

Abstract

Nonheme iron oxygenases that carry out four-electron oxidations of substrate have been proposed to employ iron(III) superoxide species to initiate this reaction [Paria, S.; Que, L.; Paine, T. K. Angew. Chem. Int. Ed.2011, 50, 11129]. Here we report experimental evidence in support of this proposal. 18O KIEs were measured for two recently discovered mononuclear nonheme iron oxygenases: hydroxyethylphosphonate dioxygenase (HEPD) and methylphosphonate synthase (MPnS). Competitive 18O KIEs measured with deuterated substrates are larger than those measured with unlabeled substrates, which indicates that C–H cleavage must occur before an irreversible reductive step at molecular oxygen. A similar observation was previously used to implicate copper(II) superoxide in the H-abstraction reactions catalyzed by dopamine β-monooxygenase [Tian, G. C.; Klinman, J. P. J. Am. Chem. Soc.1993, 115, 8891] and peptidylglycine α-hydroxylating monooxygenase [Francisco, W. A.; Blackburn, N. J.; Klinman, J. P. Biochemistry2003, 42, 1813].
Nonheme iron oxygenases are important to a wide range of biological functions that include the metabolism of xenochemicals, the biosynthesis of antibiotics, and DNA repair.1 One very large family of mononuclear iron proteins share a two His, one Glu/Asp motif at their metal binding site, referred to as the 2-His-1-carboxylate facial triad.1 Many enzymes within this class are able to activate C–H bonds of high bond dissociation energy via O2-derived iron peroxo- or oxo-intermediates.2−4 Understanding the formation and properties of activated iron–oxygen complexes not only unravels the basis of their divergent reactivities but also aids in the design of novel iron-based catalysts that are inexpensive and environmentally friendly.2
Two recently discovered mononuclear nonheme iron(II) oxygenases, 2-hydroxyethylphosphonate dioxygenase (HEPD)5 and methylphosphonate synthase (MPnS),6 have been shown to react with an unactivated aliphatic C–H bond in the absence of exogenous reductant. Both enzymes act at C-2 of a common substrate 2-hydroxyethylphosphonate (2-HEP), coupling a net C–C bond cleavage with a four electron reduction of O2, before the mechanisms bifurcate to yield different products.5−7 The Fe(II) center in HEPD is coordinated by the classical 2-His-1-carboxylate facial triad,5 whereas the precise ligand set in MPnS is not known as the MPnS sequence lacks the anticipated Glu/Asp of the triad and structural information is not available. Because these enzymes do not require an exogenous reductant, which is necessary to produce high valence iron-oxo complexes,3 HEPD and MPnS have been suggested to employ an iron(III) superoxide complex as an alternate hydrogen atom abstraction species (Scheme 1).5,8−10
Scheme 1. Proposed Mechanism of Initial H-Abstraction for HEPD and MPnS.

The role of an iron(III) superoxide in H atom abstraction has been previously demonstrated in the dinuclear nonheme iron oxygenase myo-inositol oxygenase (MIOX),11,12 and in a biomimetic inorganic model complex.13 The reactive oxygen intermediate in MIOX was characterized by Mössbauer spectroscopy,14,15 making this the only enzyme system with direct evidence for an iron(III) superoxide species that performs C–H activation.11 An iron(III) superoxide intermediate was also characterized by EPR and Mössbauer spectroscopy in homoprotocatechuate dioxygenase,16 but the subsequent chemistry was oxidation of an aromatic ring and distinct from C–H activation. While a superoxo species has also been proposed to carry out H atom abstraction in isopenicillin N synthase (IPNS), the mechanistic conclusions derive solely from structural17 and spectroscopic18 studies using NO as an analogue of O2. In general, direct experimental evidence for the existence of a superoxo intermediate in mononuclear nonheme iron(II) oxygenases has remained elusive, thus stimulating the search for an alternate means of providing support for the intermediacy of a superoxo intermediate in these enzymes.
Oxygen (18O) kinetic isotope effects (KIEs) have proven to be an effective method to interrogate the nature of activated oxygen species in a range of metalloenzymes.19 Experimentally, 18O KIEs are measured in a competitive fashion using natural abundance levels of 18O, and thus the measured KIE is on kcat/Km, noted as 18(V/K). This method has been applied to explore both oxygen binding equilibrium isotope effects (EIEs) of oxygen-transport proteins20 and KIEs of a number of oxygenases.21−2418(V/K) reflects changes in the oxygen bond order that occur in all steps from initial O2 binding up to and including the first irreversible step.23 As such, 18(V/K) can be utilized to deduce the nature of the species that carries out the H atom abstraction. In this context, the impact of the presence of a second isotope in the substrate (typically deuterium) on the magnitude of 18(V/K) serves as an important probe for the timing of H atom abstraction relative to reductive steps at O2.23−25
In this study, the 18(V/K) for all-protio-HEP and [2-2H2]-2-HEP has been measured (Table 1, left two columns). All of the KIEs are small and in the range of 1.5–2.3%. Most significantly, the 18O KIEs measured for deuterated substrates are larger than those measured with unlabeled substrates for both HEPD and MPnS. This phenomenon is a violation of the rule of the geometric mean and reflects the effect of substrate deuteration on rate limiting steps when C–H cleavage is only partially rate determining. A determination of the 2H KIE as a function of O2 with HEPD as catalyst indicates that deuterated substrate does, in fact, react more slowly than the unlabeled substrate under the condition of low [O2] (Figure S3). This apparent 2H KIE on kcat/Km(O2) contrasts with the recent report7 of an absence of 2H KIEs on either kcat or kcat/Km(2-HEP) for native HEPD. A pattern of different isotope effects on kcat and the individual kcat/Km parameters for different substrates is fairly common in enzymology (cf. ref (37)) and arises from the contribution of different steps and accompanying rate constants in each instance. As illustrated in Scheme 2, a slowing down of the hydrogen transfer step via the incorporation of deuterium unmasks an 18O isotope effect that occurs either prior to or concomitant with C–H cleavage. This observation contrasts with mechanisms where the reactive oxygen-derived species is irreversibly formed prior to H-atom abstraction.21,25 For the latter case, as anticipated in Fe(IV)=O forming enzymes, any possible fractionation at oxygen will be complete before C–H cleavage and hence insensitive to substrate deuteration. This study is the first instance of the application of a double isotope probe to iron-containing oxygenases. The results obtained for HEPD and MPnS parallel those seen earlier for the copper monooxygenases peptidylglycine α-hydroxylating monooxygenase (PHM)24 and dopamine β-monooxygenase (DβM)23 (Table 1, right two columns), where a consensus mechanism involving hydrogen atom abstraction by a copper(II) superoxide complex has emerged.26 In the case of iron-based chemistry, a large range of valence states is available to the metal center, and it is possible to conceptualize an iron(III) superoxide species as containing some contribution from a formal iron(IV) peroxide resonance structure (Scheme 3). This higher valence species is of possible interest here because of its anticipated impact on the bond order at oxygen, which could further impact the magnitude of the observed 18O KIEs. This issue is addressed further below.
Table 1. KIEs of HEPD and MPnS Compared to the Copper Enzymes PHM and DβM.
| enzyme | HEPD | MPnS | PHM | DβM |
|---|---|---|---|---|
| substrate | 2-HEP | 2-HEP | hippuric acid | dopamine |
| BDEa (kcal·mol–1) | 80b | 80b | < 82.0c | 85(2)d |
| 18(V/K)He | 1.0148(9) | 1.0158(10) | 1.0173(9)f | 1.0197(3)g |
| 18(V/K)Dh | 1.0231(26) | 1.0189(13) | 1.0212(18)f | 1.0256(3)g |
Estimated bond dissociation energy of the activated C–H bond.
Average BDE of sodium and potassium methanolate: CH2(OM)-H.36
BDE of glycine ion: NH2CH(COO–)-H.35
BDE of ethylbenzene: PhCH(CH3)-H.27
Measured with all-protio substrates.
Reference (24).
Reference (23).
Measured with substrates deuterated at the transferring hydrogen, i.e., [2-2H2]-2-HEP for HEPD and MPnS, N-[benzoyl-2-2H2]-glycine (labeled hippuric acid) for PHM, and [2-2H2]-dopamine for DβM.
Scheme 2. Impact of Substrate Deuteration on the Reaction Coordinate.
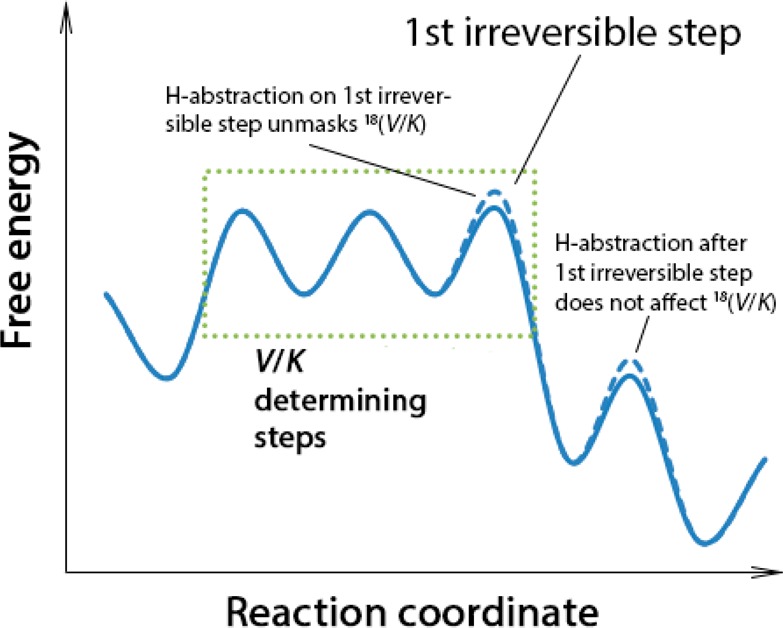
The solid line represents a typical free energy surface of an enzymatic reaction. Before the first irreversible step, there are potentially multiple steps sensitive to an 18O KIE on (V/K). When these are only partially rate determining, the 18(V/K) will become “masked” and hence smaller than the intrinsic value. The use of deuterated substrate (with its larger free energy barrier, dotted line) can unmask the 18(V/K), leading to an increased value of this parameter; by contrast, if the deuterium-sensitive step occurs after the first irreversible step, the 18(V/K) remains unchanged.
Scheme 3. Proposed H-Atom Abstraction Species.

The bond dissociation energy (BDE) of an unactivated C–H bond of a primary alcohol can be as high as 93 kcal/mol.27 Though a deprotonation of the hydroxyl group may lower the BDE to ca. 80 kcal/mol,36 this is still a significant barrier, raising the question how enzymes that employ a formal Fe(III)–O2•–, e.g., HEPD and MPnS and a recently described alkene producing fatty acid decarboxylase,28 manage the initial abstraction of a hydrogen atom from their substrates. There are three distinct conceptual frameworks for understanding these C–H bond cleavage reactions. In the context of a semiclassical transition state, both hydrogen and oxygen isotope effects arise from changes in vibrational frequencies between the ground state and transition state. In the case of a metal oxygen complex, the 18O KIE will be the product of the EIE for the formation of a metal-superoxo species (18Keq ≈ 1.004–1.005, ref (20)), together with further changes in force constants that accompany the hydrogen atom transfer; there is also the expectation of a small additional isotope effect arising from the reaction coordinate frequency. However, even in the event of a late transition state, with the property of an 18O KIE that is closer to the 18O EIE for hemerythrin than the 18O EIE of myoglobin/hemoglobin (Table 2), it is difficult to see how a direct H-transfer from substrate to Fe(III)−O2•– could elevate the 18O KIE to the observed effect of ca. 1.02.
Table 2. EIEs in Oxygen Binding Proteinsa.
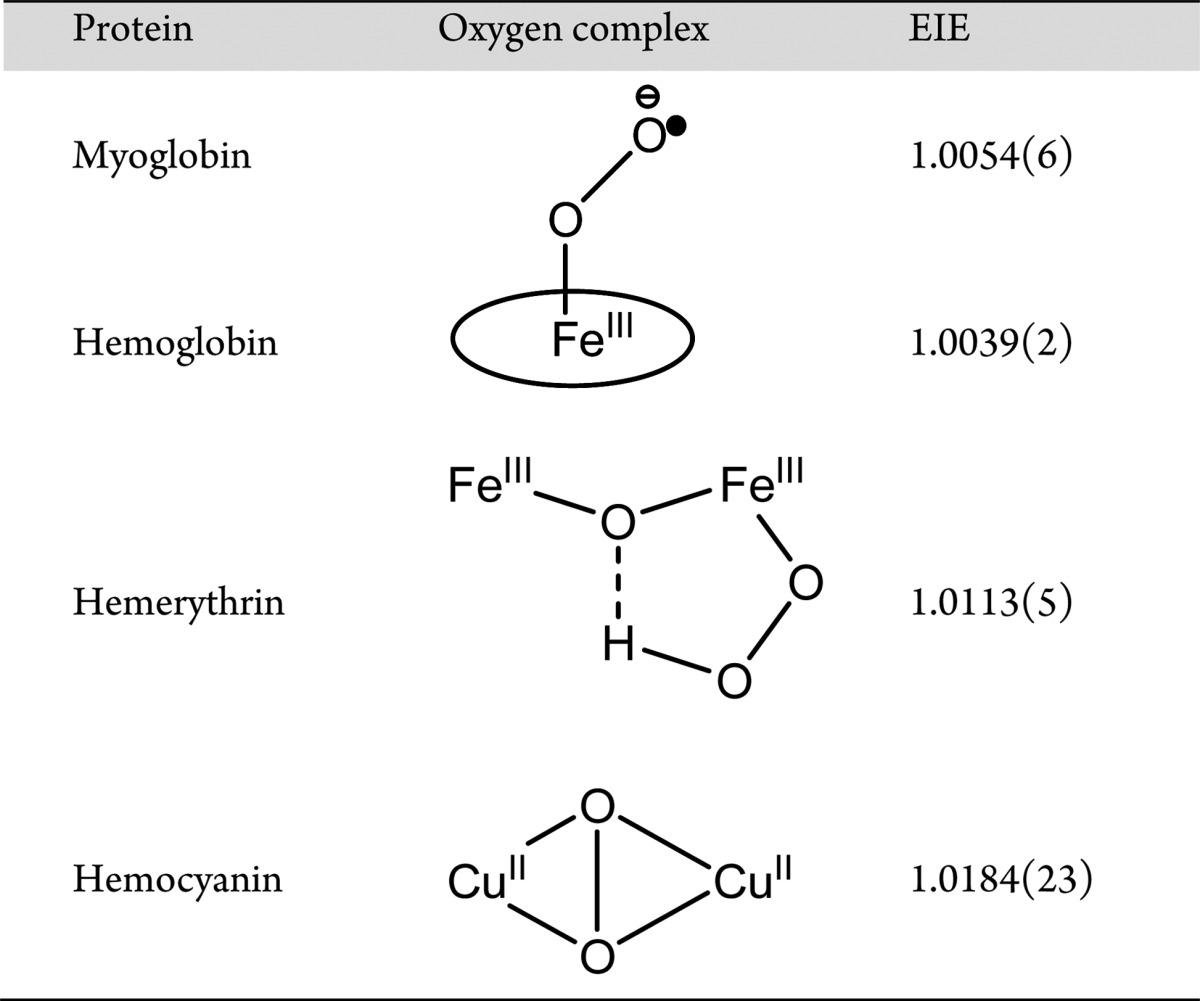
Data obtained from ref (20).
Alternatively, the observed 18O KIEs may be best rationalized in the context of a net hydrogen atom transfer that occurs by nuclear tunneling. This is a likely scenario for HEPD and MPnS, given the demonstrated importance of hydrogen tunneling in enzymatic C–H activation reactions.29−32 In this instance, the 18O KIE resides within the environmental reorganization term that establishes the appropriate bond order at the reactive oxygen center to promote hydrogenic wavefunction overlap between the hydrogen donor and acceptor wells (cf. ref (32)). In this instance, it was conceivable that the bond order at the activated oxygen center could have resembled Fe(IV)−OO2– with the expectation of an 18O KIE analogous to hemocyanin (Table 2). However, detailed EPR and Mössbauer studies of the reactive Fe(III)−O2•– in the homoprotocatechuate dioxygenase system have shown that the structure of this species is fully described as a combination of a high spin (S1 = 5/2) ferric center that is antiferromagnetically coupled to an S2 = 1/2 radical.16 On this basis, we consider any contribution from an Fe(IV)−OO2– resonance form, Scheme 3, to the ground state activated oxygen in HEPD and MPnS highly unlikely.
We therefore turn to the final possibility that the measured values for 18(V/K) (Table 1) reflect a nuclear tunneling contribution to the environmental reorganization at oxygen. This approach takes into account a role for tunneling of heavy atoms as they undergo changes in bond order in proceeding from reactant to product33,34 and has been able to successfully reproduce the unusual trend for 18(V/K) in glucose oxidase where the magnitude of the KIE is found to become smaller as the driving force is decreased.22 A similar analysis of the trend in 18(V/K), published earlier for DβM,23 is now carried out in this work (Figure S4), showing that nuclear tunneling is also needed to provide an explanation for the falloff in 18(V/K) with decreasing rate. In the absence of individual experimental rate constants for the isolated C–H bond cleavages in HEPD and MPnS, it is difficult to extend this type of analysis to the present systems under investigation. Nonetheless, the combination of 18O KIEs for HEPD and MPnS, which are both similar to DβM in magnitude and sensitivity to substrate deuteration, points toward a similar interpretation for the data presented herein. Among the three possible interpretations, the most probable is a tunneling of H• from substrate to Fe(III)–O2•–, which is accompanied by oxygen tunneling within the reactive iron-superoxide complex. Two computational studies have previously investigated the early steps of the HEPD reaction mechanism.9,10 Both studies concluded that substrate activation involved hydrogen atom abstraction from C-2 of substrate by a ferric-superoxide species. In one model the hydrogen abstraction occurred from substrate that still retained the proton of the hydroxyl group of the substrate, whereas in a second model the hydroxyl group had lost its proton prior to hydrogen atom abstraction. However, in both models the hydrogen atom transfer step from C-2 of substrate to the ferric-superoxide was rate-limiting, consistent with the observed substrate deuterium KIE on kcat/Km(O2) in this study. The absence of KIEs on kcat and kcat/Km(2-HEP) is likely a consequence of nonchemical partially rate limiting steps that involve substrate binding/conformational change or product release that are common in enzymatic reactions but not typically detected by computational methods. Neither previous computational study discussed the possibility of hydrogen (or oxygen) tunneling.
To conclude, the single most significant finding from this study is an increase in the magnitude of the 18O KIE when deuterated HEP is substituted for unlabeled substrate. This finding, in strong analogy to the properties of DβM, indicates that the H-atom abstraction step must occur either prior to or concomitant with the first irreversible step of O2 reduction (Scheme 2). We note that an initial, reversible formation of iron(III) superoxide not only satisfies the experimental findings but also satisfies the experimental exigencies of such enzymes operating in the absence of any exogenous reductant(s).
Acknowledgments
This work is supported by the National Institutes of Health (GM025765 to J.P.K.; GM077596 to W.A.V.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b03907.
Experimental procedures for the 18O KIE measurement; raw data and plots of isotope fractionation for 18O KIE measurement; reanalysis of the relationship between rates for hydrogen atom transfer and 18O KIEs in DβM (PDF)
Author Present Address
# Laboratoire d’Ingénierie des Systémes Biologiques et des Procedés, 135 Avenue de Rangueil, 31400 Toulouse, France.
Author Present Address
∇ Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Chem. Rev. 2004, 104, 939. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Que L.; Tolman W. B. Nature 2008, 455, 333. 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Fujimori D. G.; Walsh C. T.; Bollinger J. M. Jr. Acc. Chem. Res. 2007, 40, 484. 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I.; Wong S. D.; Liu L. V.; Decker A.; Chow M. S. Curr. Opin. Chem. Biol. 2009, 13, 99. 10.1016/j.cbpa.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo R. M.; Zhang H. J.; Blodgett J. A. V.; Whitteck J. T.; Li G. Y.; Nair S. K.; van der Donk W. A.; Metcalf W. W. Nature 2009, 459, 871. 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf W. W.; Griffin B. M.; Cicchillo R. M.; Gao J. T.; Janga S. C.; Cooke H. A.; Circello B. T.; Evans B. S.; Martens-Habbena W.; Stahl D. A.; van der Donk W. A. Science 2012, 337, 1104. 10.1126/science.1219875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck S. C.; Chekan J. R.; Ulrich E. C.; Nair S. K.; van der Donk W. A. J. Am. Chem. Soc. 2015, 137, 3217. 10.1021/jacs.5b00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke H. A.; Peck S. C.; Evans B. S.; van der Donk W. A. J. Am. Chem. Soc. 2012, 134, 15660. 10.1021/ja306777w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Morokuma K. J. Am. Chem. Soc. 2010, 132, 17901. 10.1021/ja108174d. [DOI] [PubMed] [Google Scholar]
- Du L.; Gao J.; Liu Y.; Liu C. J. Phys. Chem. B 2012, 116, 11837. 10.1021/jp305454m. [DOI] [PubMed] [Google Scholar]
- van der Donk W. A.; Krebs C.; Bollinger J. M. Jr. Curr. Opin. Struct. Biol. 2010, 20, 673. 10.1016/j.sbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Diao Y.; Matthews M. L.; Xing G.; Krebs C. Dalton Trans. 2009, 905. 10.1039/B811885J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paria S.; Que L.; Paine T. K. Angew. Chem., Int. Ed. 2011, 50, 11129. 10.1002/anie.201103971. [DOI] [PubMed] [Google Scholar]
- Xing G.; Barr E. W.; Diao Y. H.; Hoffart L. M.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Biochemistry 2006, 45, 5402. 10.1021/bi0526276. [DOI] [PubMed] [Google Scholar]
- Xing G.; Diao Y. H.; Hoffart L. M.; Barr E. W.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6130. 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbughuni M. M.; Chakrabarti M.; Hayden J. A.; Bominaar E. L.; Hendrich M. P.; Munck E.; Lipscomb J. D. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16788. 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzlaff N. I.; Rutledge P. J.; Clifton I. J.; Hensgens C. M. H.; Pickford M.; Adlington R. M.; Roach P. L.; Baldwin J. E. Nature 1999, 401, 721. 10.1038/44400. [DOI] [PubMed] [Google Scholar]
- Brown C. D.; Neidig M. L.; Neibergall M. B.; Lipscomb J. D.; Solomon E. I. J. Am. Chem. Soc. 2007, 129, 7427. 10.1021/ja071364v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J. P. Curr. Opin. Chem. Biol. 2007, 11, 142. 10.1016/j.cbpa.2007.01.683. [DOI] [PubMed] [Google Scholar]
- Tian G. C.; Klinman J. P. J. Am. Chem. Soc. 1993, 115, 8891. 10.1021/ja00073a001. [DOI] [Google Scholar]
- Mirica L. M.; McCusker K. P.; Munos J. W.; Liu H. W.; Klinman J. P. J. Am. Chem. Soc. 2008, 130, 8122. 10.1021/ja800265s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J. P.; Wincek R.; Nodet G.; Edmondson D. E.; McIntire W. S.; Klinman J. P. J. Am. Chem. Soc. 2004, 126, 15120. 10.1021/ja047050e. [DOI] [PubMed] [Google Scholar]
- Tian G. C.; Berry J. A.; Klinman J. P. Biochemistry 1994, 33, 226. 10.1021/bi00167a030. [DOI] [PubMed] [Google Scholar]
- Francisco W. A.; Blackburn N. J.; Klinman J. P. Biochemistry 2003, 42, 1813. 10.1021/bi020592t. [DOI] [PubMed] [Google Scholar]
- Mirica L. M.; Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1814. 10.1073/pnas.0711626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P. J. Biol. Chem. 2006, 281, 3013. 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- McMillen D. F.; Golden D. M. Annu. Rev. Phys. Chem. 1982, 33, 493. 10.1146/annurev.pc.33.100182.002425. [DOI] [Google Scholar]
- Rui Z.; Li X.; Zhu X.; Liu J.; Domigan B.; Barr I.; Cate J. H.; Zhang W. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18237. 10.1073/pnas.1419701112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco W. A.; Merkler D. J.; Blackburn N. J.; Klinman J. P. Biochemistry 1998, 37, 8244. 10.1021/bi973004y. [DOI] [PubMed] [Google Scholar]
- Miller S. M.; Klinman J. P. Biochemistry 1983, 22, 3091. 10.1021/bi00282a011. [DOI] [PubMed] [Google Scholar]
- Cha Y.; Murray C. J.; Klinman J. P. Science 1989, 243, 1325. 10.1126/science.2646716. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. Nat. Chem. Biol. 2009, 5, 543. 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- Buhks E.; Bixon M.; Jortner J. J. Phys. Chem. 1981, 85, 3763. 10.1021/j150625a011. [DOI] [Google Scholar]
- Buhks E.; Bixon M.; Jortner J.; Navon G. J. Phys. Chem. 1981, 85, 3759. 10.1021/j150625a010. [DOI] [Google Scholar]
- Luo Y.-R.Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, 2003. [Google Scholar]
- Steigerwald M. L.; Goddard W. A. III; Evans D. A. J. Am. Chem. Soc. 1979, 101, 1994. 10.1021/ja00502a011. [DOI] [Google Scholar]
- Klinman J. P. FEBS J. 2014, 281, 489. 10.1111/febs.12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.