

Abstract
Background
Profiling of 16S rRNA gene sequences is an important tool for testing hypotheses in complex microbial communities, and analysis methods must be updated and validated as sequencing technologies advance. In host-associated bacterial communities, the V1–V3 region of the 16S rRNA gene is a valuable region to profile because it provides a useful level of taxonomic resolution; however, use of Illumina MiSeq data for experiments targeting this region needs validation.
Results
Using a MiSeq machine and the version 3 (300 × 2) chemistry, we sequenced the V1–V3 region of the 16S rRNA gene within a mock community. Nineteen bacteria and one archaeon comprised the mock community, and 12 replicate amplifications of the community were performed and sequenced. Sequencing the large fragment (490 bp) that encompasses V1–V3 yielded a higher error rate (3.6 %) than has been reported when using smaller fragment sizes. This higher error rate was due to a large number of sequences that occurred only one or two times among all mock community samples. Removing sequences that occurred one time among all samples (singletons) reduced the error rate to 1.4 %. Diversity estimates of the mock community containing all sequences were inflated, whereas estimates following singleton removal more closely reflected the actual mock community membership. A higher percentage of the sequences could be taxonomically assigned after singleton and doubleton sequences were removed, and the assignments reflected the membership of the input DNA.
Conclusions
Sequencing the V1–V3 region of the 16S rRNA gene on the MiSeq platform may require additional sequence curation in silico, and improved error rates and diversity estimates show that removing low-frequency sequences is reasonable. When datasets have a high number of singletons, these singletons can be removed from the analysis without losing statistical power while reducing error and improving microbiota assessment.
Electronic supplementary material
The online version of this article (doi:10.1186/s13104-016-2172-6) contains supplementary material, which is available to authorized users.
Keywords: V1–V3, 16S rRNA gene, MiSeq, Mock community, Microbial ecology
Background
The affordability and scalability of nucleic acid sequencing have enabled researchers to conduct microbial community analyses on an unprecedented scale. One highly used method to query bacterial communities involves sequencing amplicons of the 16S rRNA gene. However, sequence and analysis of these amplicons has numerous technical limitations including chimera formation during the PCR step and errors introduced by sequencing technologies. Previous advances in validating 16S rRNA gene sequence analyses have employed various sequencing platforms, multiple sequencing centers, and both real and mock bacterial communities [1, 2]. In silico methods, such as chimera removal [3, 4], quality filtering [5], and clustering methods [6], have been developed and validated to improve analysis of amplicons and are essential to separate true data from noise [7].
The quality of 16S rRNA gene sequence data is dependent on numerous technical steps that are difficult to control, often resulting in thousands of unique sequences even after implementing quality-control steps. One approach to managing these low-frequency sequences is to implement closed-reference operational taxonomic unit (OTU)-picking, which clusters sequences into OTUs based on their assignment to a reference database [8]. However, many researchers choose not to require phylogenetic assignment prior to clustering because it could eliminate important, undescribed members of the community. An additional approach is to remove low-frequency OTUs after sequence clustering, although it is computationally expensive to retain sequencing artifacts through distance matrix creation and subsequent clustering. Unreferenced removal of sequences prior to OTU-calling improves the speed and feasibility of analyzing large datasets, as we demonstrate here by sequencing and analyzing the 16S rRNA gene, V1–V3 region, of an artificial (mock) microbial community.
The challenge of sequencing 16S rRNA gene amplicons via the MiSeq platform is choosing a variable region that both informs the microbiota of interest and results in an amplicon sufficiently short to overlap both forward and reverse reads of a paired-end reaction. The V4 region has therefore been used for this sequencing method because its short amplicon (~400 bp including primer sequences) is technically amenable to assembly with a low error rate (0.01 %) [2]. However, longer 16S gene regions are sometimes preferred for biological reasons based on the research question despite the potential higher error rates associated with longer amplicon sequences. For example, the V1–V3 region, but not the V3–V5, region can discriminate Staphylococci populations [9]. We have previously used the V1–V3 region to analyze the swine gut microbiota (e.g., [10]), thus we adapted the MiSeq amplicon method for this region by adding a singleton removal step prior to OTU clustering and validated the method by sequencing 12 technical replicates of a mock community.
Methods
Generating the mock community
Members of the mock community were selected based on three criteria: (1) diversity, (2) availability of a whole genome sequence, and (3) availability of genomic DNA either in-house or from DSMZ (Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures https://www.dsmz.de/home.html). The following equation was used to balance the contribution of each microbial genome (19 bacteria, 1 archaeon, Table 1) to the mock community: (target #16S rRNA gene copies in stock mock)/(#16S rRNA gene copies per genome × #genome copies per microgram). Genomic copies of the 16S rRNA gene from each member comprised 5 % of the total mock community. The mock community was stored in aliquots at −20 °C until 16S rRNA gene amplification.
Table 1.
16S gene composition of the mock community
| Strain | Genome sizea | # 16S genesa | Genome copies per microgramb | Reference or DSMZ catalogue numberc | |
|---|---|---|---|---|---|
| 1 | Campylobacter jejuni 11168 | 1,641,481 | 3 | 5.64E+08 | [23] |
| 2 | Salmonella enterica serovar Typhimurium SL1344 | 4,878,012 | 6 | 1.89E+08 | [24] |
| 3 | Escherichia coli mg1655 | 4,656,144 | 7 | 1.99E+08 | [25] |
| 4 | Megasphaera elsdenii LC-1 | 2,474,718 | 7 | 3.74E+08 | DSM 20460 |
| 5 | Cloacibacillus porcorum CL-84 | 3,585,187 | 3 | 2.57E+08 | [26] |
| 6 | Brachyspira hyodysenteriae | 3,050,489 | 1 | 3.04E+08 | [27] |
| 7 | Haemophilus parasuis 29755 | 2,224,137 | 2 | 4.17E+08 | [28] |
| 8 | Bordetella bronchiseptica 1289 | 5,207,899 | 3 | 1.78E+08 | [29] |
| 9 | Staphylococcus aureus USA300 | 2,872,915 | 5 | 3.22E+08 | [30] |
| 10 | Bacteroides thetaiotaomicron | 6,293,399 | 5 | 1.47E+08 | DSM 2079 |
| 11 | Faecalibacterium prausnitzii A2-165 | 3,080,849 | 3 | 3.01E+08 | DSM 17677 |
| 12 | Streptococcus parasanguinis | 2,153,652 | 4 | 4.30E+08 | DSM 6778 |
| 13 | Parabacteroides merdae | 4,431,877 | 3 | 2.09E+08 | DSM 19495 |
| 14 | Oscillibacter valericigenes | 4,470,622 | 3 | 2.07E+08 | DSM 18026 |
| 15 | Desulfovibrio gigas | 3,788,225 | 4 | 2.45E+08 | DSM 1382 |
| 16 | Lactobacillus delbrueckii subspecies bulgaricus | 1,864,998 | 9 | 4.97E+08 | DSM 20081 |
| 17 | Coriobacterium glomerans | 2,115,681 | 2 | 4.38E+08 | DSM 20642 |
| 18 | Oxalobacter formigenes BA-2 | 2,509,362 | 1 | 3.69E+08 | DSM 4420 |
| 19 | Roseburia hominis A2-183 | 3,592,125 | 4 | 2.58E+08 | DSM 16839 |
| 20 | Methanobrevibacter smithii d | 1,704,865 | 1 | 5.43E+08 | DSM 2375 |
aAll except C. porcorum were calculated by the Joint Genome Institute’s Integrated Microbial Genomes Database https://img.jgi.doe.gov/cgi-bin/w/main.cgi. C. porcorum was calculated manually (BioProject PRJNA335387)
bEstimates were calculated by URI Genomics & Sequencing Center http://cels.uri.edu/gsc/cndna.html
cGenomic DNAs were acquired from DSMZ, the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures https://www.dsmz.de/home.html
dThis archaeon was included as a control for non-specific amplification
Generating and sequencing 16S rRNA gene sequence amplicons
Twelve replicates of the mock community were added to a single well of each of 12 96-well plates alongside hundreds of experimental samples for phylotype analysis. The 16S rRNA gene V1–V3 region was amplified using previously published primers [11] fused to barcodes [2] and adaptor sequences for the MiSeq instrument (Illumina Inc., San Diego, CA). Amplicon libraries were generated according to Kozich et al. [2]. Briefly, PCRs contained the following: 17 µl AccuPrime Pfx SuperMix (Life Technologies, Grand Island, NY), 5.0 µM each of the primers i5+V3 and i7+V1 (Table 2), and 25 ng of the mock community. The following PCR conditions were used: 2 min at 95 °C, 22 cycles of [20 s at 95 °C, 15 s at 55 °C, 5 min 72 °C], 72 °C for 10 min. Libraries were normalized using the SequalPrep Normalization Plate Kit (LifeTechnologies) and quantified using both Bioanalyzer (Agilent Technologies, Santa Clara, CA) and Kapa SYBR Fast qPCR (Kapa Biosystems, Wilmington, MA). Normalized pools were sequenced using version 3 (300 × 2) chemistry on the MiSeq instrument (Illumina) according to manufacturer’s instructions [2].
Table 2.
Primers used in this study
| Primer name | Sequence (5′–3′)a |
|---|---|
| i5+V3 | AATGATACGGCGACCACCGAGATCTACAC ATCGTACGTATGGTAATTCAATTACCGCGGCTGCTGG |
| i7+V1 | CAAGCAGAAGACGGCATACGAGAT AACTCTCGAGTCAGTCAGCCGAGTTTGATCMTGGCTCAG |
| For.seq.V3 | TATGGTAATTCAATTACCGCGGCTGCTGG |
| Rev.seq.V1 | AGTCAGTCAGCCGAGTTTGATCMTGGCTCAG |
| Index.V1 | CTGAGCCAKGATCAAACTCGGCTGACTGACT |
aIllumina’s MiSeq adaptor sequences are in bold. Underlined portion denotes the barcode region. Barcodes used in this study are from the Schloss laboratory’s MiSeq SOP (http://www.mothur.org/wiki/MiSeq_SOP; [2]). Italics denotes the V1 or V3 16S rRNA gene primer [11]
Data analysis
The mothur analysis package was used to assemble contigs, align sequences, trim sequences, remove chimeras (UCHIME, [3]), and remove non-bacterial sequences (mothur versions 1.33.3 and 1.34.0, http://www.mothur.org/wiki/MiSeq_SOP, [2, 12]). Following these quality-control steps, the data were rarified to 6654 sequences/sample and analyzed in three ways: (1) all sequences together, (2) with cross-sample singletons removed, or (3) with cross-sample singletons and doubletons removed. Cross-sample singletons and doubletons were defined as sequences that occurred only once (singletons) or twice (doubletons) among all samples. Taxonomy was assigned by aligning to mothur’s implementation of the SILVA database [13]. OTUs were clustered at 97 % similarity and analyzed in mothur for community metrics. Richness, evenness, and diversity were also conducted in mothur and included the use of the program Catchall [14]. Error analysis was performed in mothur based on the alignment of the experimental mock community to a FASTA file containing the actual 16S rRNA gene sequences from the genome sequences (Additional file 1). A complete list of the commands executed in mothur is available (Additional file 2).
Results and discussion
Sequencing the V1–V3 region yields spurious sequences
The V1–V3 region of the 16S rRNA gene was sequenced from twelve technical replicates of a mock community on the MiSeq platform. The program mothur was used to make quality-checked contigs from paired sequences, align the sequences to the SILVA database, trim the sequences, and remove chimeras. Intermediate data files from processing all 12 replicates together were large, and in total contained a high percentage (35.5 %) of sequences that failed to cluster with other sequences and were unique to an individual sampling of the mock community. The relationship between cluster membership size and the frequency of cluster sizes was explored via log/log plots to stabilize the large variability (Fig. 1a). Both linear regression and lowess fitting of the data indicated that low-frequency sequences deviate from the overall trend. We aligned the sequences to the SILVA reference database and found that the sequence clusters with one or two members were either not classifiable or assigned to bacteria not present in the mock community, and therefore were either non-informative or spurious. We removed the single sequences that did not cluster or that clustered with only one other sequence, and then plotted and analyzed the data by linear regression and lowess (Fig. 1b, c). Removal of the non-informative sequences improved the overall fit of the data.
Fig. 1.
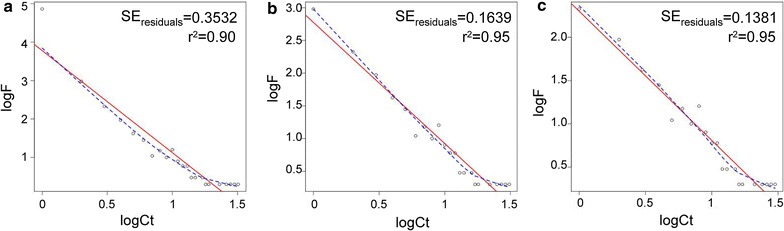
Cluster size versus frequency plot. The log10 of the cluster size (logCt) (i.e. number of sequences in each cluster) plotted against the log10 of the frequency of cluster membership sizes (i.e. the frequency of clusters that contained n sequences, where n is the cluster membership size) found among all mock community samples. Plots of all mock community data (a), data with cross-sample singletons removed (b), and data with both cross-sample singletons and doubletons removed (c) are shown. Red line regression line, blue line lowess fit line, SE standard error, r2 coefficient of determination
Removal of low-frequency sequences improves microbiota assessments
We examined the accuracy of diversity indices with and without the removal of low-frequency sequences. Removal of singletons, or singletons and doubletons, decreased the error rate and improved the accuracy of diversity metrics (Table 3). This is demonstrated by the reduction of observed and estimated number of species closer to the actual composition of the mock community. Interestingly, the two copies of the 16S rRNA gene in Haemophilus parasuis strain 29755 are only 94 % identical, so the 19 bacterial genomes in the mock community result in 20 bacterial OTUs.
Table 3.
Average diversity estimates of the mock community (n = 12, rarified to 6654 sequences per sample) with and without removing low-frequency sequences
| Mock community | Actual number of OTUsa | Observed number of OTUs | Estimated total number of OTUsb | Chao diversity index | Shannon diversity index | Inverse Simpson index | Error rate (%) | File size (Gb)c |
|---|---|---|---|---|---|---|---|---|
| All sequences | 20 | 734 ± 56 | 374,770 ± 214,807 | 21,676 ± 3273 | 3.6 ± 0.1 | 18 ± 0.8 | 3.6 | 41 |
| Singletons removed | 20 | 28 ± 0.8 | 68 ± 13 | 41 ± 3 | 2.7 ± 0.02 | 12 ± 0.3 | 1.4 | 21 |
| Single and doubletons removed | 20 | 22 ± 0.3 | 22 ± 0.3 | 23 ± 0.7 | 2.6 ± 0.02 | 12 ± 0.3 | 1.3 | 3 |
Average diversity estimates: plus or minus (±) the standard error of the mean, where appropriate
a Haemophilus parasuis has two divergent copies of the 16S rRNA gene that cluster separately
bThe estimated total number of OTUs is the number of OTUs predicted to be in the sample based on the number of OTUs observed in the sequences. The program Catchall was used to make the estimates [14]
cSize of the distance matrix file
Removing the large number of clusters with a membership size of one sequence (or one and two sequences) has a distinct advantage for streamlining the clustering of sequences into OTUs by reducing the size of the dataset without detracting from the ability to make valid comparisons. Reducing the number of sequences resulted in a 13-fold decrease in file size and exponentially reduced the time and memory space needed to create the distance matrix since the distance matrix is comprised of pairwise comparisons (Table 3).
OTUs were classified to assess how well the empirical taxonomy matched the known taxonomy of the input community. All members of the mock community were detected in the output (Fig. 2). Desulfovibrio and Coriobacterium were detected at only 0.05 and 0.23 % relative abundance, respectively, and subsequent analysis of their 16S rRNA gene sequences confirmed mismatches in the binding site for the V1 primer that would cause decreased amplification. This was not surprising since others have shown that PCR, not sequencing, is the largest source of bias in microbiota analyses [15, 16]. Also, thousands of sequences could not be classified despite their derivation from a defined community, perhaps because sequences from the MiSeq instrument have been shown to contain a small amount of cross-contamination between sequencing runs [17]. Removal of singletons or singletons and doubletons reduced the amount of unclassified sequences, decreasing their abundance from 2 % of the all reads to 0.1 % of the no-singletons dataset (Fig. 2). Our work shows that removing the large number of sequences that did not cluster and were unique to a single sample improved the taxonomic assignment of the sequences while preserving the relative abundance of real community members and facilitating the data analysis.
Fig. 2.
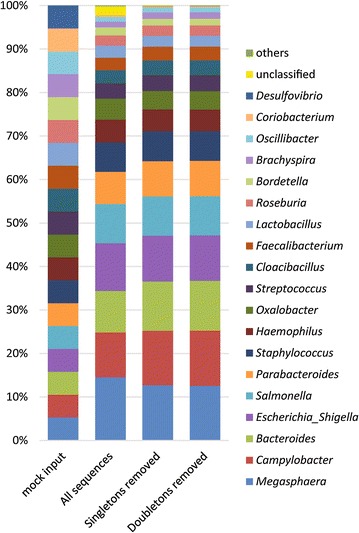
Average relative abundance of bacterial genera in 12 replicates of the mock community
Conclusions
Discarding low-frequency sequences can be concerning for those interested in the rare members of a bacterial community. Indeed, defining the rare microbiota in the context of sequencing errors has been explored [18–20]. Other work has supported the value of singleton removal [21, 22]. Our method is beneficial to researchers that use the Illumina MiSeq to sequence longer amplicons, such as the V1-V3 region of the 16S rRNA gene, from hundreds of samples. Removing the singletons and doubletons will suit the vast majority of projects seeking to analyze the abundant (>1 %) community organisms to draw biological conclusions.
Authors’ contributions
HKA analyzed data and drafted the manuscript. DOB, TL, JT, and BB performed experiments or analyses. SMDB, TN, and TAC provided strains. All authors contributed to and finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors thank Stephanie Jones, Lisa Lai, and Sarah Shore for outstanding technical support. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Competing interests
The authors declare that Diamond V, manufacturer of all-natural, microbial, fermentation-based feed additives, partially funded this work.
Availability of data and materials
The dataset supporting the conclusions of this article is available in GenBank’s Short Read Archive (SRP076629) under Bioproject PRJNA325813. The Cloacibacillus genome is available in GenBank under BioProject PRJNA335387.
Additional files
10.1186/s13104-016-2172-6 FASTA file of 16S rRNA genes contained in the mock community.
10.1186/s13104-016-2172-6 Commands executed in mothur.
Contributor Information
Heather K. Allen, Email: heather.allen@ars.usda.gov
Darrell O. Bayles, Email: darrell.bayles@ars.usda.gov
Torey Looft, Email: torey.looft@ars.usda.gov.
Julian Trachsel, Email: julian.trachsel@ars.usda.gov.
Benjamin E. Bass, Email: bbass@diamondv.com
David P. Alt, Email: david.alt@ars.usda.gov
Shawn M. D. Bearson, Email: shawn.bearson@ars.usda.gov
Tracy Nicholson, Email: tracy.nicholson@ars.usda.gov.
Thomas A. Casey, Email: thomas.casey@ars.usda.gov
References
- 1.Jumpstart Consortium Human Microbiome Project Data Generation Working Group Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One. 2012;7(6):e39315. doi: 10.1371/journal.pone.0039315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011;77(10):3219–3226. doi: 10.1128/AEM.02810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.May A, Abeln S, Crielaard W, Heringa J, Brandt BW. Unraveling the outcome of 16S rDNA-based taxonomy analysis through mock data and simulations. Bioinformatics. 2014;30(11):1530–1538. doi: 10.1093/bioinformatics/btu085. [DOI] [PubMed] [Google Scholar]
- 8.Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J. 2013;7(8):1493–1506. doi: 10.1038/ismej.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conlan S, Kong HH, Segre JA. Species-level analysis of DNA sequence data from the NIH human microbiome project. PLoS One. 2012;7(10):e47075. doi: 10.1371/journal.pone.0047075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Looft T, Allen HK, Casey TA, Alt DP, Stanton TB. Carbadox has both temporary and lasting effects on the swine gut microbiota. Front Microbiol. 2014;5:276. doi: 10.3389/fmicb.2014.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muyzer G, De Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59(3):695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunge J, Woodard L, Böhning D, Foster JA, Connolly S, Allen HK. Estimating population diversity with CatchAll. Bioinformatics. 2012;28(7):1045–1047. doi: 10.1093/bioinformatics/bts075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kennedy K, Hall MW, Lynch MD, Moreno-Hagelsieb G, Neufeld JD. Evaluating bias of Illumina-based bacterial 16S rRNA gene profiles. Appl Environ Microbiol. 2014;80(18):5717–5722. doi: 10.1128/AEM.01451-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks JP, Edwards DJ, Harwich MD, Rivera MC, Fettweis JM, Serrano MG, Reris RA, Sheth NU, Huang B, Girerd P. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015;15(1):66. doi: 10.1186/s12866-015-0351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson MC, Morrison HG, Benjamino J, Grim SL, Graf J. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS One. 2014;9(4):e94249. doi: 10.1371/journal.pone.0094249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. MBio. 2014;5(4):e1371. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12(1):118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 20.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12(7):1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 22.Flynn JM, Brown EA, Chain FJ, MacIsaac HJ, Cristescu ME. Toward accurate molecular identification of species in complex environmental samples: testing the performance of sequence filtering and clustering methods. Ecol Evol. 2015;5:2252–2266. doi: 10.1002/ece3.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403(6770):665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 24.Gulig PA, Curtiss R. Plasmid-associated virulence of Salmonella typhimurium. Infect Immun. 1987;55(12):2891–2901. doi: 10.1128/iai.55.12.2891-2901.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 26.Looft T, Levine UY, Stanton TB. Cloacibacillus porcorum sp. nov., a mucin-degrading bacterium from the swine intestinal tract and emended description of the genus Cloacibacillus. Int J Syst Evol Microbiol. 2013;63(Pt 6):1960–1966. doi: 10.1099/ijs.0.044719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris D, Kinyon JM, Mullin M, Glock R. Isolation and propagation of spirochetes from the colon of swine dysentery affected pigs. Can J Comp Med. 1972;36(1):74. [PMC free article] [PubMed] [Google Scholar]
- 28.Mullins MA, Register KB, Bayles DO, Dyer DW, Kuehn JS, Phillips GJ. Genome sequence of Haemophilus parasuis strain 29755. Stand Genom Sci. 2011;5(1):61. doi: 10.4056/sigs.2245029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Zhang Y, Buboltz AM, Zhang X, Schuster SC, Ahuja U, Liu M, Miller JF, Sebaihia M, Bentley SD. Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genom. 2012;13(1):545. doi: 10.1186/1471-2164-13-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Highlander SK, Hultén KG, Qin X, Jiang H, Yerrapragada S, Mason EO, Shang Y, Williams TM, Fortunov RM, Liu Y. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 2007;7(1):99. doi: 10.1186/1471-2180-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article is available in GenBank’s Short Read Archive (SRP076629) under Bioproject PRJNA325813. The Cloacibacillus genome is available in GenBank under BioProject PRJNA335387.