

Abstract
Detailed structural characterization of intact rough-type lipopolysaccharides (R-LPS) was accomplished using an MS3 strategy consisting of collision-induced dissociation (CID) followed by 193 ultraviolet photodissociation (UVPD) implemented on an Orbitrap Fusion mass spectrometer. Complex mixtures of R-LPS from either E. coli or S. enterica were directly infused into the mass spectrometer using static source nanoESI. An initial CID event performed on an R-LPS precursor produced spectra with abundant ions corresponding to the lipid A and core oligosaccharide (OS) substructures. Comparison of CID spectra of R-LPS ions with varying lipid A and core OS structures verify that lipid A and core OS ions are consistently produced in high abundance. The resulting lipid A and core OS ions were subsequently activated by CID, high-energy collision induced dissociation (HCD), or UVPD. For both the lipid A and core OS substructures, HCD and UVPD produced highly informative complementary spectra, with UVPD of the core OS producing an extensive array of cross-ring cleavage fragments. Successful discernment of E. coli R-LPS structures with isomeric core structures confirmed the degree to which subtle structural differences could be determined using this method.
Graphical Abstract
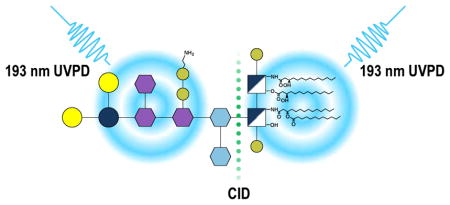
Introduction
Lipopolysaccharides (LPS), the main component of the outer membrane of most gram-negative bacteria, are complex biomolecules responsible for immune system response to pathogen invasion.1 The generic structure of LPS includes three regions: lipid A, core oligosaccharide (OS) and O-antigen. LPS molecules with and without the O-antigen are termed smooth-type (S-LPS) and rough-type (R-LPS), respectively. Although there exist R-LPS mutant strains of bacteria that normally produce an O-antigen, some bacteria, including Neisseria and Haemophilus, have naturally evolved to produce R-LPS.2 Lipid A, the hydrophobic anchor that activates the Toll-like receptor 4 (TLR4), and the core OS, largely responsible for outer membrane stability, are essential for the viability of all gram negative bacteria.3–7 Variations in the acyl chain distribution, phosphorylation, and sugar orientation and distribution in both lipid A and the core oligosaccharide greatly impact the virulence of bacteria.8–12 For example, diphosphoryl hexa-acylated lipid A induces the strongest inflammatory response of the TLR4. Alteration of the acylation pattern or phosphorylation status can reduce the activity of lipid A by more than two orders of magnitude.13 Manipulation of lipid A structure to reduce toxicity, yet still initiate an adaptive immune response, is the basis for detoxified lipid A as a vaccine adjuvant.14–17 Additionally, the presence of phosphorylations of the core oligosaccharide has shown to significantly contribute to outer membrane stability. Mutant strains of E. coli modified to lack phosphorylations in the core region have shown increased susceptibility to antibiotics and detergents.4 Considering the impact of subtle modifications to LPS, detailed structural characterization of LPS is necessary to further investigate the role of LPS in bacteria viability and to understand how LPS structure influences immune stimulation to facilitate development of bacterial vaccines.
Mass spectrometry has emerged as a powerful tool for characterization of LPS substructures. The amphiphilic nature of LPS causes micelle formation which can complicate mass spectrometric analysis. Consequently, a common approach for characterization of LPS involves hydrolysis of the glycosidic bond between lipid A and the core OS, and subsequent analysis of each substructure.10,18,19 Additionally, lipid A acyl chains can be removed by hydrolysis prior to analysis of the intact carbohydrate chain. Collisional induced dissociation (CID) is commonly used to obtain fragmentation patterns for the hydrolyzed lipid A20–25 and saccharide26 substructures. However, CID does not always provide adequate fragmentation for elucidation of subtle structural differences. Limitations of CID have been the impetus for the development of alternative activations methods including infrared multiphoton photodissociation (IRMPD), negative- electron transfer dissociation (NETD), ultraviolet photodissociation (UVPD) and activated- electron photodetachment (a-EPD) which have been employed to produce either complementary or more informative fragmentation spectra.27–29 193 nm UVPD, a fast and high energy activation method that entails irradiation of ions with 193 nm (6.4 eV) photons, has previously been shown to produce highly informative spectra for many biomolecules.29 When biological molecules are deliberately truncated or degraded prior to analysis (as is the case with “bottom up” approaches), some structural features or integrated patterns of features may be lost. This is also the case with LPS analysis, for which attention has primarily focused on characterization of the lipid A sub-structure rather than the core oligosaccharide or O-antigen. With approaches that evaluate hydrolyzed LPS substructures, not only are labile modifications potentially lost, but combinations of modifications occurring between lipid A and the core oligosaccharide are also not adequately reflected. Therefore, analysis of intact LPS molecules intact via a top-down approach offers many advantages, albeit with concomitant challenges associated with the amphiphilic characteristics of LPS.
Although there have been a few studies reported on the analysis of intact LPS, most have measured molecular weights and MS/MS spectra have not been commonly evaluated.26,30,31 Recently, we examined the fragmentation patterns of intact R-LPS using a hybrid UVPD/HCD method.32 UVPD and hybrid UVPD/HCD produced very rich fragmentation patterns arising from a diverse array of C-O, C-N, and C-C cleavages from both the lipid and oligosaccharide portions of the LPS, as illustrated for endotoxin structures from E. coli.32 The structural complexity of LPS makes interpretation of the resulting MS/MS spectra challenging. Previous studies have shown that the glycosidic bond between the 3-deoxy-D-manno-octulosonic acid (Kdo) of the core OS and the glucosamine of lipid A is particularly weak and can be easily cleaved in the gas phase.33 Considering the successful implementation of 193 nm UVPD to analyze lipid A27,28,34–36 and saccharide37 structures and the ease with which this glycosidic bond can be cleaved, multistage mass spectrometry can be implemented to interrogate intact LPS structures while at the same time simplifying data interpretation. The present study uses MS3 to cleave the glycosidic bonds between lipid A and the core OS of intact LPS transferred to the gas-phase by ESI, and subsequent UVPD of each substructure to gain detailed structural characterization of intact R-LPS with complete core.
Experimental
All R-LPS Escherichia coli (E. coli) and Salmonella enterica sv. Minnesota (S. enterica) LPS samples were purchased from Glycobiotech (Research Center Borstel, Germany) and used without further purification. As an example, the structure of S. enterica is shown in Figure 1, along with a legend displaying a key to the color coded sub-units and common neutral losses observed in the MS/MS spectra. Methanol and water were purchased from EMD Millipore (Billerica, MA) and chloroform was purchase from Sigma Aldrich (St. Louis, MO). All samples were diluted in 62:36:2 methanol:water:chloroform to 100 ug/mL. Approximately 10 μL of sample were loaded into a silver-coated pulled tip capillary (1.2 mm OD) and sprayed using a Proxeon offline nano-electrospray set-up (Thermo Scientific, San Jose, CA). Based on the concentration and volume, this corresponds to consumption of ~1 μg of LPS. The spray voltage was set to 1.1 kV. All spectra were collected in negative mode on a Thermo Scientific Orbitrap Fusion mass spectrometer (San Jose, CA) modified with a 193 nm Coherent Existar excimer laser (Santa Cruz, CA) to perform UVPD in the high pressure linear ion trap (Supplemental Figure S1).38 In brief, UVPD was implemented by adding a fused silica window at the back end of the dual linear ion trap to allow entrance of the laser beam into the high pressure trap of the instrument. A periscope and appropriate mirrors were used to guide the laser beam through the high pressure ion trap, and a pulse generator and customized instrument code were used to trigger the laser during the time of ion trapping. Data were collected with an AGC target between 5e4 and 2e5 at a resolution of 120,000. For CID and HCD 25 scans were averaged (based on collection and averaging of 25 scans via the instrumental control software), and for UVPD 100–200 scans were averaged with 2–3 μscans per scan. For CID activation at the MS2 level the NCE was set to 18. At the MS3 level CID, HCD and UVPD activation parameters were set to NCE 30, NCE 35 and five 2 mJ pulses, respectively. Default activation times were used CID and HCD. All data were analyzed in XCalibur Qual Browser and manually interpreted. For spectra that were deconvoluted to neutral forms, Xtract was used with a signal to noise threshold of 3.
Figure 1.
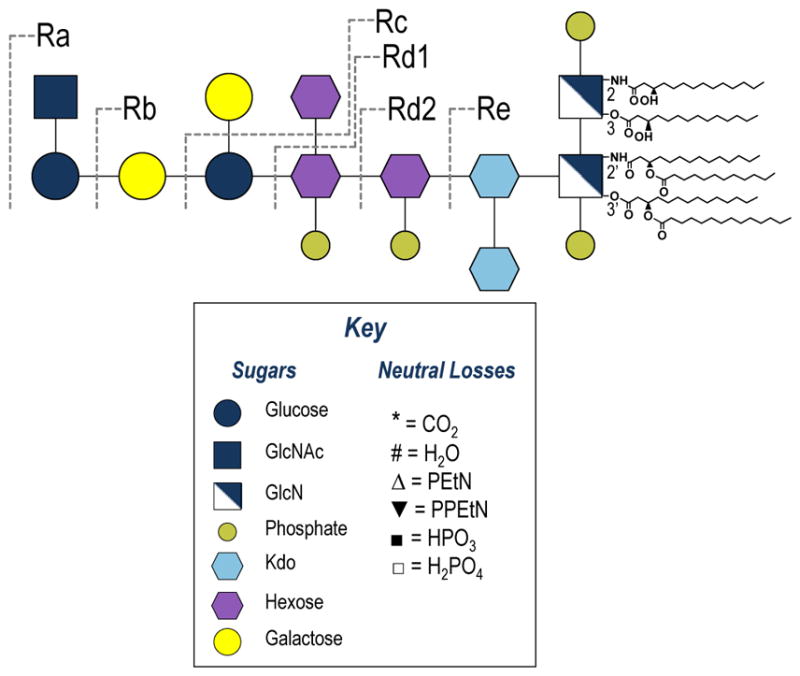
Rough-type mutant strains of S. enterica serovar Minnesota, including a key that depicts the symbols used to denote subunits and neutral losses observed in the MS/MS spectra.
Results/Discussion
Due to the complex amphipathic nature of intact LPS, a universal workflow was developed to facilitate a streamlined approach for individual characterization of the two main sub-structures: the lipid A sub-structure and the core Kdo/oligosaccharide sub-structure. As shown below, conventional CID provided the optimal means to cleave LPS into the two constituent sub-structures, and then the capabilities of CID, HCD and UVPD were compared for the subsequent characterization of the two substructures. The first step of the workflow required a robust means to transfer intact LPS into the mass spectrometer. The most common mass spectrometry approaches to LPS structural analysis generally involve deliberate degradation of LPS prior to analysis, via hydrolysis of the glycosidic bond between Kdo and glucosamine, or partial or complete removal of acyl chains. To approach analysis of intact LPS, conditions that allow dissolution of the LPS for direct analysis by nanoelectrospray were developed. Raetz et al. identified chromatographic conditions that allowed successful chromatographic separation and ionization of Kdo2-lipid A.39 Building on this earlier work, the starting gradient conditions employed in the Raetz study were used to successfully dissolve heterogeneous samples of R-LPS in the present study. LPS samples were dissolved in 62:36:2 methanol:water:chloroform to attain a concentration of approximately 100 ug/mL and sprayed without chromatographic separation via a “shotgun” approach using a static nanoESI source. Due to the large number of lipids in each sample and the production of multiple charge states upon ESI, the resulting mass spectra were quite complex for three S. enterica and two E. coli samples (Supplemental Figure S2, Supplemental Figure S3a). The charge-deconvoluted spectra confirm the high degree of heterogeneity in the samples (as exemplified in Supplemental Figure S3b for S. Enterica Rc). Although some of this heterogeneity is likely a result of the method used to isolate the LPS, the resulting array of LPS species present in each sample was advantageous for the more elaborate comparisons done in this study.
Interpretation, assignment and labelling of the MS/MS/MS spectra of LPS poses its own set of challenges, especially in the context of displaying the results in a meaningful way to others. For the MS/MS spectra presented, LPS structures are depicted using the Consortium of Functional Glycomics proposed symbol notation extension for pathogen polysaccharides.40 The complex MS/MS spectra shown in the present study are conveyed schematically as fragmentation maps, in which saccharide fragments are labeled using the nomenclature of Domon and Costello (Supplemental Figure S4)41 and acyl chain cleavages are numbered. This same system is used to label the ions in the MS/MS spectra, and for products where acyl chains are cleaved, the numbers corresponding to those particular acyl chain are shown as losses from the LPS (which is designated as “M”). For instance, Figure 2a shows the CID mass spectrum of penta-acylated S. enterica Rd1 (4-). The ion of m/z 587.32 labeled as [Y2 - 2]2− corresponds to the lipid A substructure generated upon cleavage of the Kdo-GlcN bond along with loss of the secondary 12-carbon acyl chain (laurate) which is labeled as acyl chain 2. For products corresponding to the cleaved acyl chains, the number corresponding to the cleaved bond is used. For example, in Figure 2a, the ion of m/z 199.17 corresponds to the laurate moiety and is labeled as 2.
Figure 2.

CID (NCE 18) of the 4- charge state of a) tetra-acylated S. enterica Rd1 (m/z 497.25) b) tetra-acylated S. enterica Rc (m/z 585.78) and c) tetra-acylated S. enterica Rb (m/z 765.56) (25 spectra averaged, LPS concentration 100 μg/mL in 62:36:2 CHCl3:MeOH:H2O). ■= neutral loss of HPO3; & indicates that both of the indicated cleavages occur to generated a particular fragment ion. The acyl chains are numbered, and the loss of a particular acyl chain is denoted as M – N where M represents the LPS and N represents the acyl chain that is lost.
Examples of the CID mass spectra obtained for three mutant glycoforms of tetraacylated S. enterica LPS and three lipoforms of S. enterica Rc are shown in Figures 2 and 3, respectively. (Figure 1 shows the naming convention and structures for the different mutants of S. enterica.) In all cases, glycosidic bond cleavages lead to the most prominent products for each LPS. For example, isolation and collisional activation of the tetra-acylated species from S. enterica Rc (MW = 2347.12 Da) shows predominant cleavage of the glycosidic bond between Kdo and glucosamine leading to product ions of m/z 679.41 (Y22−) and m/z 492.14 (B42−) corresponding to the lipid A and saccharide substructures, respectively (Figure 2b). The highly acidic nature of this glycosidic bond provides an explanation for its facile cleavage. This specific glycosidic cleavage is consistent with previous work from our group31 and is generally observed in MALDI spectra as well.18 Preferential cleavage of this glycosidic bond is particularly advantageous for two reasons. First, it allows general localization of structural modifications of LPS to either the lipid A or core oligosaccharide substructures based on the masses of the two products relative to the intact LPS, especially for comparisons of series of LPS like the three shown in Figure 2 that possess the same lipid A sub-structure but with different oligosaccharides. Second, the preferential glycosidic cleavage results in two well-defined and abundant products that can be interrogated individually via MS3 to allow characterization of the two sub-units (e.g. lipid A and oligosaccharide). In essence, subsequent activation of each of these products reveals more detailed structural information. To optimize the formation of the two key sub-structure products in the initial MS/MS step, the collisional energy was varied over a range of values. One of the resulting energy-variable diagrams for penta-acylated S. enterica Rc was constructed to determine the optimal normalized collision energy (NCE) to achieve maximum signal abundance for the lipid A and saccharide substructures (Supplemental Figure S5). An NCE of 18 proved to be the optimal collisional activation condition and was used throughout this study.
Figure 3.

CID (NCE 18) of the 4- charge state of a) tetra-acylated S. enterica Rc (m/z 585.78) b) penta-acylated S. enterica Rc (m/z 642.33) and c) hexa-acylated S. enterica Rc (m/z 694.88) (25 spectra averaged, concentration of LPS is 100 μg/mL in 62:36:2 CHCl3:MeOH:H2O). ■= neutral loss of HPO3; & indicates that both of the indicated cleavages occur to generated a particular fragment ion. The acyl chains are numbered, and the loss of a particular acyl chain is denoted as M – N or Y - N where M represents the LPS, Y represents the lipid A portion, and N represents the acyl chain that is lost.
To verify that the lipid A and core OS ions would be consistently formed for an array of LPS, various R-LPS structures from mutant S. enterica were subjected to CID. The effect of core OS chain length was evaluated by comparing the CID patterns of tetra-acylated S. enterica Rd1, tetra-acylated S. enterica Rc, and tetra-acylated S. enterica Rb, all with the same lipid A substructure but with variations in the core OS (Figure 2). Despite changes in the relative abundances of the lipid A and OS ions, these products are readily identified. Additionally, significantly larger number of acyl chains fragments (i.e. products labelled as [M - N]) are observed upon collisional activation of S. enterica Rd1 (Figure 2a) than for S. enterica Rc or S. enterica Rb. Similar to changes in fragmentation that occur in glycopeptides as a function of glycan structure and peptide length, the observed changes reflect changes in the size and complexity of the core OS, suggesting that acyl chain cleavages are less prominent when there are more opportunities for energetically favorable glycosidic cleavages.42 Regardless of this change in the distribution of products, the two key substructure ions are readily identified for all of the mutants. Similarly, the effect of lipid A structure was evaluated by comparing the fragmentation patterns of tetra-acylated S. enterica Rc, penta-acylated S. enterica Rc and hexa-acylated S. enterica Rc (Figure 3), three lipoforms which vary in the number of acyl chains of lipid A but all with the same OS sub-structure. In the CID spectrum of hexa-acylated S. enterica Rc (Figure 3c), although the lipid A and core OS substructure ions are readily identified (Y22− and B4), the predominant product is an ion of m/z 850.76 that corresponds to loss of the distal myristate secondary acyl chain (e.g. [M-4]3−). Previous reports have shown this acyl chain linkage at the 3′ position to be particularly labile.28 Although this alternative cleavage site diminishes the abundance of the targeted products corresponding to the full lipid A and core OS substructures, this ion can be used to confirm the presence of a secondary acyl chain at the 3′ position.
After optimization of the conditions to enhance the formation of the two substructure products by CID, the products were subsequently isolated and subjected to CID, HCD, and UVPD (MS3) to complete the characterization of LPS. Examples of the resulting MS3 spectra are shown in Figure 4 for tetra-acylated S. enterica Rb that contains a pyrophosphoethanolamine modification with the spectra for the OS (m/z 851.82, 2- charge state) on the left and the complementary spectra for the lipid A substructure (m/z 679.41, 2- charge state) on the right. All identified ions in the spectra in Figure 4 are listed in Table S1. CID-CID of the core OS substructure results in predominant losses of Kdo (Y3β and Z3β) and Kdo2 (B4α and C4α), in addition to neutral losses of CO2 and H2O (Figure 4a, Supplemental Figure S6). CID-HCD of the same OS (Figure 4b) yields a spectrum similar to the CID-CID spectrum. A notable ion of m/z 219.98 (labeled as ▼ in Figure 4b) confirms the presence of the pyrophosphoethanolamine modification on the core OS. The high resolution and high mass accuracy capabilities of the Orbitrap mass spectrometer allow this ion to be distinguished from the ion of m/z 219.05, which corresponds to the B4β ion. Additionally, fragment ions of m/z 967.25 and m/z 869.28 labeled as [C4α&Y5γ]Δ and B3α, respectively, that appear exclusively in the CID-HCD spectrum provide information regarding the saccharide branching pattern and localize the position of the pyrophosphoethanolamine modification. CID-UVPD of the ion of m/z 851.81 yields the richest mass spectrum with numerous fragments corresponding to cross-ring cleavages (X-type ions) that give saccharide linkage information (Figure 4c). For example, the 0,4X4a ion (m/z 1422.37) confirms the linkage pattern of L-glycero-D-manno-heptose (heptose) (II) and glucose to heptose (I). By combining the information from the CID-HCD spectrum and the CID-UVPD spectrum, cleavages are observed between every glycosidic bond in the core OS. This level of structural characterization is unprecedented.
Figure 4.

a) CID-CID (NCE 18- NCE 30), b) CID-HCD (NCE 18- NCE 35) and c) CID-UVPD (NCE 18- 5 pulses, 2 mJ) of the core oligosaccharide substructure of tetra-acylated S. enterica Rb containing a pyrophosphoethanolamine modification (m/z 851.81, charge state 2-). d) CID-CID (NCE 18-NCE 30), e) CID-HCD (NCE 18-NCE 35) and f) CID-UVPD (NCE 18- 5 pulses, 2 mJ) of the lipid A substructure of S. enterica Rb modification (m/z 679.41, charges state 2-). A yellow star is used to denote the precursor. Only select neutral losses are labeled on the spectra to avoid congestion: ▼= loss of pyrophosphethanolamine; △= loss of phosphethanolamine; ■= loss of HPO3; □= loss of H2PO4; & indicates that both of the indicated cleavages occur to generated a particular fragment ion. The acyl chains are numbered, and the loss of a particular acyl chain is denoted as M – N where M represents the LPS and N represents the acyl chain that is lost.
The MS3 spectra for the companion lipid A substructure (m/z 679.41, charge state 2-) of S. enterica Rb are shown on the right side of Figure 4. CID-CID and HCD-CID yield similar spectra (Figure 4d and 4e) that confirm the presence of laurate and myristate chains on the lipid A substructure based on m/z 199.19 and m/z 243.20, respectively, but do not localize their positions on the lipid A substructure. The CID-HCD spectrum displays an additional ion of m/z 242.21 that corresponds to cleavage of the proximal N-linked acyl chain. Again, high resolution and high mass accuracy allowed this ion to be distinguished from the ion of m/z 243.20 corresponding to the proximal O-linked acyl chain. CID-UVPD (Figure 4f) produces a richer array of fragment ions that assist in discerning the more subtle structural features of the lipid A substructure. For example the 1,5X1 cross ring cleavage (m/z 738.42) allows the distribution of acyl chains on the distal (containing 3′,2′ positions) and proximal (containing 3,2 positions) sides of the lipid A substructure to be determined.27,28 Additional cross ring and acyl chain cleavages allow acyl chain connectivity to be determined and hydroxyl modifications to be localized, respectively. For example, the product assigned as 0,2X1, in concert with 1,5X1, confirms that the secondary laurate acyl chain is connected to the distal N-linked myristate acyl chain. For characterization of the lipid A substructure, integration of the information from CID-HCD and CID-UVPD together offer the most comprehensive elucidation of the lipid A moiety.
Comparison of UVPD and CID-UVPD spectra of penta-acylated E. coli lipid A and penta-acylated S. enterica Rc, respectively, confirms that the integrity of the MS3 spectra collected for the lipid A substructure is maintained after the initial CID activation event (Figure 5). All identified ions in the spectra in Figure 5 are listed in Table S2. The complexity of the resulting spectra reinforces the rationale for “separating” the characterization of the lipid A and OS substructures based on the MS3 strategy. By first performing a CID event to cleave the bond between lipid A and core OS, the spectral complexity that acyl chain fragmentation would otherwise add to identification of glycosidic cleavages in core OS is eliminated, and manual interpretation of spectra becomes practical.
Figure 5.
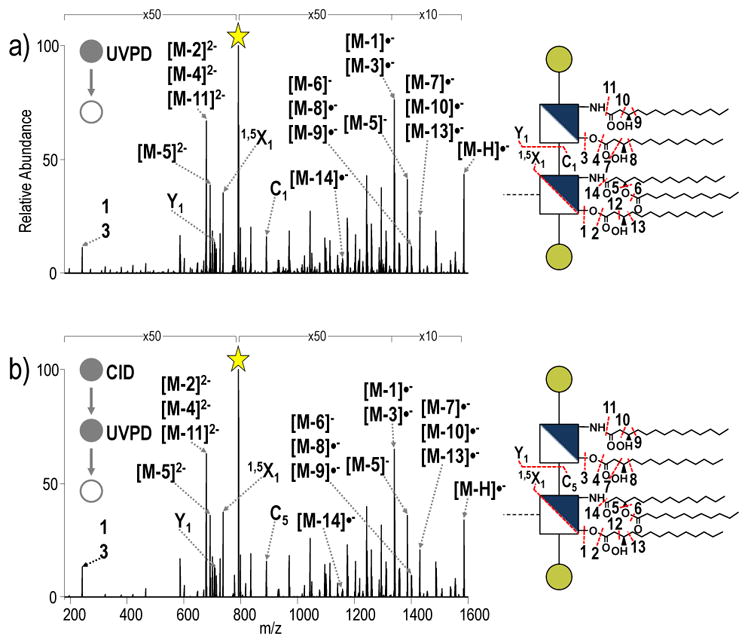
a) UVPD of penta-acylated from E. coli lipid A and b) CID-UVPD of the penta-acylated lipid A substructure from S. enterica Rc. ■= neutral loss of HPO3; the yellow star indicates the precursor ion in the spectrum. The acyl chains are numbered, and the loss of a particular acyl chain is denoted as M – N where M represents the LPS and N represents the acyl chain that is lost.
Within E. coli LPS there are five naturally occurring core types: R1–R4 and K12. These core types are very similar, all containing heptose and Kdo in their inner core (various other non-stoichiometric modifications can occur in the inner core depending on the core type) and a series of hexose carbohydrates in the outer core.43 The nature, position and linkage of the hexoses in the outer core are dependent on the core type, with some of the variation being as subtle as a difference in the kind of linkage. To further confirm the detail to which core OS structures can be determined using this multi-stage mass spectrometry method, MS3 spectra were acquired for two isomeric R-LPS structures from E. coli R2 and E. coli R3. CID of was performed on the main LPS ion (MW = 3162.16 Da, m/z 631.43, charge state 5-) in each of the E. coli R2 and E. coli R3 samples (Supplemental Figure S7). Other than small differences in ion abundances, the spectra are identical. The Y22− (lipid substructure, m/z 566.31) and B73− (OS substructure, m/z 674.84) are the two most prominent product ions in the CID spectra. The CID-HCD and CID-UVPD spectra of the lipid A substructures (Y22−, m/z 566.31) also look the same for E.coli R2 and E. coli R3 (Supplemental Figure S8), confirming that the variation in the LPS structure must be a result of core OS structure and not lipid A structure. CID-HCD and CID-UVPD spectra of the core OS substructures (m/z 674.84, Figure 6) are very similar; however, there exist fragment ions that allow the two isomeric structures to be distinguished from one another. All identified ions in the spectra in Figure 6 are listed in Table S3. In the CID-HCD spectra, the B3α product in Figure 6a and B2α and C2α products in Figure 6b correspond to glycosidic cleavages unique to the E. coli R2 and E. coli R3 cores, respectively. Similarly, in the CID-UVPD spectra, Y7α, Z7α, 2,4X5α and 1,5X7α2− ions in Figure 6c and Y7α, Z7α, Z7α2− and 1,5X6α ions in Figure 6d correspond to glycosidic and cross-ring cleavages that uniquely identify the E. coli R2 and E. coli R3 cores, respectively. CID-HCD and CID-UVPD yield complementary fragmentation patterns that enable almost complete structural elucidation of core OS structures as reflected in the comprehensive fragmentation maps shown in Figure 6e and Figure 6f.
Figure 6.

a) CID-HCD (NCE 18- NCE 30) of the core oligosaccharide substructure of E. coli R2 (m/z 674.84, charge state 3-) b) CID-HCD (NCE 18- NCE 30) of the core oligosaccharide substructure of E. coli R3 (m/z 674.84, charge state 3-), c) CID-UVPD (NCE 18- 5 pulses, 2 mJ) of the core oligosaccharide substructure of E. coli R2 (m/z 674.84, charge state 3-) and d) CID-UVPD (NCE 18- 5 pulses, 2 mJ) of the core oligosaccharide substructure of E. coli R3 (m/z 674.84, charge state, 3-). Unique ions that can be used to identify each isomer appear in red. ■= neutral loss of HPO3; the yellow star indicates the precursor ion in the spectrum; & indicates that both of the indicated cleavages occur to generated a particular fragment ion
Conclusion
An MS3 strategy utilizing UVPD has been developed to enable the detailed structural characterization of intact R-LPS. An initial CID event produced lipid A and core oligosaccharide ions that were subsequently activated in a second stage using CID, HCD or UVPD. Subsequent HCD produced ions from glycosidic cleavages that provide saccharide branching information, while UVPD produced ions from cross ring cleavages that revealed saccharide connectivity. The complementary spectra produced by HCD and UVPD enabled confident re-construction of R-LPS structures. The high resolution and high mass accuracy of the Orbitrap analyzer allowed facile differentiation of informative but nearly isobaric fragment ions. Despite the unprecedented characterization achieved in this study, the “shotgun” approach used here produces congested ESI mass spectra that can make isolation of a single LPS species challenging. Although mass isolation of a single R-LPS species was feasible in this study, smooth-type LPS has a significantly higher degree of heterogeneity as a result of the varying number of saccharide repeat units; isolation of single species is increasingly challenging. To address this concern, we are developing appropriate offline and online separation methods to facilitate analysis of smooth-type LPS in a higher throughput manner. Additionally, both online and offline separations will alleviate signal suppression (i.e. splitting or sub-division of ion current) associated with direct infusion of complex mixtures and potentially allow further diminishment of sample consumption.
Supplementary Material
Acknowledgments
Funding from the NIH (R01 GM103655) and the Welch Foundation (F-1155) is gratefully acknowledged. Technical assistance from Eugene Zhuk, Chad Weisbrod, and Jae Schwartz (Thermo Fisher Scientific Instruments) with the implementation of UVPD on the Orbitrap Fusion mass spectrometer is gratefully acknowledged.
Footnotes
Supplemental Information: Included in supplemental information are figures showing the modified Orbitrap mass spectrometer, examples of ESI mass spectra of E. coli LPS, carbohydrate fragmentation nomenclature, an energy-variable CID diagram of hepta-acylated LPS, a CID-CID mass spectrum of tetra-acylated LPS, CID spectra of two isobaric LPS, CID-HCD and CID-UVPD mass spectra of two isobaric LPS, and tables of identified fragment ions.
References
- 1.Beutler B, Rietschel ET. Nat Rev Immunol. 2003;3(2):169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 2.Preston A, Mandrell RE, Gibson BW, Apicella MA. Crit Rev Microbiol. 1996;22(3):139–180. doi: 10.3109/10408419609106458. [DOI] [PubMed] [Google Scholar]
- 3.Raetz CRH, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yethon JA, Heinrichs DE, Monteiro MA, Perry MB, Whitfield C. J Biol Chem. 1998;273(41):26310–26316. doi: 10.1074/jbc.273.41.26310. [DOI] [PubMed] [Google Scholar]
- 5.Heinrichs DE, Yethon JA, Whitfield C. Mol Microbiol. 1998;30(2):221–232. doi: 10.1046/j.1365-2958.1998.01063.x. [DOI] [PubMed] [Google Scholar]
- 6.Delcour AH. Biochim Biophys Acta. 2009;1794(5):808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silipo A, Molinaro A. Endotoxins: Structure, Function and Recognition. In: Wang X, Quinn PJ, editors. Subcellular Biochemistry. Springer; Netherlands: 2010. pp. 69–99. [Google Scholar]
- 8.Yethon JA, Vinogradov E, Perry MB, Whitfield C. J Bacteriol. 2000;182(19):5620–5623. doi: 10.1128/jb.182.19.5620-5623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS. Proc Natl Acad Sci. 2013;110(4):1464–1469. doi: 10.1073/pnas.1218080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molinaro A, Holst O, Di Lorenzo F, Callaghan M, Nurisso A, D’Errico G, Zamyatina A, Peri F, Berisio R, Jerala R, Jiménez-Barbero J, Silipo A, Martín-Santamaría S. Chem – Eur J. 2015;21(2):500–519. doi: 10.1002/chem.201403923. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Powell DA, Shaffer SA, Rasko DA, Pelletier MR, Leszyk JD, Scott AJ, Masoudi A, Goodlett DR, Wang X, Raetz CRH, Ernst RK. Proc Natl Acad Sci. 2012;109(22):8716–8721. doi: 10.1073/pnas.1202908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boll JM, Tucker AT, Klein DR, Beltran AM, Brodbelt JS, Davies BW, Trent MS. mBio. 2015;6(3):e00478–15. doi: 10.1128/mBio.00478-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. Nature. 2009;458(7242):1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 14.Zariri A, van der Ley P. Expert Rev Vaccines. 2015;14(6):861–876. doi: 10.1586/14760584.2015.1026808. [DOI] [PubMed] [Google Scholar]
- 15.Casella CR, Mitchell TC. Cell Mol Life Sci. 2008;65(20):3231–3240. doi: 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Needham BD, Trent MS. Nat Rev Microbiol. 2013;11(7):467–481. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilár A, Dörnyei Á, Kocsis B. Mass Spectrom Rev. 2013;32(2):90–117. doi: 10.1002/mas.21352. [DOI] [PubMed] [Google Scholar]
- 19.Weinbaum G, Kadis S, Ajl SJ. Bacterial Endotoxins: A Comprehensive Treatise. Elsevier; 2013. [Google Scholar]
- 20.Jones JW, Cohen IE, Tureĉek F, Goodlett DR, Ernst RK. J Am Soc Mass Spectrom. 2010;21(5):785–799. doi: 10.1016/j.jasms.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Silipo A, De Castro C, Lanzetta R, Molinaro A, Parrilli M, Vago G, Sturiale L, Messina A, Garozzo D. J Mass Spectrom. 2008;43(4):478–484. doi: 10.1002/jms.1333. [DOI] [PubMed] [Google Scholar]
- 22.Jones JW, Shaffer SA, Ernst RK, Goodlett DR, Tureĉek F. Proc Natl Acad Sci. 2008;105(35):12742–12747. doi: 10.1073/pnas.0800445105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madalinski G, Fournier F, Wind F-L, Afonso C, Tabet J-C. Int J Mass Spectrom. 2006;249–250:77–92. [Google Scholar]
- 24.Kussak A, Weintraub A. Anal Biochem. 2002;307(1):131–137. doi: 10.1016/s0003-2697(02)00004-0. [DOI] [PubMed] [Google Scholar]
- 25.Chan S, Reinhold VN. Anal Biochem. 1994;218(1):63–73. doi: 10.1006/abio.1994.1141. [DOI] [PubMed] [Google Scholar]
- 26.Kojima H, Inagaki M, Tomita T, Watanabe T. Rapid Commun Mass Spectrom. 2010;24(1):43–48. doi: 10.1002/rcm.4355. [DOI] [PubMed] [Google Scholar]
- 27.Madsen JA, Cullen TW, Trent MS, Brodbelt JS. Anal Chem. 2011;83(13):5107–5113. doi: 10.1021/ac103271w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Brien JP, Needham BD, Henderson JC, Nowicki EM, Trent MS, Brodbelt JS. Anal Chem. 2014;86(4):2138–2145. doi: 10.1021/ac403796n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brodbelt JS. Chem Soc Rev. 2014;43(8):2757. doi: 10.1039/c3cs60444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silipo A, Leone S, Molinaro A, Sturiale L, Garozzo D, Nazarenko EL, Gorshkova RP, Ivanova EP, Lanzetta R, Parrilli M. Eur J Org Chem. 2005;2005(11):2281–2291. [Google Scholar]
- 31.Banoub JH, Aneed AE, Cohen AM, Joly N. Mass Spectrom Rev. 2010;29(4):606–650. doi: 10.1002/mas.20258. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien JP, Needham BD, Brown DB, Trent MS, Brodbelt JS. Chem Sci. 2014;5(11):4291–4301. doi: 10.1039/C4SC01034E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sturiale L, Garozzo D, Silipo A, Lanzetta R, Parrilli M, Molinaro A. Rapid Commun Mass Spectrom. 2005;19(13):1829–1834. doi: 10.1002/rcm.1994. [DOI] [PubMed] [Google Scholar]
- 34.Hankins JV, Madsen JA, Giles DK, Childers BM, Klose KE, Brodbelt JS, Trent MS. Mol Microbiol. 2011;81(5):1313–1329. doi: 10.1111/j.1365-2958.2011.07765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS. Proc Natl Acad Sci. 2012;109(22):8722–8727. doi: 10.1073/pnas.1201313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowicki EM, O’Brien JP, Brodbelt JS, Trent MS. Mol Microbiol. 2015;97(1):166–178. doi: 10.1111/mmi.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko BJ, Brodbelt JS. Anal Chem. 2011;83(21):8192–8200. doi: 10.1021/ac201751u. [DOI] [PubMed] [Google Scholar]
- 38.Holden Dustin, Canterbury Jesse, Zhuk Eugene, Izgarian Nick, Schwartz Jae, Brodbelt Jennifer S. 62nd American Society for Mass Spectrometry and Allied Topics; Baltimore, MD. 2014. [Google Scholar]
- 39.Raetz CRH, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, Dennis EA. J Lipid Res. 2006;47(5):1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Berger Oren, McBride Ryan, Razi Nahid, Paulson James. Symbol Notation Entension For Pathogen Polysaccharides. http://glycomics.scripps.edu/coreD/PGAnomenclature.pdf.
- 41.Domon B, Costello CE. Glycoconj J. 1988;5(4):397–409. [Google Scholar]
- 42.Nishikaze T, Kawabata S, Tanaka K. J Am Soc Mass Spectrom. 2014;25(6):988–998. doi: 10.1007/s13361-014-0854-7. [DOI] [PubMed] [Google Scholar]
- 43.Amor K, Heinrichs DE, Frirdich E, Ziebell K, Johnson RP, Whitfield C. Infect Immun. 2000;68(3):1116–1124. doi: 10.1128/iai.68.3.1116-1124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.