

Abstract
Serine metabolism is frequently dysregulated in cancers; however, the benefit that this confers to tumors remains controversial. In many cases, extracellular serine alone is sufficient to support cancer cell proliferation, whereas some cancer cells increase serine synthesis from glucose and require de novo serine synthesis even in the presence of abundant extracellular serine. Recent studies cast new light on the role of serine metabolism in cancer, suggesting that active serine synthesis might be required to facilitate amino acid transport, nucleotide synthesis, folate metabolism, and redox homeostasis in a manner that impacts cancer.
Introduction
Most modern studies of cancer have focused on the disease from a genetic perspective. However, cancer also involves biochemical alterations, including the adaptation of metabolism to support inappropriate cell proliferation. In the past century, the biochemical pathways involved in cell metabolism have been elucidated, but how flux through these various pathways is regulated in diverse physiological settings remains an open question. Metabolic fluxes change after a cell’s transformation to a malignant state or after the expression of specific oncogenes, as well as in response to alterations in nutrient availability and tissue environment (Cairns et al., 2011; Davidson et al., 2016; Hensley et al., 2016; Mullarky et al., 2016). The importance of serine metabolism in multiple cancers is increasingly apparent, and how the metabolism of this amino acid influences cancer phenotypes is an area of active investigation. Understanding the specific metabolic needs of cancer cells holds promise for treating patients, as some of the oldest cancer therapies target metabolic vulnerabilities and are still widely used in the clinic today (Vander Heiden, 2011). In this regard, serine metabolism could yield a promising set of potentially drugable targets (Mullarky et al., 2016; Pacold et al., 2016).
This review discusses the discovery of the serine synthesis pathway and its dysregulation in cancer and summarizes the findings of many recent studies on the role of serine metabolism in cancer. Phosphoglycerate dehydrogenase (PHGDH) amplification in select cancers, the role of extracellular serine in supporting cell proliferation, potential benefits of increased serine synthesis pathway (SSP) flux, and the interaction between glycolysis, the SSP, and nucleotide biosynthesis are all discussed with a perspective on how altered serine metabolism plays a role in cancer.
Metabolic requirements of cancer cells
In nondividing cells, metabolism maintains homeostasis by fueling housekeeping processes that rely heavily on ATP; thus, a major metabolic task of nonproliferating cells is to fully oxidize nutrients to produce energy in the form of ATP. In contrast, proliferating cells must accumulate the biomass required to build a new cell. This includes nucleotides for genome replication and ribosomal RNA, lipids for membranes, amino acids for proteins, and other cellular building blocks. The biosynthesis of these macromolecules requires not only ATP but also carbon and nitrogen precursors (Lunt and Vander Heiden, 2011), as well as electron acceptors to maintain redox balance (Sullivan et al., 2015).
Increased serine biosynthesis is one of many metabolic changes that have been reported in cancer cells (Davis et al., 1970; Snell, 1984), and serine is a central node for the biosynthesis of many molecules (Fig. 1; Lehninger et al., 2013; Locasale, 2013). Serine is a precursor of the nonessential amino acids glycine and cysteine. Glycine is in turn a precursor of porphyrins and is also incorporated directly into purine nucleotide bases and into glutathione (GSH). Serine is necessary for the production of sphingolipids via the synthesis of sphingosine, and serine is a headgroup, or headgroup precursor, for phospholipids. Additionally, serine supplies carbon to the one-carbon pool, which is involved in folate metabolism (Fig. 2). The conversion of serine to glycine, catalyzed by serine hydroxymethyltransferase (SHMT), donates a one-carbon unit to tetrahydrofolate to produce 5,10-methylenetetrahydrofolate (CH2-THF). CH2-THF is used in thymidine synthesis and is a precursor of other folate species that contribute to purine synthesis. Folates can also allow the regeneration of methionine from homocysteine and thus facilitate the generation of S-adenosylmethionine (SAM), the methyl donor for both DNA and histone methylation reactions that influence the epigenetic control of gene expression. Therefore, an increase in serine availability could be valuable for proliferating cancer cells for multiple reasons. However, because some cancer cells can scavenge macromolecules (White, 2013), which molecules are most limiting for tumors in a physiological context remains an area of active study. Because serine is critical for the biosynthesis of many macromolecules required to support cell proliferation, it is important to understand how cells obtain serine.
Figure 1.

The metabolic fates of serine. This schematic illustrates the products in mammalian cells where serine is a biosynthetic precursor.
Figure 2.
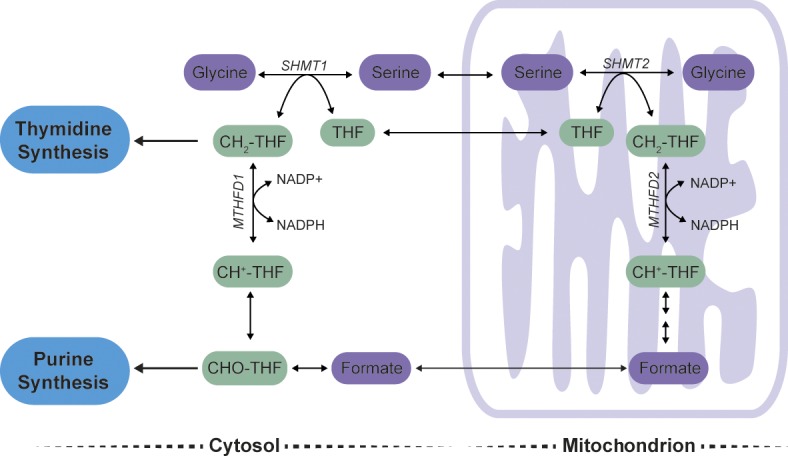
Serine and one-carbon metabolism. Serine can be cleaved into glycine and CH2-THF, a one-carbon unit, in either the cytosol or the mitochondria in a reaction catalyzed by SHMT1 or SHMT2, respectively. Cytosolic CH2-THF is needed for the biosynthesis of thymidine, and cytosolic 10-formyl-tetrahydrofolate (CHO-THF) is used in purine synthesis. One-carbon units produced by the action of SHMT2 in the mitochondria can be transported as formate to the cytosol and produce CHO-THF and CH2-THF. CH+-THF, 5,10-methenyltetrahydrofolate.
Elucidation of the serine biosynthesis pathway
Cells can obtain serine by either import from the extracellular environment or intracellular synthesis from glucose, and there is increasing evidence that serine biosynthesis from glucose is important for many cancers (Locasale, 2013; Zogg, 2014). Serine biosynthesis via the SSP was first described in the 1950s. Initially, there were two schools of thought as to how serine is synthesized. One group championed a nonphosphorylated pathway based on the observation that [14C]glyceric acid administered to rats gave rise to radioactive serine in tissues (Sallach, 1955). The opposing camp proposed that serine was derived from the glycolytic intermediate 3-phosphoglycerate (3-PG) via a phosphorylated pathway (Ichihara and Greenberg, 1955), now known to be the physiological route of serine synthesis, with the nonphosphorylated pathway representing one means of serine catabolism (Fig. 3).
Figure 3.

Pathways of serine synthesis and catabolism. Serine is synthesized from the glycolytic intermediate (3-PG) by the sequential action of PHGDH, PSAT, and PSPH. Serine can be catabolized directly to pyruvate or converted to glycerate in two steps.
The reactions of the SSP are catalyzed by the enzymes PHGDH, phosphoserine aminotransferase (PSAT), and phosphoserine phosphatase (PSPH; Fig. 3). Genetic evidence suggests that the SSP is the sole route for serine biosynthesis from glucose in all nonphotosynthetic organisms studied to date. Both Salmonella typhimurium and Escherichia coli can be converted to serine auxotrophs by single mutations in genes encoding SSP enzymes (Umbarger and Umbarger, 1962; Pizer, 1963). A genetically engineered mouse with deletion of the PHGDH gene, which encodes the first enzyme of the SSP, shows reduced serine levels in brain (Yoshida et al., 2004), and human patients with mutations in SSP genes show decreased blood serine levels (van der Crabben et al., 2013), suggesting that this pathway is also involved in serine production in mammals. Furthermore, the activity of the nonphosphorylated pathway is associated with gluconeogenesis, supporting the notion that the nonphosphorylated pathway is used for serine catabolism (Suda, 1967; Cheung et al., 1969; Snell, 1984). Based on these lines of evidence, it is clear that the SSP is the exclusive method for intracellular serine synthesis in nonphotosynthetic organisms. Given the central role of serine in cellular biosynthesis, the unique ability of the SSP to synthesize serine makes the SSP a pathway of interest for researchers studying metabolic dysregulation in cancer cells.
The discovery of dysregulated serine metabolism in cancer
The connection between cancer and serine biosynthesis was first suggested by the observations that PHGDH activity was greater in rat hepatoma cell lines passaged in rats than in normal rat liver and that, of the rat hepatoma cell lines, those with the fastest growth rates had the greatest PHGDH activity (Davis et al., 1970). Flux through the SSP was noted to be increased in many tumors relative to matched normal tissue, and of the pathway enzymes, PHGDH activity was most consistently increased and correlated with tumor growth (Snell, 1984). PHGDH activity is also increased during nonpathological proliferation, such as in the developing rat liver. However, a hepatoma cell line with the same proliferation rate as proliferating normal hepatocytes showed still higher PHGDH activity, suggesting that serine biosynthesis may have a distinct role in transformed cells (Snell and Weber, 1986).
A link between serine metabolism and nucleotide biosynthesis was hypothesized to account for the SSP requirement of cancer cells. The activity of SHMT, which converts serine into glycine and a one-carbon unit, is selectively retained in tumors (Snell, 1984). In activated lymphocytes, SHMT activity increases fourfold, and [14C]serine labels DNA with the same kinetics as [3H]thymidine, indicating that most thymidine synthesis uses serine-derived carbon (Eichler et al., 1981). Additionally, in hepatoma cell lines grown in culture, PHGDH and SHMT activities, as well as the incorporation of [14C]serine into nucleotides, are elevated during exponential growth but decrease once confluence is reached and proliferation slows (Snell et al., 1987). These data support an important role for serine in nucleotide synthesis. Originally, these data were interpreted as implying that the SSP was important for supplying carbon in the form of serine for nucleotide biosynthesis. However, much of the data are equally consistent with a role for extracellular serine in nucleotide synthesis, as labeled serine provided in the media was observed to be a major contributor to nucleotides in these cells despite increased SSP activity (Snell et al., 1987).
Collectively, the data discussed in this section suggest that cancer cells up-regulate flux through the SSP, and actively proliferating cells use serine for nucleotide biosynthesis. However, this is not sufficient to prove that the reason for up-regulating SSP flux is simply to provide carbon for nucleotide synthesis. Furthermore, these studies did not address the mechanism by which SSP flux is increased.
PHGDH amplification in human cancer
A renewed interest in the link between serine biosynthesis and cancer was sparked by the finding that a genomic region on chromosome 1p12 is amplified in a subset of human cancer samples and cell lines, most frequently in melanoma (40% based on one dataset) and breast cancer (Beroukhim et al., 2010; Locasale et al., 2011; Possemato et al., 2011). The smallest common overlapping region of copy number gain across samples contains only five genes, one of which is PHGDH. Cell lines with PHGDH copy number gain show increased PHGDH protein levels, an increase in the fraction of intracellular serine derived from glucose, and higher levels of the unique pathway intermediate, phosphoserine, all indicative of increased pathway activity (Locasale et al., 2011; Possemato et al., 2011). In addition to amplification, PHGDH expression can be up-regulated transcriptionally by activating transcription factor 4 (ATF4; Adams, 2007) and the protooncogene c-Myc (Nilsson et al., 2012) and epigenetically by the lysine methyltransferase G9A (Ding et al., 2013). ATF4-induced serine synthesis is important downstream of signaling from the pro-antioxidant transcription factor nuclear factor erythroid 2–like 2 (NRF2) and the nutrient-sensing complex mechanistic target of rapamycin complex 1 (mTORC1) and helps cells adapt to oxidative stress and amino acid deficiency (DeNicola et al., 2015; Ben-Sahra et al., 2016). Human cell lines with high PHGDH expression and pathway flux can proliferate robustly in the absence of serine, whereas those with low PHGDH expression cannot (Possemato et al., 2011).
High PHGDH expression is associated with specific subtypes of cancer. PHGDH copy number gain is more frequently found in triple-negative breast cancers (which lack expression of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 [Her2]) than in other breast cancer subtypes (Locasale et al., 2011), and PHGDH expression is higher in estrogen receptor–negative tumors compared with estrogen receptor–positive tumors (Possemato et al., 2011). Furthermore, PHGDH and PSAT protein expression is elevated in metastatic variants of estrogen receptor–negative breast cancer cells, and high expression is associated with shorter time to relapse, shorter overall survival time, higher tumor grade, and high expression of the proliferative markers proliferating cell nuclear antigen and Ki-67 (Pollari et al., 2011). PHGDH is also overexpressed in gliomas, and high expression is associated with higher tumor grade and with decreased overall survival (Liu et al., 2013). In cervical cancer, PHGDH expression is elevated compared with normal cervical epithelium, and expression positively correlates with tumor grade and size (Jing et al., 2013).
PHGDH expression and SSP flux can promote some cancer-associated phenotypes. When nontransformed breast epithelial MCF-10A cells are grown in 3D culture, cells form organoid-like spheres in which the inner cells are deprived of matrix attachment and undergo cell death, forming an empty luminal space. Expression of known driver genes in breast cancer, such as ErbB1, prevents the formation of this hollow lumen (Muthuswamy et al., 2001; Schafer et al., 2009), a phenotype also observed with wild-type PHGDH expression (Locasale et al., 2011). Expression of a hypomorphic V490M PHGDH does not promote luminal cell proliferation and survival, suggesting that increased flux through the SSP can promote the evasion of matrix detachment–induced cell death. High SSP activity has also been associated with metastasis (Piskounova et al., 2015).
PHGDH expression is necessary for the proliferation of PHGDH-amplified cell lines, as is expression of the other SSP enzymes PSAT and PSPH in at least some cell lines (Locasale et al., 2011; Possemato et al., 2011). An enzymatically inactive form of PHGDH fails to support proliferation of PHGDH-amplified cells (Mattaini et al., 2015), further indicating that flux through the SSP is required in these cells. Whether PHGDH expression is required for tumor growth in vivo is controversial, with some studies showing that PHGDH knockdown suppresses tumor growth (Possemato et al., 2011; DeNicola et al., 2015) and one study arguing that PHGDH knockdown has no effect on tumor maintenance (Chen et al., 2013). More recent data generated with small-molecule PHGDH inhibitors suggest that PHGDH activity is required to support the proliferation and survival of amplified cells, as well as tumor growth (Mullarky et al., 2016; Pacold et al., 2016). Additional work is needed to understand whether high PHGDH expression is important early in tumorigenesis or during metastasis (Piskounova et al., 2015) and to identify the tumor conditions that drive PHGDH amplification and dependence on SSP flux.
Although PHGDH amplification is associated with tumorigenesis, insufficient PHGDH expression can have deleterious effects, particularly on brain metabolism. The homozygous germline deletion of Phgdh in mice causes embryonic lethality as a result of multiple developmental defects, most notably affecting the central nervous system and attributed to the low permeability of the blood–brain barrier to serine (Yoshida et al., 2004). These defects are reminiscent of human patients with mutations in genes encoding SSP enzymes, who also have neurological symptoms (van der Crabben et al., 2013; Acuna-Hidalgo et al., 2014). Developing small-molecule inhibitors that cannot cross the blood–brain barrier may be necessary to prevent neurological complications if SSP inhibitors were to be used in the clinic as antitumor agents, although to date toxicity has not been reported in mice exposed to one PHGDH inhibitor at doses that were sufficient to suppress tumor growth (Pacold et al., 2016).
In summary, many tumors up-regulate PHGDH expression and SSP flux, and in some conditions, PHGDH activity is required for tumor growth in vivo. However, further study is required to determine at what time point tumors are susceptible to PHGDH inhibition. Also, attempts to treat patients by inhibiting PHGDH with small molecules will have to take into account possible neurological symptoms if the inhibitor crosses the blood–brain barrier. In addition, how best to identify patients likely to respond to PHGDH inhibitor treatment remains an important question because not all cancer cells are dependent on SSP flux.
Extracellular serine and tumor metabolism
As discussed earlier, cells can acquire serine by either synthesizing it internally or importing serine from the environment. Serine is a small, neutral amino acid and, as such, can be transported by one of three systems. Two of the systems are sodium dependent: the alanine/serine/cysteine/threonine transporters ASCT1 and ASCT2 (encoded by SLC1A4 and SLC1A5, respectively) and the system A transporters SAT1 and SAT2 (encoded by SLC38A1 and SLC38A2, respectively). The third is a family of neutral amino acid antiporters, the alanine/serine/cysteine transporter (ASC) system (El-Hattab, 2016). These antiporters are of particular interest because they are active even at steady state, so that for instance, one molecule of intracellular serine can be exchanged for one molecule of extracellular serine. Normally this process goes unnoticed, but a recent study (DeNicola et al., 2015) points out that it can complicate interpretation of heavy isotope–labeling experiments by setting up an exchange flux between labeled and unlabeled species across the plasma membrane.
Consistent with serine being a central precursor for biosynthetic metabolism, many tumors depend on the availability of extracellular serine. Xenografts of HCT116 colon cancer cells grow roughly half as fast when mice are fed a serine- and glycine-free diet as opposed to a normal diet (Maddocks et al., 2013). In human colon cancer and lung cancer cell lines, proliferation in medium that contains serine without glycine is indistinguishable from proliferation in medium containing both amino acids, whereas withdrawal of serine alone affects proliferation to the same degree as depletion of both amino acids (Labuschagne et al., 2014). Moreover, providing increased concentrations of glycine in the absence of serine results in even more severe suppression of proliferation than withdrawal of both serine and glycine. These findings argue that serine is a vitally important amino acid, and this conclusion is further supported by the observation that cells preferentially take up serine and excrete glycine when serine is available and consume glycine only when serine is depleted. This may explain why glycine depletion strongly correlates with proliferation rate across cancer cells (Jain et al., 2012), because the most rapidly proliferating cells will deplete serine from the media most quickly and then switch to glycine consumption (Labuschagne et al., 2014), implying that glycine loss from the media is a consequence of rapid proliferation and serine consumption.
Because the conversion of serine to glycine donates a one-carbon unit to the folate pool, the preferential consumption of serine over glycine suggests that a demand for one-carbon units might be driving serine-to-glycine conversion. Conversely, conversion of glycine to serine consumes a one-carbon unit, so a high-glycine environment might limit the availability of one-carbon units for nucleotide biosynthesis, potentially contributing to glycine toxicity. In purine biosynthesis, conversion of the precursor glycineamide ribonucleotide (GAR) to AMP or GMP requires the addition of two one-carbon units from the folate pool. Cells supplied with glycine, but not serine, show an accumulation of GAR and the depletion of both AMP and GMP, implying that they have insufficient one-carbon units. Exogenous formate can supply one-carbon units, and administration of glycine together with formate can rescue cell growth (Labuschagne et al., 2014). These data imply that the balance of serine and glycine affects folate availability and influences the ability of cancer cells to proliferate.
One-carbon units derived from serine can also be used to support SAM synthesis. The reactions by which folate metabolism donates one-carbon units to the SAM pool appear to have a low level of activity in many cancer cells, but recent work has found that serine availability is still needed to maintain SAM levels (Maddocks et al., 2016). Because serine and folate metabolism are needed for adenine synthesis, limiting the availability of these nutrients lowered the concentration of ATP without altering the ATP/AMP ratio such that SAM synthesis, which also requires ATP, was impaired. These findings illustrate the complex connections between serine metabolism, nucleotide synthesis, and SAM and represent another way in which limiting serine can have detrimental effects on tumor cells.
The presence or absence of the tumor suppressor protein p53 is a critical determinant of the ability of a cell to cope with extracellular serine depletion. Upon growth in serine- and glycine-free medium, both p53+/+ and p53−/− human colon cancer cells show an increase in serine production from glycolytic intermediates and a decrease in ATP levels (Maddocks et al., 2013). However, unlike p53−/− cells, p53+/+ cells adapt to the serine-starved state by undergoing p53- and p21-dependent, transient cell cycle arrest (Maddocks et al., 2013). Whereas p53−/− cells use the low levels of serine available to them to continue nucleotide biosynthesis, p53+/+ cells transiently divert their serine stores to the production of the antioxidant GSH. This allows p53+/+ cells to limit their oxidative stress and adapt to serine starvation. The proliferation of p53−/− cells starved of serine can be almost fully restored by providing exogenous pyruvate as a source of ATP via oxidative phosphorylation and exogenous GSH or N-acetyl cysteine, another antioxidant, to combat damage from reactive oxygen species (Maddocks et al., 2013).
A recent article (Krall et al., 2016) suggested another mechanism by which uptake of extracellular serine can affect cell proliferation. The study identified asparagine as an amino acid exchange factor, and intracellular asparagine was especially effective at promoting import of serine and threonine. In fact, in a human cervical cancer cell line deprived of extracellular asparagine and expressing an shRNA targeting asparagine synthase, SSP pathway enzymes were transcriptionally up-regulated to increase de novo serine synthesis because of impaired ability to import extracellular serine in the absence of intracellular asparagine (Krall et al., 2016).
Based on these data, it is clear that the effects of depriving cancer cells of extracellular serine are many and far-reaching. However, a growing body of data suggests that up-regulation of the SSP in cancer may play a role beyond merely providing serine itself for biosynthesis.
A serine-independent role for the SSP in cancer
A wealth of data supports a role for SSP activity in promoting tumor growth; exactly how it does so is less clear. Extracellular serine is important for cell proliferation in some contexts, and studies show that it efficiently labels nucleotides in cells (Eichler et al., 1981; Snell et al., 1987). However, abundant extracellular serine and glycine are unable to support proliferation when the SSP is inactive, and decreased proliferation after PHGDH knockdown in PHGDH-amplified human breast cancer cells cannot be rescued by adding excess serine to the growth medium (Possemato et al., 2011; Chen et al., 2013). This argues that flux through the SSP may provide a benefit to cell proliferation beyond supplying serine. Although increased SSP flux can mitigate the effects of serine depletion by providing a source of serine for cells, serine is among the most abundant amino acids present in cell culture medium and in blood (Mayers and Vander Heiden, 2015). Manipulating PHGDH activity affects the proportion of serine synthesized de novo from glucose but does not change intracellular serine concentration (Possemato et al., 2011; Chen et al., 2013). It has been argued that because the pool of newly synthesized serine is in equilibrium with extracellular serine, the requirement for newly synthesized serine might not be apparent from labeling studies (DeNicola et al., 2015). Nevertheless, cells consume large amounts of extracellular serine regardless of SSP activity (Jain et al., 2012; Hosios et al., 2016), suggesting that the SSP may still have a benefit beyond simply providing serine to be used for biosynthesis.
What then is the benefit to cells of increased flux through the SSP? Production of α-ketoglutarate (αKG) for replenishment of the tricarboxylic acid cycle (anapleurosis) via the PSAT reaction has been suggested as one benefit of increased serine synthesis (Possemato et al., 2011). This hypothesis is supported by the observation that the knockdown of SSP enzymes decreased both the fraction of αKG derived from [13C]glutamine and the intracellular αKG concentration (Possemato et al., 2011). The production of αKG through transamination by PSAT and other aminotransferases is also nitrogen neutral, unlike production of αKG from glutamate by glutamate dehydrogenase, which yields a unit of ammonia and may lead to toxicity if ammonia accumulates (Mullarky et al., 2011). However, changes in intracellular αKG concentration with PHGDH knockdown were not observed in two separate studies in human breast cancer cell lines dependent on PHGDH expression for proliferation (Locasale et al., 2011; Fan et al., 2015). Also, in many cancer cells, transamination resulting in alanine and αKG production is very active and relies on pyruvate, another intermediate in glycolysis (DeBerardinis and Cheng, 2010). Production of αKG by this method is also nitrogen neutral and does not generate ammonia. Thus, why production of serine rather that alanine would be important as a major source of αKG is not entirely clear.
In E. coli, PHGDH can produce 2-hydroxyglutarate (2-HG) from αKG in a side reaction (Zhao and Winkler, 1996). Rat PHGDH does not catalyze this reaction (Achouri et al., 1997; Dey et al., 2005), and it was initially assumed that the human enzyme also cannot produce 2-HG. However, recently Fan et al. (2015) showed that D-2-HG is produced by human PHGDH. D-2-HG is implicated in the pathogenesis of isocitrate dehydrogenase (IDH) mutant cancers, where it accumulates to millimolar levels because of a neomorphic IDH mutation, which results in IDH proteins that produce 2-HG instead of the normal product, isocitrate (Dang et al., 2009; Ward et al., 2010). However, the absolute intracellular concentrations reported in IDH wild-type, PHGDH-amplified cells are 100–1,000 times lower than those in IDH mutant cancers (Fan et al., 2015), and some data argue against a role for 2-HG production in supporting the proliferation of PHGDH-amplified cells (Mattaini et al., 2015).
Serine synthesized de novo might be used differently than imported serine, a possibility that has not been examined. Alternatively, another pathway intermediate in the SSP might be important in addition to serine. Both NADH and phosphohydroxypyruvate (PHP) are products of the PHGDH reaction, and phosphoserine is a product of the PSAT reaction. NADH production is unlikely to be limited by PHGDH levels in cancer cells, as the NAD+/NADH ratio in cancer cells is very low (Hung et al., 2011; Sullivan et al., 2015), and at most 10% of glycolytic carbon is diverted to serine synthesis even in cells with high SSP flux (Locasale et al., 2011). Therefore, if SSP flux has any appreciable effect on the NAD+/NADH ratio, increased NADH production from the SSP might be expected to exacerbate NAD+ limitation in cancer cells. Whether phosphoserine or PHP has an undiscovered biosynthetic or regulatory role in cells remains unknown and an intriguing possibility. De novo serine synthesis also plays an important regulatory role in coordinating flux of other metabolic pathways, particularly nucleotide biosynthesis, a function that cannot be recapitulated merely by increasing extracellular serine and which we discuss in more detail next.
Reciprocal regulation of glycolysis, serine synthesis, and nucleotide production
Studies of pyruvate kinase (PK) regulation have suggested a link between serine synthesis from glucose and nucleotide synthesis that might have an impact on tumor development. PK catalyzes the last step of glycolysis, coupling ATP generation to the production of pyruvate from phosphoenolpyruvate (PEP). PK has multiple different isoforms, but the M2 isoform (PKM2) is selected for in cancer, and its activity can be regulated by several signaling molecules and by metabolite levels (Israelsen and Vander Heiden, 2015). Serine acts as an allosteric activator of PKM2, with an AC50 of 1.3 mM (Ibsen and Marles, 1976; Eigenbrodt et al., 1983; Chaneton et al., 2012). PK activity also affects SSP activity, as cells with high PK activity produce a smaller fraction of serine from glucose than do cells with low PK activity (Anastasiou et al., 2012; Chaneton et al., 2012; Kung et al., 2012; Ye et al., 2012; Lunt et al., 2015). Upon PKM2 knockdown, PEP and citrate levels increase, whereas pyruvate and lactate levels decrease. These changes are mimicked by culturing cells in serine- and glycine-free medium, and it has been hypothesized that this system allows cells to adjust PK activity to match cellular serine demands (Chaneton et al., 2012). When intracellular serine is low, PKM2 activity decreases, allowing more glucose carbon to flow into the SSP to rectify the deficit. Indeed, the ability to decrease PKM2 activity has been shown to be critical for cellular proliferation (Christofk et al., 2008a,b; Hitosugi et al., 2009; Anastasiou et al., 2012; Israelsen et al., 2013), and it may be that this requirement exists in part to maintain SSP flux. However, down-regulation of PKM2 activity may confer multiple benefits on cancer cells, as serine consumption by the enzyme SHMT2 can limit activation of PKM2 and reduce oxygen demand to promote survival in conditions of metabolic stress (Kim et al., 2015).
The idea that glycolysis and serine synthesis are reciprocally regulated is supported by several observations: PKM2-expressing cells survive serine deprivation better than cells that express the constitutively active PKM1 isoform (Ye et al., 2012); the activation of PKM2 with small-molecule chemical activators depletes the intracellular serine pool in some cells (Anastasiou et al., 2012); and serine deprivation increases the toxicity of PKM2 activation (Kung et al., 2012). Alternatively, reciprocal regulation of PK activity by serine availability might allow cells to coordinate glucose flux with the availability of other nutrients for macromolecule biosynthesis (Gui et al., 2013). In support of this model, deletion of PKM2 in mouse embryonic fibroblasts results in PKM1 expression and in proliferation arrest characterized by a decrease in serine and glycine synthesis from glucose. It also causes impaired nucleotide production and the inability to synthesize DNA (Lunt et al., 2015). Nucleotide biosynthesis is limiting for proliferation in this context, despite unchanged intracellular serine and glycine concentration (Lunt et al., 2015). Exactly how cells coordinate glycolysis and SSP flux with the availability of extracellular serine and the use of serine for nucleotide synthesis remains an area of active study.
Recent advances in the study of cytosolic versus mitochondrial metabolism of serine and its downstream metabolites are informing our understanding of how serine, one-carbon metabolism, and nucleotide synthesis are linked. The direct product of serine-to-glycine conversion is CH2-THF, which acts as a substrate of cytosolic or mitochondrial methylenetetrahydrofolate dehydrogenase (MTHFD) to ultimately produce formate (Fig. 2). Although cytosolic SHMT1 can produce CH2-THF and NADPH in some cells (Fan et al., 2014), it appears that in many cells, SHMT1 operates in the glycine-to-serine direction (Herbig et al., 2002; Lewis et al., 2014; Bao et al., 2016; Ducker et al., 2016). Conversely, mitochondrial SHMT2 mediates serine-to-glycine conversion to support NADPH production in mitochondria (Ye et al., 2014). MTHFD2 is the mitochondrial enzyme responsible for the metabolism of CH2-THF, and it is also one of the most differentially expressed enzymes in tumors relative to normal tissue (Nilsson et al., 2014). This enzyme is also important for cell survival in breast cancer and acute myeloid leukemia (Nilsson et al., 2014; Pikman et al., 2016). The consumption of serine by SHMT2 also produces formate in mitochondria that is reduced in the cytosol to produce folate species for nucleotide synthesis, and the production of serine from glycine in the cytosol can consume these same folates (Fig. 2; Lewis et al., 2014). Why serine metabolism is compartmentalized has yet to be resolved. Cytosolic and mitochondrial one-carbon pathways use NAD(P)(H), and different NAD+/NADH and NADP+/NADPH ratios are present in the mitochondria and the cytosol, providing one potential explanation for compartmentalized metabolism. It is also possible that compartmentalization allows the generation of one-carbon units in both the mitochondria and the cytosol to fuel compartment-specific reactions. For example, it has been proposed that one important fate of mitochondrial folates is formylation of initiator tRNA for protein synthesis in that compartment (Tibbetts and Appling, 2010).
Recent data examining the effects of PHGDH inhibitors suggests that de novo serine synthesis may permit the productive use of extracellular serine for nucleotide synthesis (Pacold et al., 2016). Exposing PHGDH-amplified cells to a PHGDH inhibitor negatively affects cell proliferation, as well as the incorporation of glucose carbon into serine, AMP, and dTMP. Surprisingly, inhibiting PHGDH also decreases the incorporation of extracellular serine from the culture medium into AMP and dTMP. It is hypothesized that when the SSP is inhibited, SHMT1 might consume CH2-THF to generate serine at the expense of nucleotide synthesis. In support of this theory, when CRISPR/Cas9-mediated gene editing was used to produce SHMT1 null cells, no change in the incorporation of exogenous serine into nucleotides was observed upon SSP inhibition (Pacold et al., 2016). Furthermore, PHGDH knockdown reduces the total pool sizes of deoxynucleotides (DeNicola et al., 2015). These data are consistent with the notion that de novo serine synthesis inhibits SHMT1 to prevent folate wasting; however, the exact mechanism by which PHGDH activity regulates folate availability remains unknown. This ability of the SSP to regulate availability of carbon for nucleotide synthesis may represent the elusive role of the SSP that some cancer cells rely on for their ability to proliferate.
Conclusion
Data supporting the importance of extracellular serine and SSP flux in cancer continues to accumulate. In some instances, the availability of serine itself is limiting for cancer cell proliferation both in vitro and in vivo, and when serine is withheld, nucleotide precursors are depleted because of a lack of one-carbon units (Maddocks et al., 2013; Labuschagne et al., 2014). However, cells with PHGDH amplification require SSP flux even when serine is available (Possemato et al., 2011; Chen et al., 2013). In some settings, serine synthesis appears to be part of an adaptive response to oxidative stress (Maddocks et al., 2013; DeNicola et al., 2015). Key remaining questions in the field focus on how to use this knowledge in the clinic and on gaining a deeper understanding of the underlying reasons serine is important for tumor cells.
Further work is required to determine the contexts in which SSP flux and/or consumption of extracellular serine are necessary for tumor growth and to better understand how tumor dependence on the SSP and/or extracellular serine can be exploited for therapeutic benefit. In one experiment, providing a serine-deficient diet was able to slow xenograft growth (Maddocks et al., 2013), but this approach will likely be most successful in settings in which de novo serine synthesis is limited. Early studies of PHGDH inhibitors appear promising in mouse models, as the inhibitors are well tolerated and slow growth of xenografts, even when mice are fed a normal, serine-containing diet (Pacold et al., 2016). So far this has only been demonstrated in xenografts of cells with PHGDH amplification, but it is reasonable to hypothesize that cells that overexpress PHGDH or exhibit high SSP flux may also be susceptible to PHGDH inhibitors. A critical precursor to using a low-serine diet or PHGDH inhibitors in the clinic will be to develop strategies to identify which patients are likely to benefit from each method of targeting serine’s central role in cancer.
Acknowledgments
We thank members of the Vander Heiden laboratory for their thoughtful discussions and comments on the manuscript.
We acknowledge support from the American Association for Cancer Research, the Burroughs Wellcome Fund, the Koch Institute, the Ludwig Center at the Massachusetts Institute of Technology, Stand Up to Cancer, the National Science Foundation Graduate Research Fellowship Program, and the National Institutes of Health (grant R21 CA198028). M. Vander Heiden is a consultant and Science Advisory Board member for Agios Pharmaceuticals.
The authors declare no additional competing financial interests.
Footnotes
Abbreviations used:
- 2-HG
- 2-hydroxyglutarate
- 3-PG
- 3-phosphoglycerate
- αKG
- α-ketoglutarate
- ASC
- alanine/serine/cysteine transporter
- ASCT
- alanine/serine/cysteine/threonine transporter
- ATF4
- activating transcription factor 4
- CH2-THF
- 5,10-methylenetetrahydrofolate
- GAR
- glycineamide ribonucleotide
- GSH
- glutathione
- IDH
- isocitrate dehydrogenase
- MTHFD
- methylenetetrahydrofolate dehydrogenase
- PEP
- phosphoenolpyruvate
- PHGDH
- phosphoglycerate dehydrogenase
- PHP
- phosphohydroxypyruvate
- PK
- pyruvate kinase
- PKM2
- M2 isoform of PK
- PSAT
- phosphoserine aminotransferase
- PSPH
- phosphoserine phosphatase
- SAM
- S-adenosylmethionine
- SAT
- system A transporter
- SHMT
- serine hydroxymethyltransferase
- SSP
- serine synthesis pathway
References
- Achouri Y., Rider M.H., Schaftingen E.V., and Robbi M.. 1997. Cloning, sequencing and expression of rat liver 3-phosphoglycerate dehydrogenase. Biochem. J. 323:365–370. 10.1042/bj3230365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Hidalgo R., Schanze D., Kariminejad A., Nordgren A., Kariminejad M.H., Conner P., Grigelioniene G., Nilsson D., Nordenskjöld M., Wedell A., et al. . 2014. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am. J. Hum. Genet. 95:285–293. 10.1016/j.ajhg.2014.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams C.M. 2007. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J. Biol. Chem. 282:16744–16753. 10.1074/jbc.M610510200 [DOI] [PubMed] [Google Scholar]
- Anastasiou D., Yu Y., Israelsen W.J., Jiang J.K., Boxer M.B., Hong B.S., Tempel W., Dimov S., Shen M., Jha A., et al. . 2012. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8:839–847. 10.1038/nchembio.1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X.R., Ong S.E., Goldberger O., Peng J., Sharma R., Thompson D.A., Vafai S.B., Cox A.G., Marutani E., Ichinose F., et al. . 2016. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife. 5:5 10.7554/eLife.10575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I., Hoxhaj G., Ricoult S.J., Asara J.M., and Manning B.D.. 2016. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 351:728–733. 10.1126/science.aad0489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M., et al. . 2010. The landscape of somatic copy-number alteration across human cancers. Nature. 463:899–905. 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R.A., Harris I.S., and Mak T.W.. 2011. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 11:85–95. 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- Chaneton B., Hillmann P., Zheng L., Martin A.C., Maddocks O.D., Chokkathukalam A., Coyle J.E., Jankevics A., Holding F.P., Vousden K.H., et al. . 2012. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 491:458–462. 10.1038/nature11540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Chung F., Yang G., Pu M., Gao H., Jiang W., Yin H., Capka V., Kasibhatla S., Laffitte B., et al. . 2013. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget. 4:2502–2511. 10.18632/oncotarget.1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G.P., Cotropia J.P., and Sallach H.J.. 1969. The effects of dietary protein on the hepatic enzymes of serine metabolism in the rabbit. Arch. Biochem. Biophys. 129:672–682. 10.1016/0003-9861(69)90227-6 [DOI] [PubMed] [Google Scholar]
- Christofk H.R., Vander Heiden M.G., Harris M.H., Ramanathan A., Gerszten R.E., Wei R., Fleming M.D., Schreiber S.L., and Cantley L.C.. 2008a The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 452:230–233. 10.1038/nature06734 [DOI] [PubMed] [Google Scholar]
- Christofk H.R., Vander Heiden M.G., Wu N., Asara J.M., and Cantley L.C.. 2008b Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 452:181–186. 10.1038/nature06667 [DOI] [PubMed] [Google Scholar]
- Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C., et al. . 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 462:739–744. 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S.M., Papagiannakopoulos T., Olenchock B.A., Heyman J.E., Keibler M.A., Luengo A., Bauer M.R., Jha A.K., O’Brien J.P., Pierce K.A., et al. . 2016. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23:517–528. 10.1016/j.cmet.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.L., Fallon H.J., and Morris H.P.. 1970. Two enzymes of serine metabolism in rat liver and hepatomas. Cancer Res. 30:2917–2920. [PubMed] [Google Scholar]
- DeBerardinis R.J., and Cheng T.. 2010. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 29:313–324. 10.1038/onc.2009.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola G.M., Chen P.H., Mullarky E., Sudderth J.A., Hu Z., Wu D., Tang H., Xie Y., Asara J.M., Huffman K.E., et al. . 2015. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 47:1475–1481. 10.1038/ng.3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S., Hu Z., Xu X.L., Sacchettini J.C., and Grant G.A.. 2005. D-3-phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 280:14884–14891. 10.1074/jbc.M414488200 [DOI] [PubMed] [Google Scholar]
- Ding J., Li T., Wang X., Zhao E., Choi J.H., Yang L., Zha Y., Dong Z., Huang S., Asara J.M., et al. . 2013. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 18:896–907. 10.1016/j.cmet.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker G.S., Chen L., Morscher R.J., Ghergurovich J.M., Esposito M., Teng X., Kang Y., and Rabinowitz J.D.. 2016. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 23:1140–1153. 10.1016/j.cmet.2016.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler H.G., Hubbard R., and Snell K.. 1981. The role of serine hydroxymethyltransferase in cell proliferation: DNA synthesis from serine following mitogenic stimulation of lymphocytes. Biosci. Rep. 1:101–106. 10.1007/BF01117006 [DOI] [PubMed] [Google Scholar]
- Eigenbrodt E., Leib S., Krămer W., Friis R.R., and Schoner W.. 1983. Structural and kinetic differences between the M2 type pyruvate kinases from lung and various tumors. Biomed. Biochim. Acta. 42:S278–S282. [PubMed] [Google Scholar]
- El-Hattab A.W. 2016. Serine biosynthesis and transport defects. Mol. Genet. Metab. 118:153–159. 10.1016/j.ymgme.2016.04.010 [DOI] [PubMed] [Google Scholar]
- Fan J., Ye J., Kamphorst J.J., Shlomi T., Thompson C.B., and Rabinowitz J.D.. 2014. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 510:298–302. 10.1038/nature13236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Teng X., Liu L., Mattaini K.R., Looper R.E., Vander Heiden M.G., and Rabinowitz J.D.. 2015. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol. 10:510–516. 10.1021/cb500683c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui D.Y., Lewis C.A., and Vander Heiden M.G.. 2013. Allosteric regulation of PKM2 allows cellular adaptation to different physiological states. Sci. Signal. 6:pe7 10.1126/scisignal.2003925 [DOI] [PubMed] [Google Scholar]
- Hensley C.T., Faubert B., Yuan Q., Lev-Cohain N., Jin E., Kim J., Jiang L., Ko B., Skelton R., Loudat L., et al. . 2016. Metabolic heterogeneity in human lung tumors. Cell. 164:681–694. 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig K., Chiang E.P., Lee L.R., Hills J., Shane B., and Stover P.J.. 2002. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem. 277:38381–38389. 10.1074/jbc.M205000200 [DOI] [PubMed] [Google Scholar]
- Hitosugi T., Kang S., Vander Heiden M.G., Chung T.W., Elf S., Lythgoe K., Dong S., Lonial S., Wang X., Chen G.Z., et al. . 2009. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2:ra73 10.1126/scisignal.2000431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosios A.M., Hecht V.C., Danai L.V., Johnson M.O., Rathmell J.C., Steinhauser M.L., Manalis S.R., and Vander Heiden M.G.. 2016. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell. 36:540–549. 10.1016/j.devcel.2016.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung Y.P., Albeck J.G., Tantama M., and Yellen G.. 2011. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab. 14:545–554. 10.1016/j.cmet.2011.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen K.H., and Marles S.W.. 1976. Inhibition of chicken pyruvate kinases by amino acids. Biochemistry. 15:1073–1079. 10.1021/bi00650a018 [DOI] [PubMed] [Google Scholar]
- Ichihara A., and Greenberg D.M.. 1955. Pathway of serine formation from carbohydrate in rat liver. Proc. Natl. Acad. Sci. USA. 41:605–609. 10.1073/pnas.41.9.605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelsen W.J., and Vander Heiden M.G.. 2015. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 43:43–51. 10.1016/j.semcdb.2015.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelsen W.J., Dayton T.L., Davidson S.M., Fiske B.P., Hosios A.M., Bellinger G., Li J., Yu Y., Sasaki M., Horner J.W., et al. . 2013. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 155:397–409. 10.1016/j.cell.2013.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A.L., Kafri R., Kirschner M.W., Clish C.B., and Mootha V.K.. 2012. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 336:1040–1044. 10.1126/science.1218595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Z., Heng W., Aiping D., Yafei Q., and Shulan Z.. 2013. Expression and clinical significance of phosphoglycerate dehydrogenase and squamous cell carcinoma antigen in cervical cancer. Int. J. Gynecol. Cancer. 23:1465–1469. 10.1097/IGC.0b013e3182a0c068 [DOI] [PubMed] [Google Scholar]
- Kim D., Fiske B.P., Birsoy K., Freinkman E., Kami K., Possemato R.L., Chudnovsky Y., Pacold M.E., Chen W.W., Cantor J.R., et al. . 2015. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 520:363–367. 10.1038/nature14363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall A.S., Xu S., Graeber T.G., Braas D., and Christofk H.R.. 2016. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7:11457 10.1038/ncomms11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung C., Hixon J., Choe S., Marks K., Gross S., Murphy E., DeLaBarre B., Cianchetta G., Sethumadhavan S., Wang X., et al. . 2012. Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem. Biol. 19:1187–1198. 10.1016/j.chembiol.2012.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne C.F., van den Broek N.J., Mackay G.M., Vousden K.H., and Maddocks O.D.. 2014. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Reports. 7:1248–1258. 10.1016/j.celrep.2014.04.045 [DOI] [PubMed] [Google Scholar]
- Lehninger A.L., Nelson D.L., and Cox M.M.. 2013. Lehninger principles of biochemistry. W.H. Freeman, New York: 1198 pp. [Google Scholar]
- Lewis C.A., Parker S.J., Fiske B.P., McCloskey D., Gui D.Y., Green C.R., Vokes N.I., Feist A.M., Vander Heiden M.G., and Metallo C.M.. 2014. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell. 55:253–263. 10.1016/j.molcel.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Guo S., Li Q., Yang L., Xia Z., Zhang L., Huang Z., and Zhang N.. 2013. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J. Neurooncol. 111:245–255. 10.1007/s11060-012-1018-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J.W. 2013. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer. 13:572–583. 10.1038/nrc3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J.W., Grassian A.R., Melman T., Lyssiotis C.A., Mattaini K.R., Bass A.J., Heffron G., Metallo C.M., Muranen T., Sharfi H., et al. . 2011. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43:869–874. 10.1038/ng.890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt S.Y., and Vander Heiden M.G.. 2011. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27:441–464. 10.1146/annurev-cellbio-092910-154237 [DOI] [PubMed] [Google Scholar]
- Lunt S.Y., Muralidhar V., Hosios A.M., Israelsen W.J., Gui D.Y., Newhouse L., Ogrodzinski M., Hecht V., Xu K., Acevedo P.N., et al. . 2015. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol. Cell. 57:95–107. 10.1016/j.molcel.2014.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks O.D., Berkers C.R., Mason S.M., Zheng L., Blyth K., Gottlieb E., and Vousden K.H.. 2013. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 493:542–546. 10.1038/nature11743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks O.D., Labuschagne C.F., Adams P.D., and Vousden K.H.. 2016. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol. Cell. 61:210–221. 10.1016/j.molcel.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaini K.R., Brignole E.J., Kini M., Davidson S.M., Fiske B.P., Drennan C.L., and Vander Heiden M.G.. 2015. An epitope tag alters phosphoglycerate dehydrogenase structure and impairs ability to support cell proliferation. Cancer Metab. 3:5 10.1186/s40170-015-0131-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers J.R., and Vander Heiden M.G.. 2015. Famine versus feast: Understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 40:130–140. 10.1016/j.tibs.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E., Mattaini K.R., Vander Heiden M.G., Cantley L.C., and Locasale J.W.. 2011. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 24:1112–1115. 10.1111/j.1755-148X.2011.00919.x [DOI] [PubMed] [Google Scholar]
- Mullarky E., Lucki N.C., Beheshti Zavareh R., Anglin J.L., Gomes A.P., Nicolay B.N., Wong J.C., Christen S., Takahashi H., Singh P.K., et al. . 2016. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA. 113:1778–1783. 10.1073/pnas.1521548113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy S.K., Li D., Lelievre S., Bissell M.J., and Brugge J.S.. 2001. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3:785–792. 10.1038/ncb0901-785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson L.M., Forshell T.Z., Rimpi S., Kreutzer C., Pretsch W., Bornkamm G.W., and Nilsson J.A.. 2012. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 8:e1002573 10.1371/journal.pgen.1002573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson R., Jain M., Madhusudhan N., Sheppard N.G., Strittmatter L., Kampf C., Huang J., Asplund A., and Mootha V.K.. 2014. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5:3128 10.1038/ncomms4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold M.E., Brimacombe K.R., Chan S.H., Rohde J.M., Lewis C.A., Swier L.J.Y.M., Possemato R., Chen W.W., Sullivan L.B., Fiske B.P., et al. . 2016. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 12:452–458. 10.1038/nchembio.2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikman Y., Puissant A., Alexe G., Furman A., Chen L.M., Frumm S.M., Ross L., Fenouille N., Bassil C.F., Lewis C.A., et al. . 2016. Targeting MTHFD2 in acute myeloid leukemia. J. Exp. Med. 213:1285–1306. 10.1084/jem.20151574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E., Agathocleous M., Murphy M.M., Hu Z., Huddlestun S.E., Zhao Z., Leitch A.M., Johnson T.M., DeBerardinis R.J., and Morrison S.J.. 2015. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 527:186–191. 10.1038/nature15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizer L.I. 1963. The pathway and control of serine biosynthesis in Escherichia coli. J. Biol. Chem. 238:3934–3944. [PubMed] [Google Scholar]
- Pollari S., Käkönen S.M., Edgren H., Wolf M., Kohonen P., Sara H., Guise T., Nees M., and Kallioniemi O.. 2011. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 125:421–430. 10.1007/s10549-010-0848-5 [DOI] [PubMed] [Google Scholar]
- Possemato R., Marks K.M., Shaul Y.D., Pacold M.E., Kim D., Birsoy K., Sethumadhavan S., Woo H.K., Jang H.G., Jha A.K., et al. . 2011. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 476:346–350. 10.1038/nature10350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallach H.J. 1955. Evidence for a specific alanine-hydroxypyruvic transaminase. In Amino Acid Metabolism. McElroy W.D., and Glass B., editors. Johns Hopkins, Baltimore: 782–796. [Google Scholar]
- Schafer Z.T., Grassian A.R., Song L., Jiang Z., Gerhart-Hines Z., Irie H.Y., Gao S., Puigserver P., and Brugge J.S.. 2009. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 461:109–113. 10.1038/nature08268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell K. 1984. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv. Enzyme Regul. 22:325–400. 10.1016/0065-2571(84)90021-9 [DOI] [PubMed] [Google Scholar]
- Snell K., and Weber G.. 1986. Enzymic imbalance in serine metabolism in rat hepatomas. Biochem. J. 233:617–620. 10.1042/bj2330617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell K., Natsumeda Y., and Weber G.. 1987. The modulation of serine metabolism in hepatoma 3924A during different phases of cellular proliferation in culture. Biochem. J. 245:609–612. 10.1042/bj2450609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda M. 1967. A view of the comparison of the regulation of enzymes in mammalian and microbial systems. Adv. Enzyme Regul. 5:181–209. 10.1016/0065-2571(67)90016-7 [DOI] [PubMed] [Google Scholar]
- Sullivan L.B., Gui D.Y., Hosios A.M., Bush L.N., Freinkman E., and Vander Heiden M.G.. 2015. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 162:552–563. 10.1016/j.cell.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts A.S., and Appling D.R.. 2010. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30:57–81. 10.1146/annurev.nutr.012809.104810 [DOI] [PubMed] [Google Scholar]
- Umbarger H.E., and Umbarger M.A.. 1962. The biosynthetic pathway of serine in Salmonella typhimurium. Biochim. Biophys. Acta. 62:193–195. 10.1016/0006-3002(62)90515-2 [DOI] [PubMed] [Google Scholar]
- van der Crabben S.N., Verhoeven-Duif N.M., Brilstra E.H., Van Maldergem L., Coskun T., Rubio-Gozalbo E., Berger R., and de Koning T.J.. 2013. An update on serine deficiency disorders. J. Inherit. Metab. Dis. 36:613–619. 10.1007/s10545-013-9592-4 [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G. 2011. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 10:671–684. 10.1038/nrd3504 [DOI] [PubMed] [Google Scholar]
- Ward P.S., Patel J., Wise D.R., Abdel-Wahab O., Bennett B.D., Coller H.A., Cross J.R., Fantin V.R., Hedvat C.V., Perl A.E., et al. . 2010. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 17:225–234. 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. 2013. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27:2065–2071. 10.1101/gad.228122.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Mancuso A., Tong X., Ward P.S., Fan J., Rabinowitz J.D., and Thompson C.B.. 2012. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. USA. 109:6904–6909. 10.1073/pnas.1204176109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Fan J., Venneti S., Wan Y.W., Pawel B.R., Zhang J., Finley L.W., Lu C., Lindsten T., Cross J.R., et al. . 2014. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4:1406–1417. 10.1158/2159-8290.CD-14-0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K., Furuya S., Osuka S., Mitoma J., Shinoda Y., Watanabe M., Azuma N., Tanaka H., Hashikawa T., Itohara S., and Hirabayashi Y.. 2004. Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J. Biol. Chem. 279:3573–3577. 10.1074/jbc.C300507200 [DOI] [PubMed] [Google Scholar]
- Zhao G., and Winkler M.E.. 1996. A novel alpha-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J. Bacteriol. 178:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zogg C.K. 2014. Phosphoglycerate dehydrogenase: Potential therapeutic target and putative metabolic oncogene. J. Oncol. 2014:524101 10.1155/2014/524101 [DOI] [PMC free article] [PubMed] [Google Scholar]