin (5 μM) pretreatment time. (B) Model-estimated fold change and associated SE in [3H]E217G accumulation vs CTL at each indicated bafilomycin A1 (0.5 μM) pretreatment time. A generalized linear mixed model as described in the Experimental Section was fit to data (n = 3 in triplicate for both A and B). * indicates a statistically significant difference (p < 0.05) vs CTL.
Figure 6.
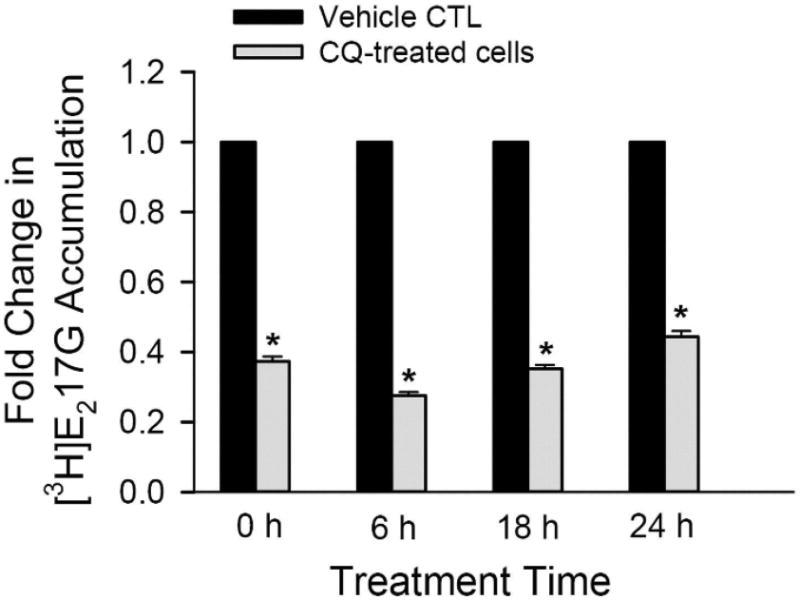
Effects of CQ on OATP1B1-mediated [3H]E217G transport after prolonged treatment in HEK293-OATP1B1 cells. HEK293-OATP1B1 cells were seeded at 0.8 × 105 cells/well in 24-well plates, and cultured for 48 h prior to treatment. Cells were preincubated with CQ-free (CTL) or 25 μM CQ-containing medium for 2 h. At the end of pre-incubation, the culture medium was removed. CTL medium preincubated cells were cultured in CQ-free medium (CTL, black bar) and 25 μM CQ-preincubated cells were cultured in medium containing 1.5 μM CQ (grey bar), for indicated times up to 24 h. [3H]E217G accumulation (1 μM, 2 min) was determined at indicated time points (n = 4 in triplicate). Fold change and SE in [3H]E217G accumulation vs CTL at each indicated time points were estimated by generalized linear mixed models, as described in the Experimental Section. * indicates a statistically significant difference (p < 0.05) vs CTL.
Figure 7.
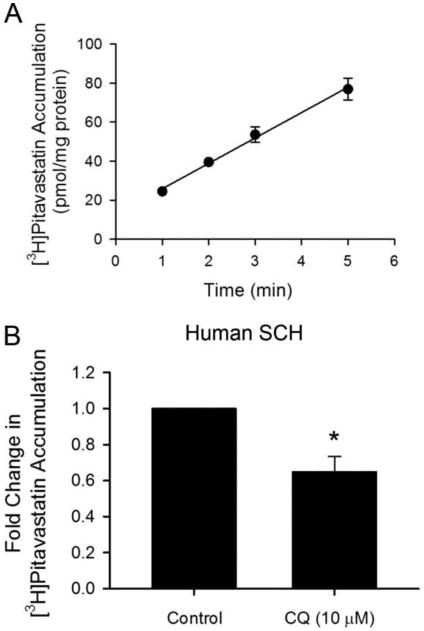
Effect of CQ pretreatment on the accumulation of [3H]pitavastatin in human SCH. (A) Time-dependent accumulation of [3H]pitavastatin (1 μM) in human SCH. Data represent the mean ± SD in triplicate from a single hepatocyte donor. (B) Model-estimated fold change and associated SE in [3H]pitavastatin accumulation vs CTL in human SCH pretreated with 10 μM CQ or CTL for 5 h. Following pretreatment in culture medium containing CQ (10 μM) or control, human SCH were rinsed three times with HBSS, and [3H]pitavastatin accumulation (1 μM, 0.5 min) was determined. Fold change and SE were estimated by a generalized linear mixed model, as described in the Experimental Section (n = 3 in triplicate). * indicates a statistically significant difference (p < 0.05) vs CTL.
Table 3. DDI-Myopathy Analysis Adjusted for Age and Sex.
| population | drugs | Na | Ma | riskb | relative risk/P-valuec |
|---|---|---|---|---|---|
| all | statinsd alone | 88,682 | 8,149 | 9.2% | 1.87/0.05 |
| CQ and statinsd | 58 | 10 | 17.2% | ||
| women | statinsd alone | 44,212 | 4,234 | 9.6% | 2.28/0.03* |
| CQ and statinsd | 32 | 7 | 21.9% | ||
| men | statinsd alone | 44,123 | 3,743 | 8.5% | 1.35/0.48 |
| CQ and statinsd | 26 | 3 | 11.5% | ||
| >50 yearse | statinsd alone | 50,322 | 5,347 | 10.6% | 1.89/0.06 |
| CQ and statinsd | 40 | 8 | 20.0% | ||
| <50 yearse | statinsd alone | 33,413 | 2,732 | 8.2% | 1.35/0.65 |
| CQ and statinsd | 18 | 2 | 11.1% |
N: Number of patients taking drug. M: Number of myopathies. N and M represent sums of counts for each individual statin.
Risk is calculated as M/N × 100%.
Relative risk is calculated as risk (CQ and statins)/risk (statins alone).
Statistical analysis was performed with the Chi-square test.
indicates a statistically significant difference (p < 0.05) (CQ and statins vs statins alone).
Statins: rosuvastatin, pravastatin, and pitavastatin.
Age.
Figure 8.
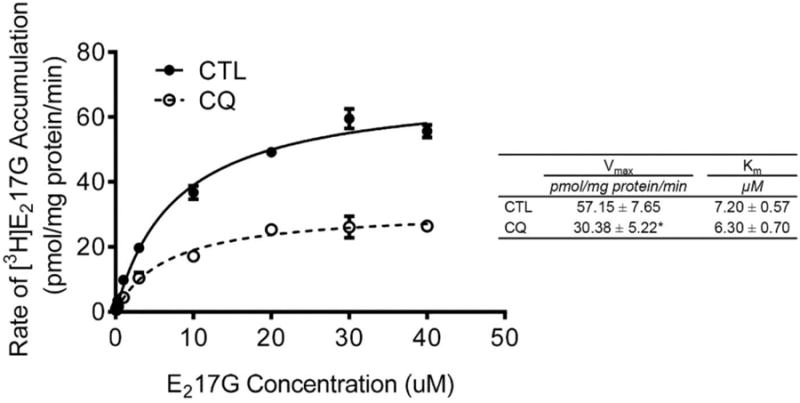
Effects of CQ on kinetic parameters of OATP1B1-mediated [3H]E217G transport. HEK293-OATP1B1 and -Mock cells were seeded at 1.1 × 105 cells/well in 24-well plates and were cultured for 48 h prior to experiment. The concentration-dependent accumulation of [3H]E217G (0.1–40 μM, 2 min) was determined in HEK293-OATP1B1 and -Mock cells pretreated with CTL or CQ (25 μM, 2 h). Values of [3H]E217G accumulation in Mock cells were subtracted from those in HEK293-OATP1B1 cells. Vmax and Km values were determined as described in the Experimental Section. Solid and dashed lines represent the best fits of the Michaelis–Menten equation to the data of CTL (black circles) and CQ (25 μM, 2 h) pretreatment (white circles), respectively. Representative graph of three independent experiments in triplicate is shown. Student's t test was conducted to compare the Vmax and Km values between CQ and CTL pretreatment. * indicates a statistically significant difference (p < 0.05; CQvs CTL).
Results
Estimation of the Maximum Unbound CQ Concentration at the Inlet to the Liver
The Iu,in,max of CQ was estimated to be 18.3–23.4, 28.2, and 56.8 μM based on data in rheumatoid arthritis patients chronically treated with CQ at a daily dose of 191 mg48 and after a 300 and 600 mg single dose of CQ in healthy volunteers,39,49 respectively (Table 2). CQ concentrations of 2.5–25 μM, which are clinically relevant, were used in subsequent experiments.
CQ Treatment Increased Total Protein Levels of OATP1B1 in HEK293-OATP1B1 Cells and in Human SCH
Initially, we tested the specificity of our custom-generated OATP1B1 antibody and optimized OATP1B1 immunoblot protocol by comparing two cell lysis methods, directly lysing the cells in the culture plate without trypsinization and lysing the cell pellets collected after trypsinization. As shown in Figure 1, OATP1B1 antibody specifically detected OATP1B1 in HEK293-OATP1B1 stable cell line but not in negative control HEK293-Mock (Mock) cells in whole cell lysates prepared with both methods. In cells that were lysed directly in the culture plates without trypsinization, the molecular weight of OATP1B1 detected by the OATP1B1 antibody was ∼90 kDa. A lower molecular weight band of OATP1B1 (∼64 kDa) was prominent in cell lysates prepared from trypsinized HEK293-OATP1B1 cells, while this band was negligible in cells lysed directly without trypsinization. Since the molecular weight of OATP1B1 (∼90 kDa) that resulted from the direct lysis method is similar to that published previously,3 this method was used to prepare whole cell lysates for immunoblot in subsequent experiments.
Figure 1.
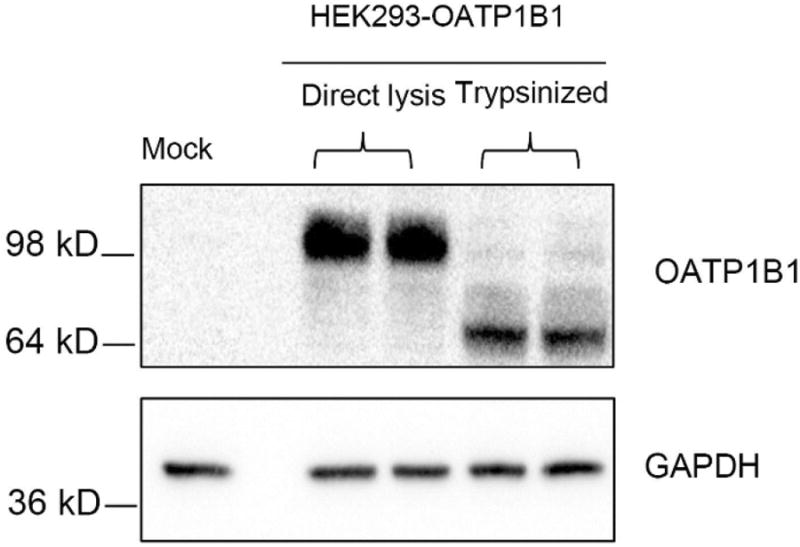
Characterization of OATP1B1 antibody and optimization of OATP1B1 immunoblot protocol. HEK293-OATP1B1 and -Mock cells were seeded at 1.1 × 105 cells/well in 24-well plates and cultured for 48 h prior to being harvested for immunoblot. Whole cell lysates (WCL) of HEK293-OATP1B1 cells were prepared either by adding ice-cold lysis buffer directly onto the culture plate after aspirating culture medium without trypsinization or by lysing the cells after trypsinization and subsequent washing. Immunoblot of OATP1B1 was performed with the custom-generated OATP1B1 antibody. GAPDH was used as the loading control. Representative images from n = 3 experiments are shown.
To determine the involvement of lysosome in degradation of OATP1B1, total protein levels of OATP1B1 were compared in HEK293-OATP1B1 cells treated with CQ or CTL by OATP1B1 immunoblotting. Treatment with 25 μM CQ for 2 and 5 h and 100 μM CQ for 5 h increased OATP1B1 total protein levels to 1.2 ± 0.1, 1.5 ± 0.1 and 1.5 ± 0.3 fold of control, respectively (Figure 2A).
Figure 2.
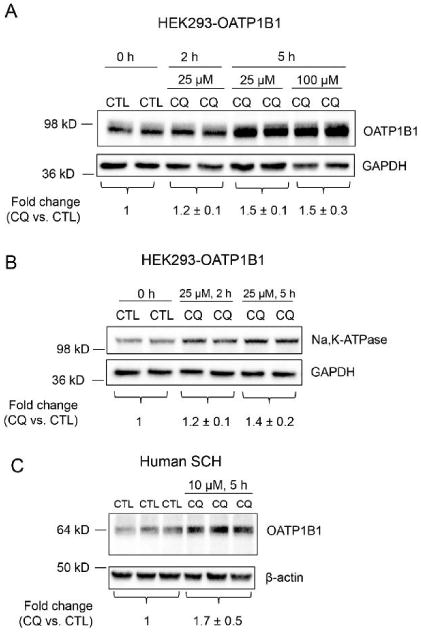
Effects of CQ on total protein levels of OATP1B1 and Na,K-ATPase in HEK293-OATP1B1 cells and on OATP1B1 total protein levels in human SCH. HEK293-OATP1B1 cells were seeded in 24-well plates at 1.1 × 105 cells/well, and were cultured for 48 h prior to treatment. Human SCH were cultured as described in the Experimental Section. WCL was prepared by directly lysing the cells in the culture plate without trypsinization. Representative immunoblot images of OATP1B1 (A) and Na,K-ATPase (B) in WCL of HEK293-OATP1B1 pretreated with CQ (25 or 100 μM) or CTL for indicated times are shown. GAPDH were used as the loading control for A and B. OATP1B1 and Na,K-ATPase protein levels determined by densitometry were normalized to levels of GAPDH. Fold changes of total protein levels of OATP1B1 in A and Na,K-ATPase in B (CQ vs CTL) are expressed as mean ± SD (n = 4 for both A and B). (C) Immunoblot of OATP1B1 and β-actin in WCL of human SCH pretreated with CQ (10 μM) or CTL for 5 h. OATP1B1 protein levels determined by densitometry were normalized to levels of β-actin. Fold changes of total protein levels of OATP1B1 (CQ vs CTL) are expressed as mean ± SD of n = 3 donor hepatocytes. Representative images are shown.
Increased total protein levels of Na,K-ATPase following lysosome inhibition by CQ treatment were reported previously.50 In the current studies, we used Na,K-ATPase as a positive control for increased total protein levels following lysosome inhibition. As shown in Figure 2B, treatment with CQ (25 μM) for 2 and 5 h increased Na,K-ATPase total protein levels to 1.2 ± 0.1 and 1.4 ± 0.2 fold of control, respectively.
Total protein levels of endogenous OATP1B1 were also compared in human SCH treated with CQ or vehicle control. As shown in Figure 2C, treatment with 10 μM CQ for 5 h increased OATP1B1 protein levels to 1.7 ± 0.5 fold of control in human SCH (ranging from 1.2–2.1-fold of control, n = 3 donors). In human SCH, a major OATP1B1 band at ∼64 kDa was observed (Figure 2C), similar as published previously.51
Colocalization of Intracellular FLAG-OATP1B1 with LAMP-2 in HEK293-FLAG-OATP1B1 Stable Cell Line
olocalization of OATP1B1 with LAMP-2, a late endosome/lysosome marker protein,52 was determined by immunofluorescence staining. Since our custom-generated OATP1B1 antibody is not suitable for immunofluorescence staining (data not shown), a stable cell line overexpressing FLAG-tagged OATP1B1 was established in HEK293 cells to overcome this limitation. Expression and transport function of FLAG-OATP1B1 in HEK293-FLAG-OATP1B1 cells were confirmed by immunoblotting and [3H]E217G accumulation, respectively. Both OATP1B1 and FLAG antibodies specifically detected the FLAG-tagged OATP1B1 in HEK293-FLAG-OATP1B1 cells, but not in the negative control HEK293-Mock cells (Figure S1A). The [3H]E217G accumulation (1 μM, 2 min) in HEK293-FLAG-OATP1B1 cells was ∼70-fold higher than in the HEK293-Mock cells (Figure S1B).
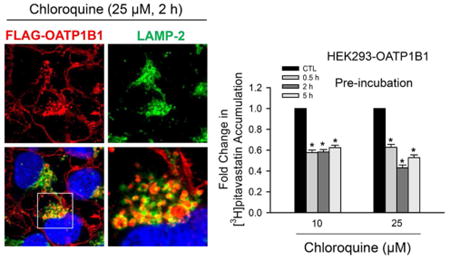
FLAG immunofluorescence staining showed specific staining in HEK293-FLAG-OATP1B1 cells but not in HEK293-Mock cells (data not shown). In both CQ-treated and vehicle control treated cells, in addition to the plasma membrane localization of FLAG-OATP1B1, intracellular localization of FLAG-OATP1B1 was also observed, which had a punctuated staining pattern reminiscent of vesicular compartments (Figure 3G–O, red). Some intracellular FLAG-OATP1B1 was detected in LAMP-2 positive vacuoles and colocalized with LAMP-2 (Figure 3J–O, as indicated by arrows and yellow coloring in merged images). Enlarged LAMP-2-positive vacuoles with FLAG-OATP1B1 protein retained inside were readily detected in cells treated with 25 μM CQ (Figure 3K,L,N,O) and 100 μM CQ (Figure S2H), consistent with inhibition of lysosomal degradation of FLAG-OATP1B1 by CQ.
Figure 3.
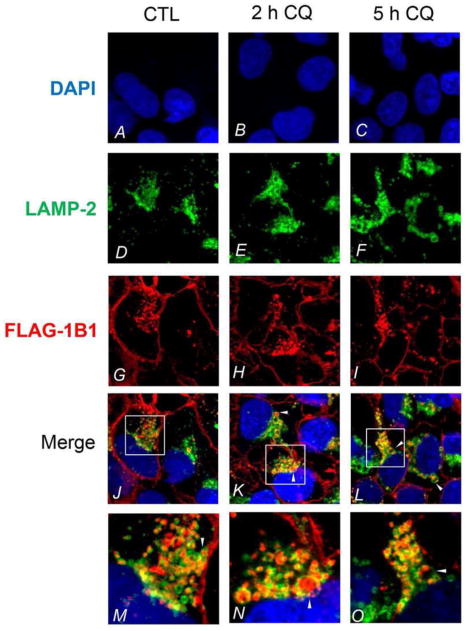
Colocalization of FLAG-OATP1B1 and LAMP-2 in HEK293-FLAG-OATP1B1 cells. Coimmunofluorescence staining of FLAG-OATP1B1 (red) and LAMP-2 (green) was performed in HEK293-FLAG-OATP1B1 cells treated with vehicle CTL or 25 μM CQ for 2 and 5 h as described in the Experimental Section. White arrow heads indicate the accumulation of FLAG-OATP1B1 inside the LAMP-2-positive vacuoles. Nuclei were counterstained with DAPI (blue). Images were taken using Olympus FV1000 confocal microscope. Representative images from the same experiments are shown (3 and 2 separate experiments for 2 h and 5 h treatment, respectively).
Pretreatment Effects and Direct Inhibition of CQ on OATP1B1-Mediated Transport
To determine the pretreatment effects of CQ on OATP1B1-mediated transport, HEK293-OATP1B1 or -FLAG-OATP1B1 was preincubated with CQ or vehicle CTL at indicated concentrations for 0.5–5 h. After washing, accumulation of [3H]E217G (1 μM, 2 min) or [3H]pitavastatin (1 μM, 0.5 min) was determined in the absence of CQ (Figure 4A–C, pre-incubation). In HEK293-OATP1B1 cells, at all CQ concentrations (2.5–100 μM) and time points determined (0.5–5 h), pretreatment with CQ significantly decreased [3H]E217G accumulation (all p < 0.001 vs control, Figure 4A). CQ pretreatment (10 and 25 μM) for 0.5–5 h also significantly decreased [3H]pitavastatin accumulation (all p < 0.001 vs control, Figure 4B). In HEK293-FLAG-OATP1B1 cells, [3H]E217G accumulation was significantly decreased following pre-incubation with CQ (10–100 μM) for 0.5–5 h (all p < 0.05 vs control, Figure 4C).
To determine the direct interaction of CQ on OATP1B1-mediated transport, experiments were conducted in HEK293-OATP1B1 cells without CQ pre-incubation. [3H]E217G accumulation (1 μM, 2 min) was determined in the presence of CQ at indicated concentrations, vehicle CTL, or positive control rifampicin (Figure 4D, co-incubation). As shown in Figure 4D, co-incubation with 25 μM rifampicin, a potent inhibitor of OATP1B1,53 significantly decreased [3H]E217G accumulation to 0.02 ± 0.021 fold of control (adjusted p < 0.01). However, there is no significant difference in [3H]E217G accumulation in the presence of CQ (5–100 μM) vs CLT (all adjusted p > 0.05).
The effect of CQ on OATP1B1-mediated [3H]E217G accumulation was further compared between two scenarios, a pre+co-incubation scenario, when [3H]E217G accumulation (1 μM, 2 min) was determined in the presence of 100 μM CQ after pre-incubation with CQ (100 μM, 5 h), and a co-incubation scenario as described above. For comparison purposes, 100 μM CQ co-incubation data in Figure 4D was replotted in Figure 4E. Under pre+co-incubation conditions, [3H]E217G accumulation decreased to 0.432 ± 0.02 fold compared to values measured under the co-incubation condition (p < 0.001, Figure 4E). The LDH assay showed negligible toxicity after 5 h treatment with 100 μM CQ in HEK293-OATP1B1 cells (Figure S3A).
Effects of Monensin and Bafilomycin A1 on OATP1B1-Mediated Transport
Three classes of compounds are frequently used to elevate the pH in endosome and other acidic organelles, which include lysosomotropic weak bases (e.g., chloroquine54), carboxylic ionophores (e.g., monensin55), and inhibitors of vacuolar H+-ATPase (e.g., bafilomycin A156) (reviewed by Huotari57). In addition to CQ, monensin and bafilomycin A1 are also reported to inhibit endosome–lysosome system acidification and lysosome activity.58,59 We determined whether pretreatment with monensin or bafilomycin A1 also affects OATP1B1-mediated [3H]E217G accumulation (1 μM, 2 min) in HEK293-OATP1B1 cells. Pretreatment with monensin (5 μM) for 0.5–2 h significantly decreased [3H]E217G accumulation ranging from 0.356 ± 0.036 to 0.512 ± 0.047 fold of CTL (all p < 0.0001 vs. CTL) (Figure 5A). Pretreatment with bafelomycin A1 (0.5 μM) for 1 and 5 h significantly decreased [3H]E217G accumulation to 0.60 ± 0.083 to 0.40 ± 0.19 fold of CTL, respectively (all p < 0.01 vs. CTL) (Figure 5B). The LDH assay showed negligible toxicity after monensin and bafilomycin A1 treatment in HEK293-OATP1B1 cells (Figure S3A).
Effects of CQ on OATP1B1-Mediated Transport after Prolonged Treatment
CQ is administered daily for long-term treatment of rheumatoid arthritis and lupus.22 In patients who had been taking CQ for at least six months, the dose normalized average unbound steady state concentration (Css) of CQ is ∼1.7 μM.48 The effect of CQ on OATP1B1-mediated transport after prolonged treatment was determined in HEK293-OATP1B1 cells. After 2 h treatment with CQ-free medium or medium containing 25 μM CQ, cells were subsequently cultured in CQ-free medium or 1.5 μM CQ-containing medium, respectively. Values of [3H]E217G accumulation in CQ-treated cells were compared to CQ-free treatment (CTL) at indicated times. A similar trend as in Figure 4A, CQ pretreatment (25 μM, 2 h) significantly decreased [3H]E217G accumulation to 0.373 ± 0.014 fold of control (Figure 6, 0 h). At each time point of 6, 18, and 24 h, [3H]E217G accumulation in 1.5 μM CQ-containing medium was significantly decreased compared to CTL (all p < 0.0001 vs CTL) (Figure 6). The LDH assay showed negligible toxicity following 24 h incubation with 1.5 μM containing medium in HEK293-OATP1B1 cells pretreated with CQ(25 μM, 2 h) (Figure S3B).
Pretreatment with CQ Significantly Decreased [3H]-Pitavastatin Accumulation in Human SCH
The effects of CQ on OATP1B1 transport function was further determined in the physiologically relevant human SCH model with [3H]pitavastatin60 as a probe substrate. [3H]Pitavastatin (1 μM) accumulation in human SCH was linear at least up to 5 min (Figure 7A). An incubation time of 0.5 min was used for subsequent uptake experiments in human SCH. Pretreatment with CQ (10 μM, 5 h) significantly decreased [3H]pitavastatin accumulation to 0.65 ± 0.084 fold of control (p < 0.01 vs control, Figure 7B). The LDH assay showed negligible toxicity after 5 h treatment with CQ at 10 μM (Figure S3C).
DDIs Associated with Increased Risk of Myopathy
To determine whether CQ plus statins (rosuvastatin, pravastatin, and pitavastatin) leads to higher myopathy risk than these statins alone, pharmacoepidemiological studies were conducted using data from the FAERS database. As shown in Table 3, among all patient data, CQ plus statins led to 17.2% myopathy risk compared to statins alone, 9.2%. The relative risk (RR) is 1.87 with a p-value of 0.05. It has been reported that the female gender and advanced age are associated with higher risk for myopathy.61 Therefore, the interaction effect between CQ and statins was further analyzed within the subpopulations. The only sub-population that showed significant CQ and statin DDIs was women. In women, CQ plus statins significantly increased the myopathy risk from 9.6% to 21.9% (Relative Risk = 2.28, p < 0.05 vs. statins alone). In other subpopulation analyses, men, >50 years, and <50 years, the CQ plus statins all showed increased myopathy risk, but were not statistically significant.
Effects of CQ on Transport Kinetics of [3H]E217G in HEK293-OATP1B1 Cells
As shown in Figure 8, CQ pretreatment (25 μM, 2 h) significantly decreased the maximum rate of [3H]E217G uptake (Vmax) values compared to vehicle CTL treatment (57.15 ± 7.65 vs 30.38 ± 5.22 pmol/mg protein/min, p < 0.05), without affecting the Km values (7.20 ± 0.57 vs 6.30 ± 0.70 μM).
Discussion
Understanding the mechanism(s) underlying altered transport activity of OATP1B1 has significant relevance in predicting potential OATP1B1-mediated DDIs. The current study demonstrates that the lysosome pathway is involved in degradation of OATP1B1, and that pre-incubation with lysosomotropic drug CQ downregulates OATP1B1-mediated transport. Pharmacoepidemiologic studies using data from the FAERS indicated that CQ plus statins (pitavastatin, rosuvastatin, and pravastatin) led to a greater myopathy risk than these statins alone.
Plasma membrane proteins are constitutively subjected to endocytosis.57 A portion of the internalized cargo is transported via late endosome for degradation by lysosomes.57 Treatment with CQ increased the total protein levels of OATP1B1 expressed exogenously in HEK293-OATP1B1 cells and endogenously in human SCH (Figure 2A,C). Increased total protein levels of membrane proteins following CQ treatment was reported previously for bone morphogenic protein type II receptor (BMPR-II),62 the transmembrane protein β-site amyloid precursor protein cleaving enzyme (BACE),63 and Na,K-ATPase.50 Increased Na,K-ATPase total protein levels following CQ treatment was also detected in current studies (Figure 2B), consistent with previous report.50 Plasma membrane localization of OATP1B1 has been detected by immunofluorescence staining in several reports.3,53,64 However, association of intracellular OATP1B1 with subcellular organelles has not been reported. Current studies demonstrated that, in addition to plasma membrane localization, the intracellular FLAG-OATP1B1 proteins were localized to LAMP-2 positive late endosomes/lysosomes in HEK293-FLAG-OATP1B1 cells without CQ treatment and under lysosome inhibition following CQ treatment (Figure 3 and Figure S2 merged images). Overall, increased total protein levels of OATP1B1 upon CQ treatment and localization of OATP1B1 with LAMP-2 suggest that the lysosome pathway is involved in the degradation of OATP1B1 proteins.65
In the current studies, we focused on determining whether clinically relevant concentrations of CQ affect OATP1B1 transport activity and statin uptake in human SCH. The estimated Iu,in,max of CQ is 18.3–56.8 μM (Table 2). The unbound Css of CQ in the plasma is ∼1.7 μM in rheumatoid arthritis patients treated with CQ daily for long-term.48 In HEK293-OATP1B1 cells, pretreatment with CQ at clinically relevant concentrations (2.5–25 μM) significantly decreased OATP1B1-mediated transport of [3H]E217G (Figure 4A,C) and [3H]pitavastatin (1 μM, 0.5 min) (Figure 4 B). CQ pretreatment (10 μM, 5 h) also significantly decreased [3H]pitavastatin accumulation (1 μM, 0.5 min) in human SCH (Figure 7B). Additionally, in HEK293-OATP1B1 cells preincubated for 2 h with 25 μM CQ (a concentration relevant to the Iu,in,max of CQ), and subsequently treated for up to 24 h with 1.5 μM CQ (a concentration relevant to the unbound Css of CQ), values of [3H]E217G accumulation were significantly less than those in the CQ-free treatment CTL (Figure 6). These findings suggest that pretreatment with CQ at clinically relevant concentrations can downregulate OATP1B1-mediated transport in OATP1B1-overexpressing cell lines and accumulation of pitavastatin in human SCH. Pitavastatin is a substrate of multiple OATP transporters, including OATP1B1, 1B3, and 2B1.60,66 Currently, there is no known specific probe substrate or inhibitor of OATP1B1 available. This is a technical challenge to specifically study the transport function of endogenous OATP1B1 in primary human hepatocytes. Therefore, from our current study, we cannot draw a conclusion regarding whether transport function of endogenous OATP1B1 is downregulated by CQ in human SCH. The current study does not exclude the possibility that CQ may affect transport activity of other transporters in human SCH. Decreased pitavastatin accumulation in human SCH following CQ pretreatment (10 μM, 5 h) may represent a net effect of CQ on hepatic transport of pitavastatin. Further studies are warranted to determine whether other OATP transporters, including the closely related family member OATP1B3, can also be regulated by CQ.
The inhibitory effects of CQ toward OATP1B1 transport activity at clinically relevant concentrations led to our hypothesis that concurrent use of CQ and statins may have a higher myopathy risk than statins alone in patients. Pharmacoepidemiologic studies using data from the FAERS was utilized to test our hypothesis. Since CQ is a substrate and inhibitor of multiple CYP enzymes,67,68 to minimize potential CYP450 mediated DDIs, we selected three statins, pitavastatin, rosuvastatin, and pravastatin, which are minimally metabolized through CYP450 enzymes,34,35,69 in our pharmacoepidemiologic studies. Increased systemic exposure of these statins has been reported in subjects bearing the V174A polymorphism of OATP1B1 (reviewed by Niemi32). Our data indicated that CQ plus these three statins is associated with increased risk of myopathy compared to these statins alone (Table 3). This result is consistent with the in vitro inhibitory effects of CQ toward OATP1B1-mediated transport and pitavastatin accumulation in human SCH following pretreatment at clinically relevant concentrations. Taken together, combination of our data from in vitro and pharmacoepidemiologic studies supports that potential OATP-mediated DDIs caused by CQ are likely.
A higher myopathy risk in women than men was observed in both the statins alone group and the CQ plus statins group (Table 3). The relative risk of myopathy (CQ plus statins vs statins alone) reaches a statistically significant level only in women, but not in men (Table 3). These findings are consistent with what we have observed in the medical record database76 and reports from others61,77 that females have higher risk for myopathy than males. Smaller vascular volumes and reduced muscle mass in females, which may result in greater tissue drug exposure per statin dose, are possible reasons of higher myopathy risk in women.61 Higher systemic exposure of statins in women was observed for pitavastatin,78 while there is no gender difference in systemic exposure of pravastatin.79 For rosuvastatin, one study reported higher AUC and Cmax in women than in men,80 while no gender difference was observed for rosuvastatin pharmacokinetics in another study.81 The gender difference in pharmacokinetics of CQ and the effects of CQ on pharmacokinetics of statins have not been reported. A clinical DDI study determining the effects of CQ on systemic exposure of statins in women and men may help to elucidate the potential mechanism underlying the higher myopathy risk in patients in whom CQ and statins are administered concurrently compared with statins alone.
Recently, the effects of pretreatment on OATP1B1-mediated transport were reported for several OATP1B1 inhibitors. Pretreatment with cyclosporine,70 saquinavir, ritonavir,71 simeprevir, asunaprevir, or daclatasvir72 significantly decreases OATP1B1-mediated transport. Pretreatment with cyclosporine decreased the apparent IC50 values toward OATP1B1 inhibition.31,73 In current studies, without CQ pre-incubation, CQ at concentrations 5–100 μM did not affect [3H]E217G accumulation (1 μM, 2 min) in HEK293-OATP1B1 cells (Figure 4D). This result is consistent with a recent report that 10 μM CQ did not affect OATP1B1-mediated estrone-3-sulfate transport.15 However, when [3H]E217G accumulation was determined in the presence of CQ following CQ pre-incubation, CQ, an apparent non-inhibitor of OATP1B1 determined without pre-incubation (Figure 4D), significantly decreased OATP1B1-mediated transport (Figure 4E). The current study reports a novel inhibitory effect of lysosomotropic drug CQ toward OATP1B1-mediated transport, where an apparent noninhibitor CQ “gained” its inhibitory effect toward OATP1B1 after pre-incubation. In clinical conditions, a perpetrator drug may be administered prior to the victim drug. A perpetrator drug and a victim drug may coexist in the body for a period of time that is longer than the in vitro incubation time used to determine the perpetrator/substrate interaction (a few minutes in many cases). Our results together with others31,73,74 highlight the importance of introducing a pre-incubation step when assessing the OATP1B1-mediated DDI potential of an investigational drug in vitro. The current approach of using the OATP1B1 inhibition tree and R-value to assess OATP-mediated DDIs in vitro assumes competitive transporter inhibition by investigational drugs.75 Our data support that mechanism(s) other than competitive inhibition of OATP1B1 transport activity may also be important in causing OATP1B1-mediated DDIs.
The finding that CQ pretreatment increased total OATP1B1 protein levels, but decreased OATP1B1 transport activity, may seem paradoxical at first. However, transporter function is not always correlated with the total protein levels of the transporters. For example, following PKC activation, the total protein levels of OATP1B3,41 OATP1A2,82 and OAT1 and OAT483,84 were not affected while their transport function was significantly decreased. CQ pretreatment decreased Vmax without affecting the Km values of OATP1B1-mediated [3H]E217G transport (Figure 8). A similar trend of decreased Vmax and unaffected Km was reported for OATP1A282 and OAT1,3 and 4,83–85 dopamine transporter,86 and sodium-glucose transporter (SGLT1)87 upon PKC activation. In these previous studies, the decreased Vmax of substrate transport was associated with decreased surface levels for OATP1A2,82 OAT1, 3 and 4,83–85 increased phosphorylation for dopamine transporter,86 or decreased turnover rate but unaltered surface levels for SGLT1.87 CQ has been reported to affect cell surface levels of several plasma membrane proteins such as the tumor necrosis factor receptor (TNF-R),88 the α-macroglobulin-protease (αM-P) surface receptors,89 and BMPR-II.62 Short-term treatment with monensin and bafilomycin A1, which have lysosome inhibition activity, also resulted in significantly decreased OATP1B1-mediated transport (Figure 5). In addition to inhibiting lysosomal activity, CQ, monensin, and bafilomycin A1 have other effects such as affecting trafficking of the membrane protein.90 The exact mechanism(s) through which CQ, monensin, and bafilomycin A1 downregulate transport function of OATP1B1 remain unknown and warrant further investigation.
In conclusion, the present studies report novel findings that CQ inhibits lysosome degradation of OATP1B1 protein; pre-incubation with CQ downregulates OATP1B1-mediated transport in stable cell lines, and decreases pitavastatin uptake in human SCH. Our in vitro data in combination with pharmacoepidemiologic studies support that CQ has potential to cause OATP-mediated DDIs.
Supplementary Material
Acknowledgments
We thank Dr. Dietrich Keppler for providing the HEK293-OATP1B1 and HEK293-Mock stable cell lines. This research was supported by NIH R01 GM094268 [W.Y.], R01 DK102694 [L.L.], GM10448301-A1 [L.L.], and R01LM011945 [L.L.]. The SCH studies were conducted under a research agreement between OUHSC and Qualyst Transporter Solutions, LLC (Durham, NC). The confocal microscopy was conducted at the Cell Biology Imaging Facility and Core Facility of OUHSC. We acknowledge John Powell's technical assistant in characterization of the OATP1B1 antibody.
Abbreviations Used
- E217G
estradiol 17β-d-glucuronide
- ABC
ATP-binding cassette
- DDIs
drug–drug interactions
- HBSS
Hanks balanced salt solution
- DMEM
Dulbecco's modified Eagle medium
- BSA
bovine serum albumin
- MEM
minimum essential medium
- NEAA
nonessential amino acids
- DPBS
Dulbecco's phosphate-buffered saline
- HEK293
human embryonic kidney 293
- LDH
lactate dehydrogenase
- OATP
organic anion transporting polypeptide
- OATs
organic anion transporters
- SLCO
solute carrier organic anion
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.5b00763.
Characterization of HEK293-FLAG-OATP1B1 stable cell line, colocalization of FLAG-OATP1B1 with LAMP-2, treated with CQ (100 μM, 5 h) and CTL, and cytotoxicity in HEK293-OATP1B1 cells and human SCH (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Zhang L, Zhang YD, Zhao P, Huang SM. Predicting drug-drug interactions: an FDA perspective. AAPS J. 2009;11(2):300–306. doi: 10.1208/s12248-009-9106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pfluegers Arch. 2004;447(5):653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 3.König J, Cui Y, Nies AT, Keppler D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver Physiol. 2000;278(1):G156–G164. doi: 10.1152/ajpgi.2000.278.1.G156. [DOI] [PubMed] [Google Scholar]
- 4.König J. Uptake transporters of the human OATP family: molecular characteristics, substrates, their role in drug-drug interactions, and functional consequences of polymorphisms. Handb Exp Pharmacol. 2011;201:1–28. doi: 10.1007/978-3-642-14541-4_1. [DOI] [PubMed] [Google Scholar]
- 5.Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/Drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. JAIDS, J Acquired Immune Defic Syndr. 2008;47(5):570–578. doi: 10.1097/QAI.0b013e318160a542. [DOI] [PubMed] [Google Scholar]
- 6.Shitara Y, Itoh T, Sato H, Li AP, Sugiyama Y. Inhibition of transporter-mediated hepatic uptake as a mechanism for drug-drug interaction between cerivastatin and cyclosporin A. J Pharmacol Exp Ther. 2003;304(2):610–616. doi: 10.1124/jpet.102.041921. [DOI] [PubMed] [Google Scholar]
- 7.SEARCH Collaborative Group. SLCO1B1 variants and statin-induced myopathy–a genomewide study. N Engl J Med. 2008;359(8):789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 8.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discovery. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981;90(3):665–669. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanoni C, Massari S, Losa M, Carrega P, Perego C, Conforti L, Pietrini G. Increased internalisation and degradation of GLT-1 glial glutamate transporter in a cell model for familial amyotrophic lateral sclerosis (ALS) J Cell Sci. 2004;117(Part 22):5417–26. doi: 10.1242/jcs.01411. [DOI] [PubMed] [Google Scholar]
- 11.Rosa SC, Goncalves J, Judas F, Mobasheri A, Lopes C, Mendes AF. Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res Ther. 2009;11(3):R80. doi: 10.1186/ar2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou JC, Williams D, Vicogne J, Pessin JE. The glucose transporter 2 undergoes plasma membrane endocytosis and lysosomal degradation in a secretagogue-dependent manner. Endocrinology. 2009;150(9):4056–64. doi: 10.1210/en.2008-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller F, Konig J, Glaeser H, Schmidt I, Zolk O, Fromm MF, Maas R. Molecular mechanism of renal tubular secretion of the antimalarial drug chloroquine. Antimicrob Agents Chemother. 2011;55(7):3091–3098. doi: 10.1128/AAC.01835-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rijpma SR, van den Heuvel JJ, van der Velden M, Sauerwein RW, Russel FG, Koenderink JB. Atovaquone and quinine anti-malarials inhibit ATP binding cassette transporter activity. Malar J. 2014;13:359–366. doi: 10.1186/1475-2875-13-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu C, Zhu L, Chan T, Lu X, Shen W, Madigan MC, Gillies MC, Zhou F. Chloroquine and Hydroxychloroquine Are Novel Inhibitors of Human Organic Anion Transporting Polypeptide 1A2. J Pharm Sci. 2016 doi: 10.1002/jps.24663. [DOI] [PubMed] [Google Scholar]
- 16.Ferslew BC, Brouwer KL. Identification of hepatic phospholipidosis inducers in sandwich-cultured rat hepatocytes, a physiologically relevant model, reveals altered basolateral uptake and biliary excretion of anionic probe substrates. Toxicol Sci. 2014;139(1):99–107. doi: 10.1093/toxsci/kfu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008;36(10):2014–23. doi: 10.1124/dmd.108.021410. [DOI] [PubMed] [Google Scholar]
- 18.Sanofi-Aventis Aralen® (Chloroquine Phosphate) Tablets Prescribing Information, 2013. 2014 Dec 20; http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/006002s043lbl.pdf.
- 19.Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxy-chloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol. 2012;42(2):145–153. doi: 10.1007/s12016-010-8243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bezerra EL, Vilar MJ, da Trindade Neto PB, Sato EI. Double-blind, randomized, controlled clinical trial of clofazimine compared with chloroquine in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52(10):3073–3078. doi: 10.1002/art.21358. [DOI] [PubMed] [Google Scholar]
- 21.Meier FM, Frerix M, Hermann W, Muller-Ladner U. Current immunotherapy in rheumatoid arthritis. Immunotherapy. 2013;5(9):955–74. doi: 10.2217/imt.13.94. [DOI] [PubMed] [Google Scholar]
- 22.Avloclor Tablets-Summary of Product Characteristics Electronic Medicines Compendium. Avloclor® Tablets-Summary of Product Charateristics. 2015 Jan 6; http://www.medicines.org.uk/emc/medicine/2272.
- 23.Inova Health Care Services Study of the Efficacy of Chloroquine in the Treatment of Ductal Carcinoma in Situ (The PINC Trial), 2009. 2015 Jan 15; https://clinicaltrials.gov/ct2/show/study/NCT01023477.
- 24.Maastricht Radiation Oncology Chloroquine as an Anti-autophagic Radiosensitizing Drug in Stage I-III Small Cell Lung Cancer, 2012. 2015 Jan 15; https://clinicaltrials.gov/ct2/show/NCT01575782.
- 25.Maastricht Radiation Oncology Chloroquine as an Anti-Autophagy Drug in Stage IV Small Cell Lung Cancer (SCLC) Patients (Chloroquine IV), 2009. 2015 Jan 15; https://clinicaltrials.gov/ct2/show/NCT00969306.
- 26.The Methodist Hospital System Chloroquine With Taxane Chemotherapy for Advanced or Metastatic Breast Cancer Patients Who Have Failed an Anthracycline (CAT), 2011. 2015 Jan 15; https://clinicaltrials.gov/ct2/show/NCT01446016.
- 27.University of Cincinnati Chloroquine in Combination With Carboplatin/Gemcitabine in Advanced Solid Tumors, 2014. 2015 Jan 15; https://clinicaltrials.gov/ct2/show/NCT02071537.
- 28.US Food and Drug Administration FDA Adverse Event Reporting System (FAERS) web site. 2015 Aug 31; http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/
- 29.Han X, Quinney SK, Wang Z, Zhang P, Duke J, Desta Z, Elmendorf JS, Flockhart DA, Li L. Identification and Mechanistic Investigation of Drug-Drug Interactions Associated With Myopathy: A Translational Approach. Clin Pharmacol Ther. 2015;98(3):321–7. doi: 10.1002/cpt.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raschi E, Poluzzi E, Koci A, Salvo F, Pariente A, Biselli M, Moretti U, Moore N, De Ponti F. Liver injury with novel oral anticoagulants: assessing post-marketing reports in the US Food and Drug Administration adverse event reporting system. British journal of clinical pharmacology. 2015;80(2):285–93. doi: 10.1111/bcp.12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Izumi S, Nozaki Y, Maeda K, Komori T, Takenaka O, Kusuhara H, Sugiyama Y. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab Dispos. 2015;43(2):235–247. doi: 10.1124/dmd.114.059105. [DOI] [PubMed] [Google Scholar]
- 32.Niemi M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics. 2007;8(7):787–802. doi: 10.2217/14622416.8.7.787. [DOI] [PubMed] [Google Scholar]
- 33.McTaggart F, Buckett L, Davidson R, Holdgate G, McCormick A, Schneck D, Smith G, Warwick M. Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol. 2001;87(5A):28B–32B. doi: 10.1016/s0002-9149(01)01454-0. [DOI] [PubMed] [Google Scholar]
- 34.Baker WL, Datta R. Pitavastatin: a new 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor for the treatment of hyperlipidemia. Adv Ther. 2011;28(1):13–27. doi: 10.1007/s12325-010-0092-8. [DOI] [PubMed] [Google Scholar]
- 35.Jacobsen W, Kirchner G, Hallensleben K, Mancinelli L, Deters M, Hackbarth I, Benet LZ, Sewing KF, Christians U. Comparison of cytochrome P-450-dependent metabolism and drug interactions of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors lovastatin and pravastatin in the liver. Drug Metab Dispos. 1999;27(2):173–9. [PubMed] [Google Scholar]
- 36.Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev. 1998;50(3):387–412. [PubMed] [Google Scholar]
- 37.Zhao Q, Tensfeldt TG, Chandra R, Mould DR. Population pharmacokinetics of azithromycin and chloroquine in healthy adults and paediatric malaria subjects following oral administration of fixed-dose azithromycin and chloroquine combination tablets. Malar J. 2014;13:36. doi: 10.1186/1475-2875-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bradley SE, Ingelfinger FJ, et al. The estimation of hepatic blood flow in man. J Clin Invest. 1945;24:890–7. [PubMed] [Google Scholar]
- 39.Gustafsson LL, Walker O, Alvan G, Beermann B, Estevez F, Gleisner L, Lindstrom B, Sjoqvist F. Disposition of chloroquine in man after single intravenous and oral doses. British journal of clinical pharmacology. 1983;15(4):471–9. doi: 10.1111/j.1365-2125.1983.tb01532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swift B, Pfeifer ND, Brouwer KL. Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev. 2010;42(3):446–471. doi: 10.3109/03602530903491881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powell J, Farasyn T, Kock K, Meng X, Pahwa S, Brouwer KL, Yue W. Novel mechanism of impaired function of organic anion-transporting polypeptide 1B3 in human hepatocytes: post-translational regulation of OATP1B3 by protein kinase C activation. Drug Metab Dispos. 2014;42(11):1964–1970. doi: 10.1124/dmd.114.056945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kock K, Xie Y, Hawke RL, Oberlies NH, Brouwer KLR. Interaction of silymarin flavonolignans with organic anion-transporting polypeptides. Drug Metab Dispos. 2013;41:958–965. doi: 10.1124/dmd.112.048272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.König J, Cui Y, Nies AT, Keppler D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J Biol Chem. 2000;275(30):23161–23168. doi: 10.1074/jbc.M001448200. [DOI] [PubMed] [Google Scholar]
- 44.Köck K, Koenen A, Giese B, Fraunholz M, May K, Siegmund W, Hammer E, Völker U, Jedlitschky G, Kroemer HK, Grube M. Rapid modulation of the organic anion transporting polypeptide 2B1 (OATP2B1, SLCO2B1) function by protein kinase C-mediated internalization. J Biol Chem. 2010;285(15):11336–11347. doi: 10.1074/jbc.M109.056457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian X, Zamek-Gliszczynski MJ, Zhang P, Brouwer KLR. Modulation of multidrug resistance-associated protein 2 (Mrp2) and Mrp3 expression and function with small interfering RNA in sandwich-cultured rat hepatocytes. Mol Pharmacol. 2004;66(4):1004–1010. doi: 10.1124/mol.66.4.. [DOI] [PubMed] [Google Scholar]
- 46.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42(Database issue):D1091–7. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schelleman H, Han X, Brensinger CM, Quinney SK, Bilker WB, Flockhart DA, Li L, Hennessy S. Pharmacoepidemiologic and in vitro evaluation of potential drug-drug interactions of sulfonylureas with fibrates and statins. British journal of clinical pharmacology. 2014;78(3):639–48. doi: 10.1111/bcp.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Augustijns P, Geusens P, Verbeke N. Chloroquine levels in blood during chronic treatment of patients with rheumatoid arthritis. Eur J Clin Pharmacol. 1992;42(4):429–33. doi: 10.1007/BF00280130. [DOI] [PubMed] [Google Scholar]
- 49.Walker O, Salako LA, Alvan G, Ericsson O, Sjoqvist F. The disposition of chloroquine in healthy Nigerians after single intravenous and oral doses. British journal of clinical pharmacology. 1987;23(3):295–301. doi: 10.1111/j.1365-2125.1987.tb03048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang YB, Leroy V, Maunsbach AB, Doucet A, Hasler U, Dizin E, Ernandez T, de Seigneux S, Martin PY, Féraille E. Sodium Transport Is Modulated byp38 Kinase–Dependent Cross-Talk between ENaC and Na,K-ATPase in Collecting Duct Principal Cells. J Am Soc Nephrol. 2014;25(2):250–259. doi: 10.1681/ASN.2013040429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmaster KA, Turncliff RZ, LeCluyse EL, Kim RB, Meier PJ, Brouwer KL. P-glycoprotein expression, localization, and function in sandwich-cultured primary rat and human hepatocytes: relevance to the hepatobiliary disposition of a model opioid peptide. Pharm Res. 2004;21(7):1294–302. doi: 10.1023/b:pham.0000033018.97745.0d. [DOI] [PubMed] [Google Scholar]
- 52.Mello AS, Goldim MP, Mezzalira J, Garcia CS, Daitz VV, Castilhos CD, Viegas MS, Vieira OV, Coelho JC. LAMP2 as a marker of EBV-mediated B lymphocyte transformation in the study of lysosomal storage diseases. Mol Cell Biochem. 2014;385(1–2):1–6. doi: 10.1007/s11010-013-1806-4. [DOI] [PubMed] [Google Scholar]
- 53.Gui C, Miao Y, Thompson L, Wahlgren B, Mock M, Stieger B, Hagenbuch B. Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur J Pharmacol. 2008;584(1):57–65. doi: 10.1016/j.ejphar.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A. 1978;75(7):3327–31. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reijngoud DJ, Oud PS, Tager JM. Effect of ionophores on intralysosomal pH. Biochim Biophys Acta, Biomembr. 1976;448(2):303–13. doi: 10.1016/0005-2736(76)90244-3. [DOI] [PubMed] [Google Scholar]
- 56.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85(21):7972–6. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30(17):3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Misinzo G, Delputte PL, Nauwynck HJ. Inhibition of endosome-lysosome system acidification enhances porcine circovirus 2 infection of porcine epithelial cells. J Virol. 2008;82(3):1128–35. doi: 10.1128/JVI.01229-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266(26):17707–12. [PubMed] [Google Scholar]
- 60.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311(1):139–146. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- 61.Feng Q, Wilke RA, Baye TM. Individualized risk for statin-induced myopathy: current knowledge, emerging challenges and potential solutions. Pharmacogenomics. 2012;13(5):579–94. doi: 10.2217/pgs.12.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunmore BJ, Drake KM, Upton PD, Toshner MR, Aldred MA, Morrell NW. The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum Mol Genet. 2013;22(18):3667–3679. doi: 10.1093/hmg/ddt216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koh YH, von Arnim CA, Hyman BT, Tanzi RE, Tesco G. BACE is degraded via the lysosomal pathway. J Biol Chem. 2005;280(37):32499–504. doi: 10.1074/jbc.M506199200. [DOI] [PubMed] [Google Scholar]
- 64.van de Steeg E, Greupink R, Schreurs M, Nooijen IH, Verhoeckx KC, Hanemaaijer R, Ripken D, Monshouwer M, Vlaming ML, DeGroot J, Verwei M, Russel FG, Huisman MT, Wortelboer HM. Drug-drug interactions between rosuvastatin and oral antidiabetic drugs occurring at the level of OATP1B1. Drug Metab Dispos. 2013;41(3):592–601. doi: 10.1124/dmd.112.049023. [DOI] [PubMed] [Google Scholar]
- 65.Xia X, Roundtree M, Merikhi A, Lu X, Shentu S, Lesage G. Degradation of the apical sodium-dependent bile acid transporter by the ubiquitin-proteasome pathway in cholangiocytes. J Biol Chem. 2004;279(43):44931–7. doi: 10.1074/jbc.M400969200. [DOI] [PubMed] [Google Scholar]
- 66.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34(7):1229–36. doi: 10.1124/dmd.106.009290. [DOI] [PubMed] [Google Scholar]
- 67.Projean D, Baune B, Farinotti R, Flinois JP, Beaune P, Taburet AM, Ducharme J. In vitro metabolism of chloroquine: identification of CYP2C8, CYP3A4, and CYP2D6 as the main isoforms catalyzing N-desethylchloroquine formation. Drug Metab Dispos. 2003;31(6):748–54. doi: 10.1124/dmd.31.6.748. [DOI] [PubMed] [Google Scholar]
- 68.Simooya OO, Sijumbil G, Lennard MS, Tucker GT. Halofantrine and chloroquine inhibit CYP2D6 activity in healthy Zambians. Br J Clin Pharmacol. 1998;45(3):315–7. doi: 10.1046/j.1365-2125.1998.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20(4):303–28. doi: 10.1111/j.1527-3466.2002.tb00099.x. [DOI] [PubMed] [Google Scholar]
- 70.Amundsen R, Christensen H, Zabihyan B, Asberg A. Cyclosporine A, but not tacrolimus, shows relevant inhibition of organic anion-transporting protein 1B1-mediated transport of atorvastatin. Drug Metab Dispos. 2010;38(9):1499–1504. doi: 10.1124/dmd.110.032268. [DOI] [PubMed] [Google Scholar]
- 71.Shitara Y, Takeuchi K, Horie T. Long-lasting inhibitory effects of saquinavir and ritonavir on OATP1B1-mediated uptake. J Pharm Sci. 2013;102(9):3427–3435. doi: 10.1002/jps.23477. [DOI] [PubMed] [Google Scholar]
- 72.Furihata T, Matsumoto S, Fu Z, Tsubota A, Sun Y, Matsumoto S, Kobayashi K, Chiba K. Different interaction profiles of direct-acting anti-hepatitis C virus agents with human organic anion transporting polypeptides. Antimicrob Agents Chemother. 2014;58(8):4555–4564. doi: 10.1128/AAC.02724-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gertz M, Cartwright CM, Hobbs MJ, Kenworthy KE, Rowland M, Houston JB, Galetin A. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug-drug interaction potential. Pharm Res. 2013;30(3):761–780. doi: 10.1007/s11095-012-0918-y. [DOI] [PubMed] [Google Scholar]
- 74.Tweedie D, Polli JW, Berglund EG, Huang SM, Zhang L, Poirier A, Chu X, Feng B International Transporter, C. Transporter studies in drug development: experience to date and follow-up on decision trees from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94(1):113–125. doi: 10.1038/clpt.2013.77. [DOI] [PubMed] [Google Scholar]
- 75.Tweedie D, Polli JW, Berglund EG, Huang SM, Zhang L, Poirier A, Chu X, Feng B International Transporter, C. Transporter studies in drug development: experience to date and follow-up on decision trees from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94(1):113–25. doi: 10.1038/clpt.2013.77. [DOI] [PubMed] [Google Scholar]
- 76.Duke JD, Han X, Wang Z, Subhadarshini A, Karnik SD, Li X, Hall SD, Jin Y, Callaghan JT, Overhage MJ, Flockhart DA, Strother RM, Quinney SK, Li L. Literature based drug interaction prediction with clinical assessment using electronic medical records: novel myopathy associated drug interactions. PLoS Comput Biol. 2012;8(8):e1002614. doi: 10.1371/journal.pcbi.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bhardwaj S, Selvarajah S, Schneider EB. Muscular effects of statins in the elderly female: a review. Clin Interventions Aging. 2013;8:47–59. doi: 10.2147/CIA.S29686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kowa Pharmaceuticals America, I. LIVALO® (pitavastatin) Tablet Package Insert 2013.
- 79.Pan HY, Waclawski AP, Funke PT, Whigan D. Pharmacokinetics of pravastatin in elderly versus young men and women. Ann Pharmacother. 1993;27(9):1029–33. doi: 10.1177/106002809302700902. [DOI] [PubMed] [Google Scholar]
- 80.Nazir S, Iqbal Z, Shah Y, Ahmad L, Khan A. Pharmacokinetic study of rosuvastatin in males and females. Eur J Drug Metab Pharmacokinet. 2015;40(3):313–8. doi: 10.1007/s13318-014-0211-z. [DOI] [PubMed] [Google Scholar]
- 81.Martin PD, Dane AL, Nwose OM, Schneck DW, Warwick MJ. No effect of age or gender on the pharmacokinetics of rosuvastatin: a new HMG-CoA reductase inhibitor. J Clin Pharmacol. 2002;42(10):1116–21. doi: 10.1177/009127002401382722. [DOI] [PubMed] [Google Scholar]
- 82.Zhou F, Lee AC, Krafczyk K, Zhu L, Murray M. Protein kinase C regulates the internalization and function of the human organic anion transporting polypeptide 1A2. British journal of pharmacology. 2011;162(6):1380–8. doi: 10.1111/j.1476-5381.2010.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou F, Illsley NP, You G. Functional characterization of a human organic anion transporter hOAT4 in placental BeWo cells. Eur J Pharm Sci. 2006;27(5):518–23. doi: 10.1016/j.ejps.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Q, Hong M, Duan P, Pan Z, Ma J, You G. Organic anion transporter OAT1 undergoes constitutive and protein kinase C-regulated trafficking through a dynamin- and clathrin-dependent pathway. J Biol Chem. 2008;283(47):32570–32579. doi: 10.1074/jbc.M800298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duan P, Li S, You G. Angiotensin II inhibits activity of human organic anion transporter 3 through activation of protein kinase Calpha: accelerating endocytosis of the transporter. Eur J Pharmacol. 2010;627(1-3):49–55. doi: 10.1016/j.ejphar.2009.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huff RA, Vaughan RA, Kuhar MJ. Phorbol esters increase dopamine transporter phosphorylation and decrease transport Vmax. J Neurochem. 1997;68(1):225–232. doi: 10.1046/j.1471-4159.1997.68010225.x. [DOI] [PubMed] [Google Scholar]
- 87.Vayro S, Silverman M. PKC regulates turnover rate of rabbit intestinal Na+-glucose transporter expressed in COS-7 cells. Am J Physiol. 1999;276(5 Part1):C1053–60. doi: 10.1152/ajpcell.1999.276.5.C1053. [DOI] [PubMed] [Google Scholar]
- 88.Jeong JY, Choi JW, Jeon KI, Jue DM. Chloroquine decreases cell-surface expression of tumour necrosis factor receptors in human histiocytic U-937 cells. Immunology. 2002;105(1):83–91. doi: 10.1046/j.0019-2805.2001.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaplan J, Keogh EA. Analysis of the effect of amines on inhibition of receptor-mediated and fluid-phase pinocytosis in rabbit alveolar macrophages. Cell. 1981;24(3):925–932. doi: 10.1016/0092-8674(81)90118-5. [DOI] [PubMed] [Google Scholar]
- 90.Weisz OA. Acidification and protein traffic. Int Rev Cytol. 2003;226:259–319. doi: 10.1016/s0074-7696(03)01005-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.