

Abstract
Background and Aims Sphagnum-dominated peatlands contain approx. 30 % of the terrestrial carbon pool in the form of partially decomposed plant material (peat), and, as a consequence, Sphagnum is currently a focus of studies on biogeochemistry and control of global climate. Sphagnum species differ in ecologically important traits that scale up to impact ecosystem function, and sequencing of the genome from selected Sphagnum species is currently underway. As an emerging model system, these resources for Sphagnum will facilitate linking nucleotide variation to plant functional traits, and through those traits to ecosystem processes. A solid phylogenetic framework for Sphagnum is crucial to comparative analyses of species-specific traits, but relationships among major clades within Sphagnum have been recalcitrant to resolution because the genus underwent a rapid radiation. Herein a well-supported hypothesis for phylogenetic relationships among major clades within Sphagnum based on organellar genome sequences (plastid, mitochondrial) is provided.
Methods We obtained nucleotide sequences (273 753 nucleotides in total) from the two organellar genomes from 38 species (including three outgroups). Phylogenetic analyses were conducted using a variety of methods applied to nucleotide and amino acid sequences. The Sphagnum phylogeny was rooted with sequences from the related Sphagnopsida genera, Eosphagnum and Flatbergium.
Key Results Phylogenetic analyses of the data converge on the following subgeneric relationships: (Rigida (((Subsecunda) (Cuspidata)) ((Sphagnum) (Acutifolia))). All relationships were strongly supported. Species in the two major clades (i.e. Subsecunda + Cuspidata and Sphagnum + Acutifolia), which include >90 % of all Sphagnum species, differ in ecological niches and these differences correlate with other functional traits that impact biogeochemical cycling. Mitochondrial intron presence/absence are variable among species and genera of the Sphagnopsida. Two new nomenclatural combinations are made, in the genera Eosphagnum and Flatbergium.
Conclusions Newly resolved relationships now permit phylogenetic analyses of morphological, biochemical and ecological traits among Sphagnum species. The results clarify long-standing disagreements about subgeneric relationships and intrageneric classification.
Keywords: Bryophytes, mitochondrial genome, organellar genomes, peatmoss, phylogenomics, plastid genome, Sphagnum
INTRODUCTION
Peatmosses (the genus Sphagnum) are unique plants in virtually every aspect of their morphology and ecology. Although Sphagnum species occur on every continent apart from Antarctica, they are especially prominent and diverse in the boreal zone where they are keystone species in the vegetation of peatlands that form on poorly drained substrates (Rydin and Jeglum, 2013), and are important ecosystem engineers in those communities (van Breeman, 1995). As many as 20 or more sympatric Sphagnum species can co-occur in boreal peatlands and are differentiated with respect to chemical and hydrological gradients of their microhabitat (Rydin and Jeglum, 2013; Johnson et al., 2015). For these reasons, and because Sphagnum-dominated peatlands are relatively tractable low-diversity communities, they have long served as a model for studying community structure and interspecific interactions (e.g. Vitt and Slack, 1975, 1984; Hajkova and Hajek, 2007; Robroek et al., 2007).
More recently, Sphagnum has also been the focus of extensive microevolutionary research, including intraspecific phylogeography (e.g. Stenøien et al., 2011a, b; Szövenyi et al., 2008, 2012; Shaw et al., 2014a, b), polyploidy (Såstad et al., 1999, 2000; Ricca and Shaw, 2010; Ricca et al., 2011; Karlin et al., 2014), phenotypic plasticity (Såstad, 1999) and reproductive biology (Cronberg, 1991; Sundberg and Rydin, 1998, 2002; Natcheva and Cronberg, 2007; Szövényi et al., 2009; Johnson and Shaw, 2015). It is fair to say that more population genetic and molecular systematic work has been done on Sphagnum than on any other genus of bryophytes.
The biogeochemistry of Sphagnum-dominated peatlands is currently another focus for intensive research. Approximately 30 % (455–547 Gt) of the earth’s organic carbon pool is stored in boreal peatlands (Gorham, 1991; Yu, 2012). Northern peatlands currently function as a carbon sink and have done so for thousands of years, but they are also a significant source of atmospheric methane and release some 276 Tg of carbon annually as carbon dioxide and methane (Yu, 2012). Warmer temperatures, permafrost melting, increased fire frequency and changing plant–microbe interactions associated with global climate change may result in peatlands transitioning from carbon sinks to carbon sources (Zhuang et al., 2006; McGuire et al., 2009). Species of Sphagnum differ in functional traits that impact biogeochemical processes (e.g. Turetsky et al., 2008), so changing community membership, and possibly changing intraspecific gene pools, will probably scale up to impact global climate further. There is consequently much interest in the biogeochemistry of peatlands and the Sphagnum traits that impact biogeochemical cycling.
Although species of Sphagnum are notoriously difficult to separate, the genus is readily distinguished from any other moss (see Fig. 1). The gametophytes are relatively large, typically with fascicles of lateral branches that are differentiated as so-called spreading and pendent types. Branches near the stem apex are clustered as a dense terminal capitulum; capitulum morphology affects the shape of the colony ‘canopy’ and therefore water relations and other ecological processes (Rice et al., 2008). Sphagnum gametophytes lack roots (like all other mosses), and they also lack rhizoids, which anchor other mosses to their substrates. The leaves of Sphagnum gametophytes are unistratose and composed of dimorphic cells, with large, empty, hyaline cells enclosed in networks of narrow chlorophyllose cells. The hyaline cells store water, and also various microbes (Bragina et al., 2014) including nitrogen fixers, methanogens and a variety of small eukaryotes (Hingley, 1993). Sporophytes of Sphagnum are raised on a pseudopodium of (maternal) gametophyte origin, and the sporophytes themselves consist of little more than a sporangium attached to the pseudopodium by a swollen foot. The subgenera of Sphagnum are generally well marked morphologically; traits for distinguishing subgenera include the shapes of chlorophyllose cells in transverse section, and the size, number and arrangement of hyaline cell pores. Several boreal species are known to be inter-subgeneric allopolyploids (Karlin et al., 2010). Inter-subgeneric allopolyploids are also known from the Southern Hemisphere (Karlin et al., 2009, 2014).
Fig. 1.

Morphological diversity among species of Bryophyta, class Sphagnopsida. Subgeneric classification of each Sphagnum species in parentheses. (A) Flatbergium novo-caledoniae; (B) Eosphagnum rigescens; (C) S. strictum and S. compactum (Rigida); (D) S. compactum (Rigida); (E) S. microporum (Subsecunda); (F) S. lescurii (Subsecunda); (G) S. contortum (Subsecunda); (H) S. luzonense (Subsecunda); (I) S. cuspidatum (Cuspidata); (J) S. riparium (Cuspidata); (K) S. obtusum (Cuspidata); (L) S. balticum (Cuspidata); (M) S. magellanicum (Sphagnum); (N) S. austinii (Sphagnum); (O) S. alaskense (Sphagnum); (P) S. papillosum (Sphagnum); (Q) S. wulfianum (Acutifolia); (R) S. teres (Acutifolia); (S) S. aongstroemii (Acutifolia); (T) S. fuscum (brown) and S. rubellum (red) (Acutifolia). All photos by B. Shaw.
To facilitate ecological and evolutionary research on peatmosses and peatlands, we need to know much more about Sphagnum biochemistry and physiology, the genetic basis of differences among species and individual plants, and phylogenetic relationships among Sphagnum species. Toward that end, the Joint Genome Institute (JGI; US Department of Energy) recently approved a proposal to sequence the genomes and transcriptomes of representative Sphagnum species (Principal Investigators, A. J. Shaw and D. J. Weston). It is well known that phylogenetic relationships must be taken into account to distinguish traits inherited from a unique common ancestor vs. those acquired independently and correlated with environmental parameters (Felsenstein, 1985). Thus, resolution of phylogenetic relationships within Sphagnum is a critical component of establishing and empowering the genus as a model for ecological and climate research (Johnson et al., 2015; Weston et al., 2015).
A number of recent papers have focused on phylogenetic relationships among closely related Sphagnum species (e.g. Shaw et al., 2004, 2008, 2012, 2015), but the genus seems to have diversified rapidly (Shaw et al., 2010b) and, as a consequence, relationships among major clades (subgenera) have been recalcitrant to resolution because of short internal branches in phylogenetic reconstructions based on limited numbers of loci (Shaw et al., 2003, 2010a). Here we analyse organellar genome sequences (plastid and mitochondrial) to resolve deep clades within Sphagnum based on nearly complete plastid and mitochondrial sequences (273 753 nucleotides in total). Whole organellar genome sequences have been used to resolve genome evolution and phylogenetic relationships in bryophytes (Li et al., 2009; Wang et al., 2009; Forrest et al., 2011; Liu et al., 2011, 2012a, 2014b; Xue et al., 2010), within the angiosperms (e.g. Jansen et al., 2007; Liu et al., 2012; Barrett et al., 2014; Yang et al., 2013) and across land plants (Liu et al., 2012b, 2014a; Cox et al., 2014) or seed plants (e.g. Xi et al., 2013; Zhong et al., 2013).
Our primary goal in the present study was to resolve deep relationships (i.e. among the major clades/subgenera) within Sphagnum. Although we did not obtain complete sequences for organellar genomes, we were also able to assess whether gene composition and order in the plastid and mitochondrial genomes of Sphagnum species conform to those documented in other bryophyte lineages. Finally, we used the phylogenetic reconstruction for Sphagnum to infer patterns of ecological evolution in the genus.
MATERIALS AND METHODS
Accessions used for sequencing
Thirty-five Sphagnum species plus three outgroups were included in the analyses (Supplementary Data Table S1). Each of the five subgenera recognized by Shaw et al. (2010a) is represented by 2–11 species, and the three outgroups are Eosphagnum rigescens, Flatbergium sericeum and F. novo-caledoniae. All three outgroup taxa had always been classified within Sphagnum, each in its own monospecific subgenus (or section) based on morphology (Warnstorf, 1911; Crum, 1990). However, multigene phylogenetic analyses (Shaw et al., 2010a) showed that Flatbergium and Eosphagnum (as well as Ambuchanania leucobryoides, not included in the present study) are within the Sphagnopsida, but are highly divergent from all known Sphagnum species and are outside the Sphagnum clade. Eosphagnum rigescens is an earlier name for E. inretortum and we herein (below) make the new combination in Eosphagnum. We also below transfer Sphagnum novo-caledoniae to Flatbergium based on phylogenetic analyses presented herein.
Sphagnum species were selected for the analyses to represent the subgenera recognized by Shaw et al. (2010a), as well as some enigmatic species that have in the past sometimes been included in their own subgenus [or section – most authors before Shaw et al. (2010a) used sections rather than subgenera for the major intrageneric Sphagnum clades]. Sphagnum wulfianum represents the monospecific sect. Polyclada, and S. aongstroemii is the sole member of sect. Insulosa (e.g. Crum, 1984). Shaw et al. (2010a) recognized the sections Polyclada and Squarrosa (the latter with about four species worldwide and represented here by S. squarrosum and S. teres) as sections within the subg. Acutifolia. One other monospecific section, Mollusca (for S. tenellum), has been resolved with strong support within the subg. Cuspidata; S. tenellum is not included in the present analyses. Also not included in the present analyses are S. macrophyllum and S. cribrosum, which were separated in the genus Isocladus (Lindberg, 1862), or section Isocladus by Crum (1984) and McQueen and Andrus (2007), but were shown to be nested within the subgenus Subsecunda by Shaw et al. (2004). Voucher specimen information for all specimens included in the analyses are provided as Table S1.
Table 1.
Mitochondrial cox1 introns in Sphagnaceae and other selected land plants
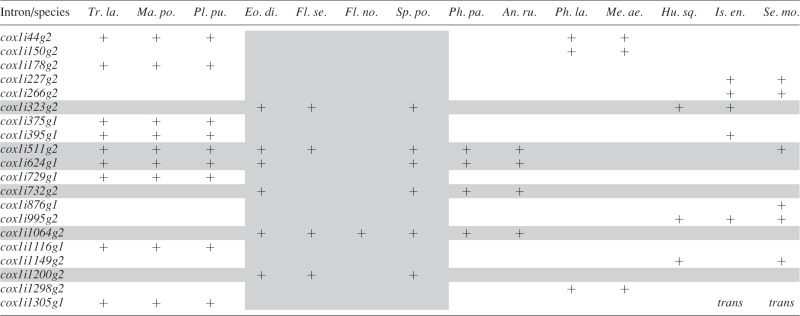 |
Species names are as follows (in the order in which they appear): Treubia lacunosa, Marchantia polymorpha, Pleurozia purpurea, Eosphagnum rigescens, Flatbergium sericeum, F. novo-caledoniae, Sphagnum portoricense, Physcomitrella patens, Anomodon rugelii, Phaeoceros laevis, Megaceros aenigmaticus, Huperzia squarrosa, Isoetes engelmannii and Selaginella moellendorffii.
‘+’ indicates the presence of an intron, and ‘trans’ denotes a trans-spliced intron. Intron nomenclature follows Dombrovska and Qiu (2004) and Knoop (2004).
Of the 35 Sphagnum species included in this study, 28 have haploid gametophytes and six are allodiploids based on heterozygosity at multiple microsatellite loci (Såstad et al., 1999, 2000, 2001; Ricca and Shaw, 2010). Sphagnum australe and S. falcatulum include both allodiploid and allotripoid populations (Karlin et al., 2009). One gametophytically haploid species, S. contortum, was previously hypothesized to be a homoploid hybrid based on incongruence between plastid and nuclear DNA sequences (Shaw et al., 2008). Our results below indicate that S. contortum has the organellar genomes of subg. Cuspidata, although the species is resolved with species in the subg. Subsecunda based on nuclear genes and is classified in that subgenus based on morphology.
DNA extraction and next-generation sequencing
Approximately 0·5 g of dried gametophytic tissue was harvested from each of the 38 samples. The plant material was ground in liquid nitrogen and total genomic DNA was extracted using Macherey-Nagel’s NucleoSpin plant II Midi kit (Macherey-Nagel, Germany). After extraction, DNA concentrations were quantified using the Qubit fluorometer system with a Quant-iT™ ds-DNA BR Assay (Invitrogen, San Diego, CA, USA). For each sample, 100 ng of genomic DNA was used for Illumina library preparation using the TrueSeq Nano DNA kit (http://www.illumina.com/science/education/truseq.html). Paired-end libraries were generated for each sample, and libraries were indexed to allow multiplexing of samples. Twenty-four samples were pooled in equimolar concentrations before sequencing on one HiSeq2500 rapid flow cell, while the remaining 14 samples were pooled and sequenced on one lane of an Illumina HiSeq2000 flow cell. All Illumina sequencing was done at The Duke Center for Genomic and Computational Biology and generated 100 bp paired-end sequences. All assembled plastid and mitochondrial genomes were deposited in GenBank (see Table S1 for GenBank accession numbers).
De novo assembly and mapping to reference plastid and mitochondrial genomes
The 3-prime end of each read was trimmed based on quality using the FASTQ Quality Trimmer (FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/) with a quality score threshold of 25 (Sanger/Illumina 1·9 encoding). Illumina sequencing adaptors, when present, were clipped off using FASTQ/A Clipper (FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/) and only paired-end reads for which both sequences were longer than 40 bp after trimming and clipping were kept for de novo assembly or mapping to the reference. To produce a reference sequence for both the plastid and mitochondrial genomes to which raw reads could be mapped, raw sequences from sample ND2735 (S. palustre) were assembled de novo into contigs using CLC Genomic Workbench (vers. 6.5; CLCbio www.clcbio.com) and default parameters. The de novo assembled plastid and mitochondrial genomes were subsequently annotated using Geneious (vers. 5.4.4; http://www.geneious.com; Biomatters Ltd, Auckland, New Zealand) and used as reference for mapping raw reads from the remaining 37 samples/species. Raw reads were mapped against the plastid and mitochondrial reference genomes using the mapping algorithm in CLC Genomic Workbench. The consensus plastid and mitochondrial sequences resulting from the mapping of the raw reads were exported for each sample with ‘N’ inserted where the coverage of the reference sequence was under a threshold of four reads.
Sequence alignment and phylogenetic reconstruction
Six data sets were constructed, each with 38 taxa including the three outgroups: (1) plastid genome sequences (130 953 nucleotides); (2) 86 plastid proteins (24 741 amino acids); (3) mitochondrial genome sequences (142 800 nucleotides); (4) 37 mitochondrial proteins (10 315 amino acids); (5) combined plastid and mitochondrial genome sequences (273 753 nucleotides); and (6) combined plastid and mitochondrial proteins (123 proteins, 35 056 amino acids).
Genome sequences, both plastid and mitochondrial, were aligned using CLC Genomic Workbench and adjusted by eye. Protein-coding genes were aligned using TranslatorX (Abascal et al., 2010) and combined using ad-hoc Python scripts using P4 (vers. 0.89.r234; Foster, 2004) and Biopython (vers. 1.59; Cock et al., 2009) libraries. Optimal substitution models were determined using Modelgenerator (Keane et al., 2006) applying the Akaike Information Criterion (AIC1) as the selection criterion. Additional empirical protein models specific to streptophytes, namely gcpREV (Cox and Foster, 2013) and stmtREV (Liu et al., 2014a), were also employed for the analysis of the plastid and mitochondrial proteins respectively. Maximum likelihood (ML) analyses were performed with RAxML (vers. 7.0.4 MPI; Stamatakis, 2006) with 400 bootstrap replicates, each with two search replicates. Bayesian inference was performed with MrBayes (Ronquist and Huelsenbeck, 2003), Phylobayes (Lartillot and Philippe, 2004) and P4. Markov chain Monte Carlo (MCMC) chains were run in duplicate for 5 million generations in MrBayes and 2 million generations in P4, and until convergence diagnostics (maximum clade difference between runs of < 0·3) were met. Convergence between independent runs of MrBayes and P4 was determined by calculating an average standard deviation of split support (ASDOSS): a value <0·01 was considered sufficiently low to signify convergence between separate runs and the results were combined. Auxiliary model parameters in addition to the substitution rates included a proportion of invariant sites (I) and among-site rate variation modelled with a gamma distribution approximated by four discrete rates (Γ4). MCMC analyses using P4 employed a polytomy prior (PP; Lewis et al., 2005), and a relative rate parameter (RR) was used in combined chloroplast and mitochondrial data analyses. Model composition was either estimated (Fest) by maximum likelihood, or fixed to model specifications (Fmod). Tree-heterogenous composition analyses were performed in P4 using the NDCH model (Cox et al., 2008) by including addition composition vectors (CVs), and goodness-of-fit of the NDCH model to the data was determined by a posterior predictive probability test using the χ2 statistic of composition homogeneity (Foster, 2004). The NDCH analyses allow a pre-defined number of analytically determined compositions to evolve across the tree, thereby modelling lineage-specific composition heterogeneity. Marginal likelihoods of all MCMC chains were calculated using the harmonic mean estimator of Newton and Raftery (1994: equation 16). Details of the model and change characteristics for individual analyses are presented in the legends of Supplementary Data Figs S2–S33.
Given the long outgroup branch lengths and comparatively short ingroup branches, we considered it possible that alternative rootings on the ingroup might be statistically indistinguishable from the optimal rooting (to sect. Rigida) that was resolved in the combined genome analyses. Alternative topologies were assessed using Consel (vers. 1.20; Shimodaira and Hasegawa, 2001), aided by methods present in P4, based on the combined plastid and mitochondrial genome data set (data set 5 above) and the topology resulting from the ML bootstrap analysis of the same data (Supplementary Data Fig. S27). The topologies representing the rooting points of each alternative ingroup were assessed using all default P-value estimators of Consel, namely the Approximately Unbiased test, bootstrap probability, Bayesian posterior probability, unweighted and weighted Kishino–Hasegawa tests, and unweighted and weighted Shimodaira–Hasegawa tests.
Abiotic microhabitat niche data were compiled for selected species representing the two major clades within Sphagnum resolved by phylogenetic analyses. The data are derived from the studies of Gignac et al. (2004); Pouliot et al. (2011) and Tahvanainen et al. (2002), and were utilized by Johnson et al. (2015) in their phylogenetic analyses of niche evolution in Sphagnum. See Johnson et al. (2015) for formal analyses of the ecological data relative to phylogeny; they are included here for heuristic purposes to help briefly summarize ecological correlates of subgeneric relationships.
RESULTS
Genome architecture
Our sequencing effort yielded sufficiently large contigs to recover all genes for all taxa and most of the genomes, but did not allow the assemblies of complete organellar chromosomes. However, the coverage is sufficient to address exon and intron content of the plastid and mitochondrial genomes. Plastid genomes are uniform in Sphagnum (and outgroups) in terms of both gene and intron content, with as far as can be estimated from the contigs no gene relocation compared with other mosses (e.g. Bell et al., 2014; Lewis et al., 2016). They all contain 37 tRNA genes, eight rRNA genes and 87 protein-coding genes, of which 20 harbour a total of 22 introns. All 38 mitochondrial genomes include 24 tRNA genes, three rRNA genes and 40 protein-coding genes, with the gene order in the contigs matching the known sequence of genes in mosses (Liu et al., 2014b). The mitochondrial genomes of the 35 Sphagnum species have 30 introns distributed among 17 genes. The mitochondrial genomes of the three outgroup taxa, however, carry fewer introns in two genes. Eosphagnum lacks rrn18i839, a group I intron in the ribosomal small subunit gene (rrn18), present in all other Sphagnopsida. Flatbergium novo-caledoniae contains only one, and F. sericeum four of the six introns present in cox1 of Sphagnum and Eosphagnum (Table 1).
Subgeneric phylogenetic relationships: separate plastid and mitochondrial genome analyses
We conducted a series of analyses using different software, optimality criteria and substitution parameters – marginal likelihoods, chain diagnostics and tree lengths of each analysis can be found in the legends of Supplementary Data Figs S2–S31. Alternative analytical approaches for nucleotide sequences from the plastid genome alone provide highly resolved, well-supported, and congruent estimates of phylogenetic relationships (Figs S2–S6). Mitochondrial sequences alone provide less resolution (Figs S12–S17). ML analyses resolve groups of species that correspond to groups resolved by plastid sequences, but several clades with one or two species are not included in the larger clades (Fig. S14). Bayesian reconstructions (Figs S13–S17) yield some relationships that appear to conflict with those resolved by the plastid genome, but these are supported by low posterior probability values. The Bayesian MCMC analysis using Phylobayes, on the other hand, resolved relationships fully congruent with those from the plastid genome, though less completely resolved (Fig. S16).
Plastid (Supplementary Data Figs S7–S11) and mitochondrial protein (Figs S18–S24) sequences provide results that were largely congruent with those based on nucleotide sequences, though less resolved with regard to some relationships, and with lower support. Apparent conflicts, for instance with both S. contortum and S. australe seemingly well supported as early-diverging lineages in some Bayesian analyses of the mitochondrial proteins (Figs S19–S23), were not supported by the better fitting (higher marginal likelihood) Phylobayes CAT model analyses (Fig. S24).
Subgeneric phylogenetic relationships: combined plastid and mitochondrial genome analyses
Conflicts between plastid and mitochondrial analyses when using the best-fitting models were not statistically significant – no conflicting inter-subgeneric clades >70 % bootstrap proportions or > 0·95 posterior probability – and hence data from the two organellar genomes were combined (Supplementary Data Figs S25–S31). As with the single genome analyses, reconstructions based on concatenated plastid and mitochondrial protein sequences are congruent with those resolved by nucleotide sequences, except among a few poorly supported nodes.
Because of the absence of well-supported conflicts among data sets and analytical approaches, we present an ML tree based on concatenated plastid and mitochondrial nucleotide sequences to illustrate the consensus reconstruction (Fig. 2). Using Flatbergium and Eosphagnum as outgroups, the reconstruction resolves subg. Rigida as sister to the remaining species of Sphagnum. Within the rest of the genus, two major clades are resolved with maximum support by three alternative analyses: using ML (RAxML) and Bayesian MCMC [both MrBayes and P4 (CV2)]. One clade includes subg. Cuspidata and Subsecunda and the other, subg. Sphagnum and Acutifolia (Fig. 2). All except four nodes across the tree are supported with 100 % bootstrap percentages and maximum posterior probabilities. Only one node is truly ambiguous, namely the uniquely shared ancestry of S. pylaesii, S. orientale and S. lenense within subg. Subsecunda.
Fig. 2.

Maximum likelihood (ML) reconstruction for Sphagnum (rooted with Eosphagnum and Flatbergium as outgroups). All nodes other than those marked by dots are supported by 100 % ML bootstraps and 100 % posterior probabilities. For the three nodes marked, the left value is the ML bootstrap percentage (RAxML; see Supplementary Dat Fig. S25) and the right value is the posterior probability (MrBayes; see Fig. S26). On the left figure, note that branch lengths are scaled differently before and after the node marking the origin of Sphagnum. The smaller figure on the right shows how divergent the outgroups are from Sphagnum, as well as the relatively high divergence among species of Eosphagnum and Flatbergium. *, homoploid hybrid; **, allopolyploid (diploid gametophytes); ***, allopolyploid (diploid and triploid gametophytes).
Phylogenetic reconstructions based on concatenated nucleotide sequences (Fig. 2; Supplementary Data Figs S27–S31) consistently resolve subg. Rigida as sister to the rest of the genus, but we further tested this rooting using Consel. Six alternative roots (Supplementary Data Fig. S32) were compared with the optimal ML reconstruction and rejected by all statistical measures except the Shimodaira–Hasegawa test; the latter was unable to reject any of the alternative rooting points. Statistics comparing alternative rooting schemes with the ML tree are provided in Fig. S33.
Phylogenetic placement of reticulating taxa
Sphagnum falcatulum, S. majus, S. australe, S. papillosum and S. palustre are known from previous genetic and cytological analyses to be gametophytic allodiploids. Sphagnum lenense has never been recognized as an allodiploid, but heterozygous microsatellite profiles suggest that it too is an allodiploid (A. J. Shaw, unpubl. res.). Sphagnum falcatulum and S. lenense are generally classified in subg. Cuspidata, but they both have the organellar genomes of subg. Subsecunda (Fig. 2). Sphagnum australe includes both allodiploid and allotripoid populations, but the species is part of a strongly supported subg. Sphagnum, albeit sister to the remaining species included in our analyses. Two other species, S. palustre and S. papillosum, are allodiploids whose organellar genomes match their typical classification in subg. Sphagnum. One haploid species, S. contortum, is resolved as part of the subg. Cuspidata, although it is universally classified in subg. Subsecunda.
DISCUSSION
Shotgun sequencing of total genomic extracts allowed for the recovery of nearly complete organellar genomes for all Sphagnopsida. Although the reads obtained did not cover 100 % of either the mitochondrial or chloroplast genome, the coverage is sufficient to reveal (1) that the genic composition of these genomes is identical to that of other mosses (e.g. Liu et al., 2014b; Lewis et al., 2016); and (2) that these genes are aligned, at least within the recovered contigs, in both chromosomes in the same order as in other mosses (Wicke et al., 2011; Liu et al., 2014b). We have indeed recovered all genes known from both the plastid and mitochondrial genomes of mosses and did not find any evidence of gene gains. The perfect alignment of the chromosomes to the plastid genomes of another early diverging moss, Tetraphis pellucida (Bell et al., 2014), or to the mitochondrial genomes of other mosses (Liu et al., 2014a, b) confirms the stability and conservation of both genomes in the Bryophyta not only throughout their long evolutionary history and but also following rapid diversifications, such as that of the genus Sphagnum in the late Tertiary (Shaw et al., 2010b). This stability contrasts with patterns found in some other plant clades (e.g. Wu and Chaw, 2014).
The intron content is constant within the chloroplast genome but variable within the mitochondrial genome among the Sphagnospida. All species of Sphagnum have 30 introns distributed among 17 mitochondrial genes. Eosphagnum is the only Sphagnopsida lacking rrn18i839, a group I intron in the rrn18 gene, and Flatbergium novo-caledoniae and F. sericeum lack five and two introns, respectively, in the cox1 gene compared with Sphagnum and Eosphagnum, which have six introns (Table 1). The cox1i511g2, cox1i624g1, cox1i732g2 and cox1i1064g2 introns also occur in all other mosses (Liu et al., 2014b). The first and second of these introns are shared by liverworts and mosses, but are lacking in F. novo-caledoniae or both species of Flatbergium, respectively (Table 1). The latter two introns are unique to mosses, with the cox1i732g2 intron absent in both species of Flatbergium. Only cox1i1064g2 may be unique to and shared by all mosses (Liu et al., 2014b). Intron cox1i1200g2 is unique to the Sphagnopsida although it is absent in F. novo-caledoniae, and finally cox1i323g2 has an identical distribution among mosses but is not unique as it is also present in Huperzia (Liu et al., 2012b) and Isoetes (Grewe et al., 2009). Intron distributions in the mitochondrial genomes of mosses is thus homoplastic and the phylogenetic utility of introns within these lineages is ambiguous.
Subgeneric classification and phylogenetic relationships
Sphagnum is one of the larger genera of mosses, and a variety of intrageneric classification schemes have been proposed over the last 150 years for its approx. 250–400 species. Linneaus (1753 – the starting point for Sphagnum nomenclature, though not for other mosses) recognized a single species in the genus, S. palustre (subg. Sphagnum). An early attempt to sub-divide Sphagnum by Lindberg (1882) distinguished three groups: Eusphagnum with almost all species of the genus, Isocladus for S. macrophyllum only (including S. cribrosum) and Hemitheca for S. pylaisii and S. cyclophyllum. Russow (1887) proposed a primary division of the genus into two groups, Inophloea and Litophloea. The former is equivalent to subg. Sphagnum but Russow (1887) further divided the Litophloea into several sub-groups and these correspond more or less to sections or subgenera distinguished by later authors. In the only worldwide monograph of Sphagnum to date, Warnstorf (1911) recognized the Inophloea and Litophloea groups as sections and divided the Litophloea into nine sub-sections. Crum (1990) added the section Inretorta when he described S. inretortum as a new species from Bolivia. Based on molecular data, Shaw et al. (2010a) separated this species as the genus Eosphagnum.
Phylogenetic inferences presented here are the first to yield a well-supported hypothesis for relationships among major intrageneric clades within Sphagnum. Shaw et al. (2003) provided a reconstruction based on 15 nuclear and organellar genes that resolved relationships among some of the major Sphagnum clades with strong support, but that conflict with the present results. We do not have an explanation for that incongruence, but consider the present results to be more reliable as they are based on a much larger data set from (presumably) non-recombining organellar genomes. Our current hypothesis agrees with previous ideas in some regards and also proposes some novel relationships. The hypothesis that subg. Rigida is sister to the rest of the genus does not seem to have been proposed previously. The assertion by Andrews (1911), like Russow (1887), that the subg. Sphagnum is so distinct from all other subgenera that the two groups, Inophloea and Litophloea, might deserve generic status is not supported by our analyses. Andrews (1911) argued that separating the subg. Cuspidata and Subsecunda is ‘arbitrary’ because of the intermediate morphology of S. mendocinum from the Pacific coast of North America. However, Karlin et al. (2010) showed that S. mendocinum is an allopolyploid hybrid between the two subgenera, and our results indicate that Cuspidata and Subsecunda are reciprocally monophyletic. While some authors (e.g. Andrews, 1911; Eddy, 1977) have suggested that distinctions between these subgenera break down in tropical regions, phylogenetic analyses based on a worldwide sampling of species (A. J. Shaw, unpubl. res.) suggest otherwise, notwithstanding scattered inter-subgeneric allopolyploids. In a subjectively constructed diagram illustrating their concepts of relationships in Sphagnum, Daniels and Eddy (1990) considered S. wulfianum (sect. Polyclada here) sister to a clade including subg. Sphagnum, Acutifolia and Squarrosa, whereas our results indicate that S. wulfianum is sister to the rest of subg. Acutifolia including species generally separated as sect. Squarrosa, and this inclusive clade is sister to subg. Sphagnum.
Allopolyploidy in Sphagnum
Seven species included in the present study were resolved in clades (subgenera) that conflict with their traditional classification based on morphology. Five were known allopolyploids and their placement agrees with previous hypotheses about parentage.
Sphagnum falcatulum and S. australe have complex allopolyploid histories and both comprise (gametophytically) allodiploid and allotriploid cytotypes (Karlin et al., 2009). Karlin (2014) suggested that allodiploids of S. australe originated at least twice, and one of the allodiploids was one parent of allotriploid plants. Sequences from the plastid trnG locus resolved four samples of S. australe in a well-supported clade but, because the phylogeny was unresolved at deeper levels, monophyly with any other subgenus could not be excluded. Karlin (2014) hypothesized that a haploid species of subg. Rigida crossed with a species that represents an unsampled lineage, and the resulting allodiploid crossed with S. fimbriatum (subg. Acutifolia) to form allotriploid S. australe. Plastid and mitochondrial genome sequences provide strong evidence that Karlin’s (2014 ‘Cryptosphagnum’) mystery lineage was a species of subg. Sphagnum. Sphagnum australe is characterized by a mosaic of morphological traits that include features typical of subg. Sphagnum, Rigida and possibly Acutifolia (Karlin, 2014). It lacks fibrils in the stem and branch cortical cells, a synapomorphy for subg. Sphagnum.
Sphagnum majus and S. jensenii are known to be polyploid hybrids between species of subg. Cuspidata (Såstad et al., 2000) and we confirm that S. majus has the organellar genomes of that subgenus. (S. jensenii was not included in our analyses.) Our resolution of the arctic species, S. lenense, in subg. Subsecunda was not expected because this species has not been recognized as a hybrid and is classified in subg. Cuspidata. However, S. lenense is heterozygous at two of four microsatellite loci (A. J. Shaw, unpubl. res.), consistent with the interpretation that it is an allopolyploid. Our results suggest that it represents a hybrid between parents in the subg. Cuspidata and Subsecunda. The known allopolyploid, S. palustre, was resolved in our analyses within the subg. Sphagnum (where it is classified based on morphology), consistent with the interpretation that it represents an intra-subgeneric hybrid. The most surprising case is S. contortum, a haploid species. Shaw et al. (2008) suggested that this species may have hybridity in its ancestry, but assumed that the reticulating parents belong to subg. Subsecunda, where this species is classified. Our results show that the (presumed) maternal parent of S. contortum belongs to subg. Cuspidata. Its subg. Cuspidata organellar genomes could reflect hybridization and introgression subsequent to the origin of S. contortum by divergent speciation, or S. contortum could represent the first case of homoploid (without change in chromosome number) hybrid speciation known in mosses. We also cannot eliminate the possibility that S. contortum is an allopolyploid with subsequent genome reduction. In contrast to most species of subg. Subsecunda, the branch leaves of S. contortum have relatively few pores, a feature characteristic of subg. Cuspidata, possibly reflecting its hybrid ancestry.
Ecological correlates
Sphagnum-dominated peatlands do not appear in the fossil record until the late Tertiary (Greb et al., 2006), matching Miocene estimates for the timing of peatmoss diversification from molecular phylogenetic analyses (Shaw et al., 2010b). Twenty or more Sphagnum species sometimes occur sympatrically at local scales and sort themselves relative to abiotic environmental gradients (Rydin and Jeglum, 2013). Important gradients include substrate pH, ionic concentrations and height above the water table (HWT). The latter is especially prominent, even over centimetre scales, and hummock–hollow microtopography is characteristic of most boreal peatlands. The subgenera Acutifolia and Sphagnum are characteristically hummock-forming species, whereas the Subsecunda and Cuspidata typically grow in hollows (Johnson et al., 2015) and divergence between these two major clades occurred relatively early in the diversification of Sphagnum (Fig. 2). Based on the same data as utilized by Johnson et al. (2015), species in the Acutifolia + Sphagnum clade typically occur more than twice as high above the water table than those in the Cuspidata + Subsecunda clade, 10·0 ± 7·5 vs. 25·2 ± 13·2 cm, respectively. Nevertheless, more recent evolutionary niche shifts have also occurred within the two clades and interspecific divergence relative to HWT has occurred at a faster rate in hummock species than among hollow species (Johnson et al., 2015).
Hummock-forming vs. hollow-inhabiting Sphagnum species differ in a number of other ecologically important traits. For example, hummock species appear generally to decompose more slowly (under common-garden conditions) (Clymo and Hayward, 1982) and appear to differ in other morphological and chemical traits (see summary table in Rydin et al., 2006). The importance of Sphagnum-dominated peatlands for global biogeochemical cycling hinges on the relationship between production of new biomass and decomposition, so the evolution of these traits during Sphagnum phylogeny is critical to understanding the evolution and genetic basis of the traits that underlie peat formation and accumulation.
The phylogenetic reconstruction resolved in this study also implies a qualitative scenario of peatmoss niche evolution (Bryophyta class Sphagnopsida). The outgroup taxa, Eosphagnum and Flatbergium, do not occur in peatlands and do not accumulate significant peat. Species of subg. Rigida, here resolved as sister to the rest of the genus, do sometimes occur in temperate to boreal peatlands in addition to other sites (e.g. wet soil or rocks), but are never significant components of peatland communities. The major peat-formers within Sphagnum belong to the two major clades (with two subgenera each) that are crown groups in peatmoss diversification. This study is consistent with the view that Sphagnum diversified rapidly, as late Tertiary cooling prompted the formation of new types of cold-environment communities (notwithstanding much earlier global cold intervals before extant Sphagnum diversified). Early diversification of peatmosses appears to have emphasized niche differentiation relative to abiotic gradients within wetlands.
Nomenclature
Eosphagnum rigescens (Warnst.) A. J. Shaw & Flatberg, comb. nov. Basionym: Sphagnum rigescens Warnst., Botanisches Centralblatt 76: 387. 1898. Type: ‘Südamerika. Feuerländische Inselgruppe, Puerto Angusto im März 1896 leg. P. Dusén no. 273’ (lectotype nov: S-B231605!). Syn: Sphagnum inretortum H. A. Crum, Bryologist 93: 283, f. 1–8. 1990 (holotype: MICH!). Sphagnum lapazense H. A. Crum, Contributions from the University of Michigan Herbarium 23: 107. f. 1: a–d. 2001 (holotype: MICH!). Warnstorf’s protocol of S. rigescens designates only one type collection, and is thus a holotype (ICN, Melbourne code; Art. 61). The holotype is missing, but we have traced and confirmed the taxonomic conspecificity of two isotype specimens, one in herb. H, the other in herb. S. Since Dusén’s primary bryophyte collections are at the Stockholm herbarium, we select this isotype specimen (S-B231605) as the lectotype of the name Sphagnum rigescens.
Flatbergium novo-caledoniae (Paris & Warnst. in Warnst.) A. J. Shaw & Flatberg, comb. nov. Basionym: Sphagnum novo-caledoniae Paris & Warnst. in Warnstorf, Sphagnologia Universalis 297. 1911. ‘Monsungebiet: Araucarien-Provinz: Neu-Kaledonien, Plateau de Dogny 1100 m ü. d. M. (Louise Le Rat; Herb Paris!; nördl. Neu-Kaledonien 100–600 m ü. d. M. (Franc – 1. 1910; Herb Thériot!)’. ‘Nov. Caledon. [Nouvelle-Caledoniae]. in jugo Dogny. 1072 m, Julio 09 [July 1909]. Leg. L. Le Rat.’ Lectotype nov.: PC 0167758.
The ICN (Melbourne code; Art. 61) states that confusingly similar names based on the same type are to be treated as orthographical variants. Sphagnum Novae Caledonieae Paris & Warnst., sensu Brotherus (1911: 1) and Sphagnum novo-caledoniae Paris & Warnst. (1911: 297) are based on the same original material and are orthographic variants of the same name. Brotherus’s name is a nomen nudum, and Brotherus’s publication was not cited by Warnstorf. The ICN (Art. 61) allows that a later author can publish a valid scientific name based on a nomen nudum. Warnstorf’s description of S. novo-caledoniae (1911) fulfils all requirements for valid publication. We therefore consider Sphagnum novo-caledoniae Paris & Warnst. in Warnstorf (1911: 297) to be the correct basionym of this species. Brotherus (1911: 1) gave the following information about the new species from New Caledonia: ‘Sphagnum Novae Caledoniae Par. et Warnst. n. sp. In jugo Dogny, alt. 1072 m (L. Le Rat)’. Warnstorf (1911: 299) cited two localities for S. novo-caledoniae (see above). The Paris herbarium (PC) contains four original specimens of S. novo-caledoniae from Plateau de Dogny [in jugo Dogny] collected [Leg.] by Louise Le Rat; three of the labels list an altitude of 1072 m and the collecting date as July 1909 [Julio 09], the fourth is without altitude and collecting date. Warnstorf (1911) failed to give a collecting date. There are no specimens located from northern parts of New Caledonia in herb. PA. We select the Paris specimen PC0167758 from Plateau de Dogny as the lectotype of the name Sphagnum novo-caledoniae.
Conclusions
Sphagnum (and more broadly, the Sphagnopsida) represents a unique model for linking genome evolution, phenotypic traits and ecosystem function, and its value is enhanced by the wealth of ecological data currently available, and newly developing genomic resources. Peatmosses have particular value for two areas linking ecological genomics and phylogenetics: the evolution of niche differences, and the genomic architecture of phenotypic traits that scale up to impact global biogeochemistry. Previous studies that have attempted to resolve genus-wide evolutionary history have been hindered by ambiguity at the level of subgeneric sister-group relationships. This study has yielded a high resolution, highly supported hypothesis of phylogenetic relationships across major Sphagnum clades.
SUPPLEMENTARY DATA
Supplementary data are available online at www.aob.oxfordjournals.org and consist of the following. Table S1: list of samples used in this study with voucher information. Figs S2–S33: phylogenetic analyses of plastid and mitochondrial sequences, translated protein sequences from each genome, combined plastid and mitochondrial genome sequences and proteins, and rooting analyses using Consel.
ACKNOWLEDGEMENTS
This research was supported by the National Science Foundation [grant no. DEB-0918998 to A.J.S. and B.S., and DEB-1240045 to B.G.].
LITERATURE CITED
- Abascal F, Zardoya R, Telford MJ. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Research 38: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AL. 1911. Notes on North American Sphagnum. I. The groups. The Bryologist 14: 72–75. [Google Scholar]
- Barrett CF, Specht CD, Leebens-Mack J, Stevenson DW, Zomlefer WB, Davis JI. 2014. Resolving ancient radiations: can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Annals of Botany 113: 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell NE, Boore JL, Mishler BD, Hyvönen J. 2014. Organellar genomes of the four-toothed moss, Tetraphis pellucida. BMC Genomics 15: 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragina A, Oberauner-Wappis L, Zachow C, et al. 2014. The Sphagnum microbiome supports bog ecosystem functioning under extreme conditions. Molecular Ecology 23: 4498–4510. [DOI] [PubMed] [Google Scholar]
- van Breemen N. 1995. How Sphagnum bogs down other plants. Trends in Ecology and Evolution 10: 270–275. [DOI] [PubMed] [Google Scholar]
- Brotherus VF. 1911. Contribution á la flore bryologique de la Nouvelle Calédonie. III. Öfversigt af Finska Vetenskaps-Societetens Förhandlingar 53A: 1–43. [Google Scholar]
- Clymo RS, Hayward PM. 1982. The ecology of Sphagnum In: AJE Smith, ed. Bryophyte ecology. New York: Chapman and Hall, 229–289. [Google Scholar]
- Cock PJA, Antao T, Chang JT, et al. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25: 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CJ, Foster PG. 2013. A 20-state empirical amino-acid substitution model for green plant chloroplasts. Molecular Phylogenetics and Evolution 68: 218–220. [DOI] [PubMed] [Google Scholar]
- Cox CJ, Foster PG, Hirt RP, Harris SR, Embley MT. 2008. The archaebacterial origins of eukaryotes. Proceedings of the National Academy of Sciences, USA 105: 20356–20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CJ, Li B, Foster PG, Embley TM, Civáň P. 2014. Conflicting phylogenies for early land plants are caused by composition biases among synonymous substitutions. Systematic Biology 63: 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronberg N. 1991. Reproductive biology of Sphagnum. Lindbergia 17: 69–82. [Google Scholar]
- Crum HA. 1984. Sphagnopsida, Sphagnaceae. North American Flora, series 2, part 11. New York: New York Botanical Garden, 1–180. [Google Scholar]
- Crum HA. 1990. Sphagnum inretortum, a new species in a new section from Bolivia. The Bryologist 93: 283–285. [Google Scholar]
- Daniels RE, Eddy A. 1990. Handbook of European Sphagna. London: HMSO. [Google Scholar]
- Dombrovska O, Qiu YL. 2004. Distribution of introns in the mitochondrial gene nad1 in land plants: phylogenetic and molecular evolutionary implications. Molecular Phylogenetics and Evolution 32: 246–263. [DOI] [PubMed] [Google Scholar]
- Eddy A, 1977. Sphagnales of tropical Asia. Bulletin of the British Museum (Natural History). Historical Series 5: 359–445. [Google Scholar]
- Felsenstein J. 1985. Phylogenies and the comparative method. American Naturalist 125: 1–15. [DOI] [PubMed] [Google Scholar]
- Forrest LL, Wickett NJ, Cox CJ, Goffinet B. 2011. Deep sequencing of Ptilidium (Ptilidiaceae) suggests evolutionary stasis in liverwort plastid genome structure. Plant Ecology and Evolution 144: 29–43. [Google Scholar]
- Foster P. 2004. Modeling compositional heterogeneity. Systematic Biology 53: 485–495. [DOI] [PubMed] [Google Scholar]
- Gignac LD, Gauthier R, Rochefort L, Bubier J. 2004. Distribution and habitat niches of 37 peatland Cyperaceae species across a broad geographic range in Canada. Canadian Journal of Botany 82: 1292–1313. [Google Scholar]
- Gorham E. 1991. Northern peatlands: role in the carbon cycle and probable responses to climatic warming. Ecological Applications 1: 182–195. [DOI] [PubMed] [Google Scholar]
- Greb SF, DiMichele WA, Gastaldo RA. 2006. Evolution and importance of wetlands in earth history In: SF Greb, WA DiMichele, eds. Wetlands through time. Geological Society of America Special Paper 399, 1–40. [Google Scholar]
- Grewe F, Viehoever P, Weisshaar B, Knoop V. 2009. A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Research 15: 5093–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Hajek M. 2007. Sphagnum distribution patterns along environmental gradients in Bulgaria. Journal of Bryology 29: 18–26. [Google Scholar]
- Hingley M. 1993. Microscopic life in Sphagnum. Exeter, UK: Pelagic Publishing. [Google Scholar]
- Jansen RK, Cai Z, Raubeson LA, et al. 2007. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proceedings of the National Academy of Sciences, USA 104: 19369–19374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MG, Shaw AJ. 2015. Genetic diversity, sexual condition, and microhabitat preference determine mating patterns in Sphagnum (Sphagnaceae) peatmosses. Biological Journal of the Linnean Society 15: 96–113. [Google Scholar]
- Johnson MJ, Granath G, Teemu T, et al. 2015. Evolution of niche preference in Sphagnum peat mosses. Evolution 69: 90–103. [DOI] [PubMed] [Google Scholar]
- Karlin EF. 2014. Subgenome analysis of two southern hemisphere allotriploid species in Sphagnum (Sphagnaceae). Journal of Bryology 36: 165–179. [Google Scholar]
- Karlin EF, Boles SB, Ricca M, Temsch E, Greilhuber J, Shaw AJ. 2009. Three-genome mosses: complex double allopolyploid origins for triploid gametophytes in Sphagnum. Molecular Ecology 18: 1439–1454. [DOI] [PubMed] [Google Scholar]
- Karlin EF, Gardner GP, Lukshis K, Boles SB, Shaw AJ. 2010. Allopolyploidy in Sphagnum mendocinum and S. papillosum (Sphagnaceae). The Bryologist 113: 114–119. [Google Scholar]
- Karlin EF, Temsch EM, Bizuru E, et al. 2014. Invisible in plain sight: recurrent double allopolyploidy in the African Sphagnum × planifolium (Sphagnaceae). The Bryologist 117: 187–201. [Google Scholar]
- Keane TM, Creevey CJ, Pentony MM, Naughton TJ, Mclnerney JO. 2006. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evolutionary Biology 6: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoop V. 2004. The mitochondrial DNA of land plants: peculiarities in phylogenetic perspective. Current Genetics 46: 123–139. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. 2004. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Molecular Biology and Evolution 21: 1095–1109. [DOI] [PubMed] [Google Scholar]
- Lewis PO, Holder M, Holsinger KE. 2005. Polytomies and Bayesian phylogenetic inference. Systematic Biology 54: 241–253. [DOI] [PubMed] [Google Scholar]
- Lewis L, Liu Y, Rozzi R, Goffinet B. 2016. Infraspecific variation within and across complete organellar genomes and nuclear ribosomal repeats in a moss. Molecular Phylogenetics and Evolution 96: 195–199. [DOI] [PubMed] [Google Scholar]
- Li L, Wang B, Liu Y, Qiu Y-L. 2009. The complete mitochondrial genome sequence of the hornwort Megaceros aenigmaticus shows a mixed mode of conservative yet dynamic evolution in early land plant mitochondrial genomes. Journal of Molecular Evolution 68: 665–678. [DOI] [PubMed] [Google Scholar]
- Lindberg SO. 1862. Torfmossornas byggnad, utbredning och systematiska uppställning. Öfversigt af Forhandlingar: Kongliga Svenska Vetenskaps-Akademien 19: 113–156. [Google Scholar]
- Lindberg SO. 1882. Europas och Nord Amerikas Hvitmossor (Sphagna). Helsinki: J. C. Frenckell. [Google Scholar]
- Linnaeus C. 1753. Species Plantarum. Stockholm: Laurentii Salvii. [Google Scholar]
- Liu J, Qi Z-C, Zhao Y-P, Fu C-X, Xiang Q-Y(Jenny) 2012. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales – influences of gene partitions and taxon sampling. Molecular Phylogenetics and Evolution 64: 545–562. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xue J-Y., Wang B, Li L, Qiu Y-L. 2011. The mitochondrial genomes of the early land plants Treubia lacunosa and Anomodon rugelii: dynamic and conservative evolution. PLoS One 6: e25836. doi:10.1371/journal.pone.0025836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang B, Li L, Qiu YL, Xue JY. 2012a. Conservative and dynamic evolution of mitochondrial genomes in early land plants In: R Bock, V Knoop, eds. Genomics of chloroplasts and mitochondria. Dordrecht, The Netherlands: Springer, 159–174. [Google Scholar]
- Liu Y, Wang B, Cui P, et al. 2012b. The mitochondrial genome of the lycophyte Huperzia squarrosa: the most archaic form in vascular plants. PLoS One 7: e35168. doi:10.1371/journal.pone.0035168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cox CJ, Wang W, Goffinet B. 2014a. Mitochondrial phylogenomics of early land plants: mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Systematic Biology 63: 862–878. [DOI] [PubMed] [Google Scholar]
- Liu Y, Medina R, Goffinet B. 2014b. 350 my of mitochondrial genome stasis in mosses, an early land plant lineage. Molecular Biology and Evolution 31: 2586–2591. [DOI] [PubMed] [Google Scholar]
- McGuire AD, Anderson LG, Christensen TR, et al. 2009. Sensitivity of the carbon cycle in the Arctic to climate change. Ecological Monographs 79: 523–555. [Google Scholar]
- McQueen CB, Andrus RE. 2007. Sphagnaceae Dumortier In: Flora of North America Editorial Committee, ed. Flora of North America, Vol. 27: Bryophytes: mosses, part 1. New York: Oxford University Press, 45–101. [Google Scholar]
- Natcheva R, Cronberg N. 2007. Recombination and introgression of nuclear and chloroplast genomes between the peat mosses, Sphagnum capillifolium and Sphagnum quinquefarium. Molecular Ecology 16: 811–818. [DOI] [PubMed] [Google Scholar]
- Newton MA, Raftery AE. 1994. Approximate Bayesian inference with the weighted likelihood bootstrap. Journal of the Royal Statistical Society B: Methodological 56: 3–48. [Google Scholar]
- Pouliot R, Rochefort L, Karofeld E. 2011. Initiation of microtopography in revegetated cutover peatlands. Applied Vegetation Science 14: 158–171. [Google Scholar]
- Ricca M, Shaw AJ. 2010. Allopolyploidy and homoploid hybridization in the Sphagnum subsecundum complex (Sphagnaceae: Bryophyta). Biological Journal of the Linnaean Society 99: 135–151. [Google Scholar]
- Ricca M, Szövényi P, Shaw AJ. 2011. Interploidal hybridization and mating patterns in the Sphagnum subsecundum complex. Molecular Ecology 20: 3202–3218. [DOI] [PubMed] [Google Scholar]
- Rice SK, Aclander L, Hanson DT. 2008. Do bryophyte shoot systems function like vascular plant leaves or canopies? Functional trait relationships in Sphagnum mosses (Sphagnaceae). American Journal of Botany 95: 1366–1374. [DOI] [PubMed] [Google Scholar]
- Robroek BJM, Limpens J, Breeuwer A, Crushell PH, Schouten MGC. 2007. Interspecific competition between Sphagnum mosses at different water tables. Functional Ecology 21: 805–812. [Google Scholar]
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- Rydin H, Jeglum J. 2013. The biology of peatlands, 2nd edn Oxford: Oxford University Press. [Google Scholar]
- Rydin H, Gunnarsson G, Sundberg S. 2006. The role of Sphagnum in peatland development and persistence In: RK Wider, DH Vitt, eds. Boreal peatland ecosystems. Ecological Studies Vol. 188. Berlin: Springer-Verlag, 47–65. [Google Scholar]
- Russow E. 1887. Zur Anatomie resp. physiologischen und vergleichenden Anatomie der Torfmoose. Dorpat: Naturforscher-Gesellschaft bei der Universität Dorpat. [Google Scholar]
- Såstad SM. 1999. Genetic and environmental sources of variation in leaf morphology of Sphagnum fallax and Sphagnum isoviitae (Bryopsida): comparison of experiments conducted in the field and laboratory. Canadian Journal of Botany 77: 1–10. [Google Scholar]
- Såstad SM, Flatberg KI, Cronberg N. 1999. Electrophoretic evidence supporting a theory of allopolyploid origin of Sphagnum jensenii. Nordic Journal of Botany 19: 355–362. [Google Scholar]
- Såstad SM, Flatberg KI, Hanssen L. 2000. Origin, taxonomy and genetic structure of the allopolyploid peat moss Sphagnum majus. Plant Systematics and Evolution 225: 73–84. [Google Scholar]
- Såstad SM, Stenøien H, Flatberg KI, Bakken S. 2001. The narrow endemic Sphagnum troendelagicum is an allopolyploid derivative of the widespread S. balticum and S. tenellum . Systematic Botany 26: 66–74. [Google Scholar]
- Shaw AJ, Cox CJ, Boles SB. 2003. Polarity of peatmoss (Sphagnum) evolution: who says mosses have no roots? American Journal of Botany 90: 1777–1787. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, Boles SB, Shaw B. 2008. A phylogenetic delineation of the ‘Sphagnum subsecundum complex’ (Bryophyta). American Journal of Botany 95: 731–744. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, Cox CJ, Buck WR, et al. 2010a. Newly resolved relationships in an early land plant lineage: Bryophyta class Sphagnopsida (peat mosses). American Journal of Botany 97: 1511–1531. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, Devos N, Cox CJ, et al. 2010b. Peatmoss (Sphagnum) diversification associated with Miocene Northern Hemisphere climatic cooling? Molecular Phylogenetics and Evolution 55: 1139–1145. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, Flatberg KI, Szövényi P, et al. 2012. Systematics of the Sphagnum fimbriatum complex: phylogenetic relationships, morphological variation, and allopolyploidy. Systematic Botany 37: 15–30. [Google Scholar]
- Shaw AJ, Golinski GK, Clark EG, Shaw B, Stenøien HK, Flatberg KI. 2014a. Intercontinental genetic structure in the amphi-Pacific peatmoss Sphagnum miyabeanum (Bryophyta: Sphagnaceae). Biological Journal of the Linnean Society 111:17–37. [Google Scholar]
- Shaw AJ, Shaw B, Stenøien HK, Golinski GK, Hassel K, Flatberg KI. 2014b. Pleistocene survival, regional genetic structure and interspecific gene flow among three northern peat-mosses: Sphagnum inexspectatum, S. orientale and S. miyabeanum. Journal of Biogeography 42:364–376. [Google Scholar]
- Shaw AJ, Shaw B, Johnson MG, Devos N, Carter B. 2015. Genetic and phylogenetic structure of the Pacific Rim clade of Sphagnum subg. Subsecunda: haploid and allodiploid taxa. Biological Journal of the Linnaean Society 116: 295–311. [Google Scholar]
- Shaw S, Cox CJ, Boles SB. 2004. Phylogenetic relationships among Sphagnum sections, Hemitheca, Isocladus, and Subsecunda. The Bryologist 107:189–196. [Google Scholar]
- Shimodaira H, Hasegawa M. 2001. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics Application Note 17: 1246–1247. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stenøien HK, Shaw AJ, Shaw B, Hassel K, Gunnarsson U. 2011a. North American origin and recent European establishment of the amphi-Atlantic peat moss Sphagnum angermanicum. Evolution 65: 1181–1194. [DOI] [PubMed] [Google Scholar]
- Stenøien HK, Shaw AJ, Stengrundet K, Flatberg KI. 2011b. The narrow endemic Norwegian peat moss Sphagnum troendelagicum originated before the last glacial maximum. Heredity 106: 370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg S, Rydin H. 1998. Spore number in Sphagnum and its dependence on spore and capsule size. Journal of Bryology 20: 1–16. [Google Scholar]
- Sundberg S, Rydin H. 2002. Habitat requirements for establishment of Sphagnum from spores. Journal of Ecology 90: 268–278. [Google Scholar]
- Szövényi P, Terracciano S, Ricca M, Shaw AJ. 2008. Recent divergence, intercontinental dispersal and shared polymorphisms are shaping the genetic structure of amphi-Atlantic peatmoss populations. Molecular Ecology 17: 5364–5377. [DOI] [PubMed] [Google Scholar]
- Szövényi P, Ricca M, Shaw AJ. 2009. Multiple paternity and sporophytic inbreeding depression in a dioicous moss species. Heredity 103: 394–403. [DOI] [PubMed] [Google Scholar]
- Szövényi P, Sundberg S, Shaw AJ. 2012. Long-distance dispersal and genetic structure of natural populations: an assessment of the inverse isolation hypothesis in peat mosses. Molecular Ecology 21: 5461–5472. [DOI] [PubMed] [Google Scholar]
- Tahvanainen T, Sallantaus T, Heikkila R, Tolonen T. 2002. Spatial variation of mire surface water chemistry and vegetation in northeastern Finland. Annales Botanici Fennici 39: 235–251. [Google Scholar]
- Turetsky MR, Crow SE, Evans RJ, Vitt DH, Wieder RK. 2008. Tradeoffs in resource allocation among moss species control decomposition in boreal peatlands. Journal of Ecology 96: 1297–1305. [Google Scholar]
- Vitt DH, Slack NG. 1975. An analysis of the vegetation of Sphagnum-dominated kettle-hole bogs in relation to environmental gradients. Canadian Journal of Botany 53: 332–359. [Google Scholar]
- Vitt DH, Slack NG. 1984. Niche diversification of Sphagnum relative to environmental factors in northern Minnesota peatlands. Canadian Journal of Botany 62: 1409– 1430. [Google Scholar]
- Wang B, Xue J-Y, Li L, Liu L, Qiu Y-L. 2009. The complete mitochondrial genome sequence of the liverwort Pleurozia purpurea reveals extremely conservative mitochondrial genome evolution in liverworts. Current Genetics 55: 601–609. [DOI] [PubMed] [Google Scholar]
- Warnstorf C. 1911. Sphagnales–Sphagnaceae. Sphagnologia universalis. (ed. by A. Engler). Das Pflanzenreich 51: 1–546. [Google Scholar]
- Weston DJ, Timm CM, Walker AP, et al. 2015. Sphagnum physiology in the context of changing climate: emergent influences of genomics, modelling and host–microbiome interactions on understanding ecosystem function. Plant, Cell and Environment 38: 1737–1751. [DOI] [PubMed] [Google Scholar]
- Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. 2011. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology 76: 273–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-S, Chaw S-M. 2014. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): evolution towards shorter intergenic spacers. Plant Biotechnology Journal 12: 344–353. [DOI] [PubMed] [Google Scholar]
- Xi Z, Rest JS, Davis CC. 2013. Phylogenomics and coalescent analyses resolve extant seed plant relationships. PLoS One 8: e80870. doi:10.1371/journal.pone.0080870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J-Y, Liu Y, Li L, Wang B, Qiu Y-L. 2010. The complete mitochondrial genome sequence of the hornwort Phaeoceros laevis: retention of many ancient pseudogenes and conservative evolution of mitochondrial genomes in hornworts. Current Genetics 56: 53–61. [DOI] [PubMed] [Google Scholar]
- Yang J-B, Yang S-X, Li H-T, Yang J, Li D-Z. 2013. Comparative chloroplast genomes of Camellia species. PLoS One 8: e73053. doi:10.1371/journal.pone.0073053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZC. 2012. Northern peatland carbon stocks and dynamics: a review. Biogeosciences 9: 4071–4085. [Google Scholar]
- Zhuang QL, Melillo JM, Sarofim et al. 2006. CO2 and CH4 exchanges between land eco-systems and the atmosphere in northern high latitudes over the 21st century. Geophysical Research Letters 33: L17403. [Google Scholar]
- Zhong B, Yonezawa T, Zhong Y, Hasegawa M. 2013. The position of Gnetales among seed plants: overcoming pitfalls of chloroplast phylogenomics. Molecular Biology and Evolution 27: 2855–2863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.