

Abstract
Atherosclerosis, cancer and various chronic fibrotic conditions are characterized by an increase in the migratory behavior of resident cells and the enhanced invasion of assorted exogenous cells across a stiffened extracellular matrix. This stiffened scaffold aberrantly engages cellular mechanosignaling networks in cells, which promotes the assembly of invadosomes and lamella for cell invasion and migration. Accordingly, deciphering the conserved molecular mechanisms whereby matrix stiffness fosters invadosome and lamella formation could identify therapeutic targets to treat fibrotic conditions, and reducing extracellular matrix stiffness could ameliorate disease progression.
Keywords: extracellular matrix stiffness, migration, invasion, cancer, fibrosis, cardiovascular disease
A biomechanical view of fibrotic disease
The extracellular matrix (ECM; see Glossary) regulates cellular differentiation, function and homeostasis through a dynamic biochemical and biophysical interplay between the resident cells and their microenvironment [1]. Far from an inert scaffold, the three-dimensional ECM undergoes dynamic remodeling to maintain tissue homeostasis while simultaneously providing physical support for tissue integrity. Conversely, loss of tissue homeostasis and dysregulation of ECM remodeling participates intimately with pathological fibrotic conditions characterized by excessive ECM production, deposition and accumulation without reciprocally-balanced degradation [2]. Unresolved fibrosis has been linked to an elevated risk of cancer [3, 4], cardiovascular disease [5], organ failure [6], and osteoarthritis [7]. The ECM of a fibrotic tissue is stiffer, and a pathologically stiff ECM promotes aberrant cellular mechanotransduction, the process of sensing and converting extracellular mechanical stimuli into downstream intracellular signaling changes. In response, cytoskeletal remodeling and elevated Rho GTPase-dependent cellular tension remodel and further stiffen the ECM. The mechanosensing process involves assessment of the mechanics of the ECM by the cells through integrins and the actomyosin cytoskeleton, and is followed by a mechanoregulation process, which includes the deposition, rearrangement or removal of the ECM to maintain overall form and function.
Both acute and chronic fibrotic conditions are characterized by an increase in the migratory behaviour of resident cells, higher numbers of infiltrating immune, vascular, mesenchymal stem cells and smooth muscle actin-positive cells, and inappropriate cellular invasion into the parenchyma [8–11]. Fibrotic tissues promote this inappropriate cellular invasion and migration by fostering the assembly of invadosomes and lamella (see Glossary), which are specialized actin-rich structures that combine adhesion and localized degradation to facilitate cellular movement through the surrounding ECM [12, 13]. In this review, we discuss how diverse pathological states characterized by chronic fibrosis exploit conserved mechanically-directed molecular mechanisms to enhance cell invasion and migration to drive disease progression. We describe commonalities in atherosclerosis, cancer and various chronic fibrotic conditions across length scales from the tissue, to the cell and ultimately to the subcellular and molecular level whereby mechanical cues initiate and elaborate cell invasion and guide cell migration. We discuss how cells sense and assimilate biomechanical cues by regulating cell-ECM adhesions that can modify growth factor signalling to direct the assembly and dynamics of invadosomes and lamella that facilitate cell invasion and migration. Given that all cells use conserved mechanotransduction pathways to drive their invasion and migration, we conclude with a discussion of therapeutic opportunities that could be applied to either inhibit cell tension or reduce fibrotic accumulation to ameliorate disease pathology.
Elevated stiffness: a mechanical hallmark of fibrotic lesions
Cardiovascular disease and many cancers are characterized by fibrosis, and similar to conditions of chronic fibrosis, they typically have high amounts of infiltrating exogenous cells and exhibit an enhanced motility of resident cells linked to the pathology [8, 11, 14, 15]. Chronic fibrosis is characterized by an increase in ECM deposition, turnover and post-translational modifications that progressively stiffen the non-cellular stroma to support the enhanced migratory and invasive phenotypes observed in these conditions. Elevated levels of fibrillar collagen are found in the stroma surrounding solid tumors including those of the breast, pancreas, and prostate [16–18] (Fig. 1A). Likewise, atherosclerotic lesions contain more collagen types I, III, IV, V, and VI [19, 20] (Fig. 1B). Lung and liver fibrosis are also characterized by the progressive replacement of the normal parenchymal tissue with fibrillar type collagens [11, 21]. Type I collagen, which is the predominant collagen deposited in chronic fibrosis, is a major contributor to the mechanical properties of the ECM, suggesting the increased collagen content of these fibrotic conditions reflects a denser stroma. In addition to fibrillar collagens, fibrotic tissues contain a mixture of other ECM proteins such as tenascin and fibronectin that are heterogeneously dispersed throughout the tissue to generate a diverse and varying ECM landscape. Yet, the higher amount of ECM protein in a fibrotic tissue also stiffens the stroma, and this effect can be greatly enhanced by crosslinking through enzymes such as lysyl oxidase (LOX; see Glossary) and transglutaminase, which are both frequently increased in fibrotic conditions [22]. Increased ECM deposition, decreased degradation and elevated levels of various matrix crosslinking enzymes such as LOX and transglutaminase contribute to the hallmark elevated stiffness observed in fibrotic lesions [2, 22]. Indeed, mechanical testing of atherosclerotic plaques, solid tumors and other fibrotic lesions demonstrates that the ECM within the diseased lesion is stiffer than the corresponding healthy tissue (Table 1).
Figure 1.
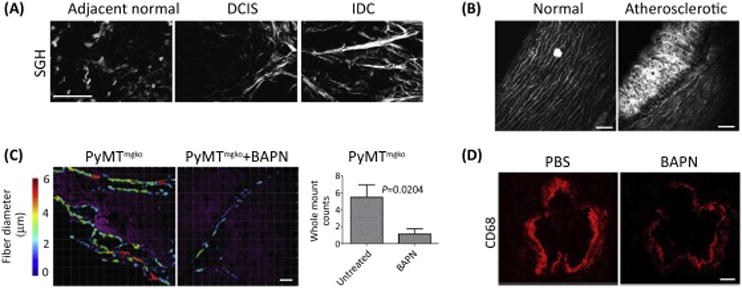
Increased collagen deposition and thickening in breast cancer and cardiovascular disease promotes cell invasion and migration. (A) Second harmonics generation (SHG) imaging of human breast tumor biopsies containing normal adjacent tissue, ductal carcinoma in situ (DCIS), and invasive ductal carcinoma (IDC) showed that breast cancer transformation is accompanied by a progressive increase in interstitial collagen fibrils. Scale bar = 40 μm. Figure is modified, with permission, from [16]. (B) SHG imaging of healthy and atherosclerotic arteries showed atherosclerotic lesions have an increase in collagen fibrils. Scale bar = 75 μm. Figure is modified, with permission, from [20]. (C) LOX Inhibition using BAPN significantly reduced collagen thickening (color-coded fibrillar collagen diameter) and concurrently decreased the number of lung metastases in Polyoma middle-T-induced carcionomas lacking type II-TGF-beta (PyMTmgko). Scale bar = 30 μm. Figure is modified, with permission, from [24]. (D) The number of inflammatory macrophages (anti-CD68; red) in aortic root lesions of atherosclerotic apoE-null mice was significantly decreased in the BAPN-treated mice. Scale bar = 200μm. Figure is modified, with permission, from [14].
Table 1.
Alterations in Cell Behaviors in Pathological Fibrotic Conditions and Stiffened ECMs.
| Conditions | Stiffness range | Predominant ECM ligands | Cell phenotypes | Ref |
|---|---|---|---|---|
| Pathological tissues | ||||
| Breast cancer | 0.2–2.5kPa | Collagen | Enhanced tumor cell invasion into surrounding
ECM; Enhanced circulating tumor cells and lung metastasis; Enhanced macrophage infiltration. |
[15] [24] [16] |
| Pancreatic cancer | Und. | Collagen | Enhanced cancer cell invasion surrounding stroma; enhanced macrophage and neutrophil infiltration | [23] |
| Atherosclerotic Artery | 2–20kPa | Collagen and fibronectin | Enhanced monocyte/macrophage infiltration | [14] |
| Lung fibrosis | Und. | Collagen | Increased resident lung fibroblasts, lymphocytes, macrophages, plasma cells, eosinophils and neutrophils | [11] |
| Liver fibrosis | Und. | Collagen | Increased neutrophil, mast cells, lymphocytes, and natural killer cells | [21] |
| Osteoarthristis | 4–31kPa | Collagen | Altered chondrocyte metabolism (e.g. increased LOX expression) | [7] |
| Model culture systems | ||||
| Epithelial cells (NRK) and Fibroblasts (3T3) | 0.03–0.3% | Collagen (2D) | Stiff substrates promote cell spreading and stable focal adhesion formation | [25] |
| Bovine aortic VSMCs | 5–80 kPa | Collagen (2D) | Stiff substrates promote cell spreading, polarization, and migration; durotaxis increases with increasing magnitude of stiffness gradient | [26] |
| MDA-MB 231 breast carcinoma cells | 0.3–2.4 mg/ml | Collagen (3D) | Stiff substrates promote cell invasion (when pore size >5μm) | [28] |
| U373-MG glioma cells | 0.4–120kPa | Fibronectin (3D) | Stiff ECMs increase cell migration speed | [29] |
| NIH/3T3 fibroblasts and bovine pulmonary arterial endothelial cells | 1.8–34 kPa 12 kPa–2.5 MPa |
Fibronectin (3D) | Cells migrate to stiffer regions | [36] |
| Oncogene-initiated mammary tumor (MMTV-Neu mice) | 0.2–2.5kPa | Collagen (3D) | Stiff ECMs promote tumor cell invasion into surrounding ECM | [15] |
| Isolated tumor cells from MMTV PyMT transgenic mammary tumor mouse model | 0.4–4kPa | Collagen (3D) | Stiff ECMs enhance tumor cell invasion and migration | [30] |
| Ha-ras premalignant mammary organoids | 0.2–2.5kPa | Collagen (3D) | Stiff ECMs promote tumor cell invasion into surrounding ECM | [15], [31] |
Und.: undetermined
These and other experimental results imply that the elevated ECM stiffness that accompanies the development of fibrosis plays a causative, rather than consequential, role in the pathogenesis of disease. In support of this concept, transgenic mice engineered to develop genetically-driven mammary or pancreatic tumors that were treated with inhibitors targeting LOX to prevent type I collagen cross-linking and ECM stiffening, developed tumors later. Moreover, the tumors that these mice developed were more amenable to therapy and overall tumor incidence and metastasis were reduced, possibly reflecting the less invasive nature of the tumor cells within the treated tissue [15, 23, 24] (Fig. 1C). Similarly, when LOX was inhibited to reduce collagen cross-linking and ECM stiffening, mice fed a high-cholesterol diet to induce cardiovascular disease had reduced arterial rigidity and fibrotic lesion formation that was linked to decreased inflammatory cell infiltration (Fig. 1D) [14]. Interestingly, these same studies showed that ameliorating tissue fibrosis and ECM stiffening also reduced the invasion and migration of resident and infiltrating exogenous immune cells into the tissue. These findings suggest a stiff, fibrotic ECM fosters disease progression by enhancing cell invasion and migration.
ECM stiffness regulates cell invasion and migration
While chemotaxis during disease progression is a widely recognized stimulator of cell invasion and migration, in vitro studies have also provided compelling evidence to show that substrate stiffness, itself, can modulate cell invasion and migration (see Table 1). Pelham and Wang were the first to employ synthetic polyacrylamide (PA) gel substrates with tuned stiffness to demonstrate that a stiffer substrate significantly enhances serum-stimulated cell migration [25]. Their observations motivated a new field of inquiry regarding the impact of physical properties of the ECM on cell migration, and have since been confirmed and mechanistically elaborated in diverse cell types [26, 27]. Importantly, the relationship between ECM stiffness and cell migration and invasion appears to be maintained in more physiologically relevant three-dimensional (3D) tissue-like microenvironments. Data obtained using breast cancer and glioblastoma cells embedded within a 3D collagen gel or fibronectin-conjugated 3D micro-channels similarly attest to the strong impact of substrate stiffness on migration speed [28, 29]. Indeed, non-transformed, pre-malignant and transformed cancer cells not only invade in greater numbers but also migrate more persistently within a stiffer 3D type I collagen gel [15, 30, 31]. In this respect, ECM density and composition can impose physical constraints to restrict cell movement through reducing pore size, necessitating a requirement for the cells to degrade the matrix or undergo transdifferentiation (epithelial-mesenchymal transition) to be able to invade and migrate [32, 33]. Nonetheless, work conducted using 3D self-assembling peptide gels and those employing a unique collagen hydrogel bioreactor, in which the ECM can be stiffened without changing pore size or ECM composition or concentration, definitively demonstrate that ECM stiffness can directly promote cell invasion and migration, even in a 3D ECM [30, 34]. These findings have been further elaborated to include data showing that cell directionality in 2D and 3D formats is also guided by substrate stiffness [30, 35]. For instance, PA or polydimethylsiloxane gel studies demonstrated that fibroblasts and endothelial cells [36], as well as vascular smooth muscle cells (VSMC) [26] each preferentially migrate up a 2D stiffness gradient. Importantly, a recent study using a unique bioreactor showed that breast tumor cells migrate towards a stiffened 3D collagen ECM [30], although it has yet to be determined if their migration velocity is also affected. In this regard, the migration velocity of VSMC [26] and mesenchymal stem cells [37] does increase in combination with the strength of the 2D stiffness gradient. Such directional cell movement in response to ECM stiffness is described as durotactic behavior, and may explain why diverse cell types preferentially migrate towards and accumulate in stiff fibrotic tissues. The phenomenon could also explain why inhibiting ECM stiffening effectively impairs exogenous and resident cell invasion and migration in fibrotic lesions.
ECM stiffness promotes invadosome and lamella formation
Cells invade and migrate into the interstitial stroma of a tissue by assembling distinct actin-rich protrusive structures at their leading edge termed invadosomes and lamella [12, 13]. Invadopodia and podosomes, both members of the invadosome family, comprise an actin-rich core containing the actin-nucleating Arp2/3 complex, the actin-regulating WASP and cortactin proteins, and the adaptor proteins Tks4 and Tks5 [38]. Additionally, proteolytic enzymes, such as MT1 and the matrix metalloproteinase (MMP; see Glossary) family, are surrounded by this adhesion protein complex, which is localized to the ventral plasma membrane in invading cells. In mammalian systems, invadopodia are found in multiple cancer cell types, whereas podosomes are found in non-transformed, highly motile cells of mesenchymal and myelomonocytic lineage such as macrophages, smooth muscle cells, endothelial cells, and fibroblasts [12]. Lamella describe two subcellular structures, including both lamellipodia and filopodia (see Glossary), that are comprised of highly branched actin meshwork and parallel actin bundles respectively, to drive membrane protrusions during cell migration.
In vitro analysis indicate that invadosomes facilitate localized MMP-mediated degradation of underlying ECM substrates [12], while in vivo studies illustrate that invadosomes are essential for MMP-dependent invasion of cells across ECM barriers. For instance, in C elegans development, vulval organogenesis is facilitated by the ability of anchor cells to assemble invadosome structures so they can transmigrate across two basement membranes [39]. Similarly, orthotopic tumors require invadopodia to intravasate and metastasize to lung as knockdown of N-WASP (see Glossary), a key component of invadosomes that promote Arp2/3 complex (see Glossary) nucleation activity [40] and trafficking MMP to the invadopodia [41], abolished both invadopodia formation and lung metastasis [42]. Consistently, high-resolution intravital imaging using a chick embryo chorioallantoic membrane model demonstrated that breast cancer cells exploit invadopodia to breach the endothelium [43]. These findings suggest that invadosomes are critical subcellular structures required for cell invasion through an ECM barrier.
Lamella fragments isolated from fish epidermal keratinocytes, even in the absence of a nucleus and microtubules, are able to move persistently, implying that the lamella is the minimal system required for cell migration [44]. It is now appreciated that two actin-rich protrusive structures assembled at the leading edge of the lamella, filopodia and lamellipodia, are important for directional cell migration. Filopodia formation in migrating fibroblasts typically precedes the emergence of the nascent lamellipodia, such that inhibition of filopodia formation impairs fibroblast movement towards a gradient of surface-bound fibronectin [45]. Indeed, genetic knockout of Arp2/3 complex subunits, which are required for the assembly of the highly branched actin network that powers membrane protrusions within lamellipodia, revealed that while lamellipodia are not required for cell migration, they are essential for directional cell movement along an ECM ligand gradient [46]. In this manner, lamellipodia and filopodia may cooperate to promote directional cell migration.
The mechanical properties of the ECM can have a profound impact on the formation and activity of both invadosomes and lamella, thereby influencing cell invasion and migration. Experiments using mechanically-tuned PA gels demonstrated that a stiff ECM increases the local density of podosomes [47]. Similarly, the number of invadosomes in breast carcinoma cells and macrophages were found to increase in tandem with ECM-coated PA gel stiffness (Fig. 2A) [48–50]. Substrate stiffness can also modify the stability of invadosomes since a stiff ECM PA gel can prolong the lifespan of invadosomes in fibroblasts [47]. The mechanosensing ability of invadosomes has likewise been implicated as indicated by evidence provided by early studies, which showed that lymphocytes extend and retract invadosomes to palpate the underlying microvascular monolayer prior to breaching the endothelial barrier [51]. Indeed, traction force microscopy studies revealed that podosomes in fibroblasts can exert traction on the underlying ECM matrix, and showed that the traction generated beneath the podosome increases proportionately with the stiffness of the substrate [52]. Using Protrusion Force Microscopy (PFM), a derivation of AFM that can quantify the protrusive force of a single podosome, experiments demonstrated that cells tune the magnitude of the oscillating force in a podosome to the stiffness of the underlying substrate [49].
Figure 2.
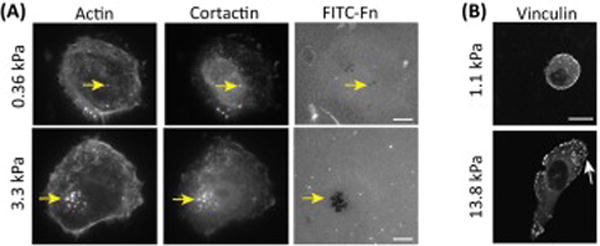
Stiff substrates promote invadosome and lamella formation. (A) Breast carcinoma cells cultured on stiff gels formed a higher number of invadosomes and exhibited more proteolytic activity. Invadosomes are identified by colocalization of actin and cortactin and proteolytic activity of invadosomes can be visualized as the loss of fluorescently labelled fibronectin (Arrow: Invadopodia). Scale bar=10μm. Figure is modified, with permission, from [48]. (B) Nonmalignant mammary epithelial cells (MEC) cultured on stiff gels assembled larger focal adhesions (marker: vinculin) and had larger lamella (arrow). Figure is modified, with permission, from [31].
Substrate stiffness also profoundly impacts lamella formation and directionality. For instance, non-malignant mammary epithelial cells (MECs) display large lamella when cultured on a classic rigid 2D tissue culture plastic or glass substrate (~108 Pa), or even on a laminin-conjugated stiff PA gel (5 kPa), but spread minimally when in contact with a compliant PA gel (~400 Pa range) (Fig. 2B) [31, 53]. Likewise, fibroblasts and endothelial cells spread more and form large lamella when plated on a stiff fibronectin-coated substrate (180Pa vs. 16000Pa) [54]. These findings are consistent with data showing that during cell migration, lamellae undergo cycles of leading edge protrusion and retraction to interrogate the biophysical properties of substrate rigidity. Indeed, periodic contraction of lamella is governed by substrate stiffness since cells on a stiff substrate typically exhibit periodic oscillation of lamella but demonstrate only minimal activity on a soft fibronectin-coated PA gel [55]. Cells on a softer matrix also spend more time retracting lamella than extending protrusions, and the net result of this behavior is a decrease in protrusion formation [56]. Intriguingly, experimental work has demonstrated that the filopodia extended at the leading edge of the cell, in the vicinity of the lamella, may function as “mechanical antennae” to assess the rigidity of the ECM beyond the border of the cell prior to lamellipodia extension. Using substrates with micropatterned rigidity, fibroblast filopodia were found to exhibit a strong preference for the more rigid regions of the substrate, whereas by contrast they retracted more frequently on the more compliant areas [57]. These data suggest that substrate stiffness not only promotes productive protrusions that increase the migratory phenotype of cells, but also likely favors the directionality of lamella to foster durotaxis. Given that diseases with chronic fibrosis develop a stiffened ECM it is perhaps not surprising that these conditions are also characterized by the enhanced invasion of endogenous and exogenous cells into the tissue. Whether the invasion of the cells into the tissue depends upon directed lamella extension and invadosome formation, however, remains unclear. Regardless, such cumulative observations may explain the directed migration of macrophages towards the stiffer intimal plaque, and the persistent migration of breast tumor cells along rigid collagen tracts and their subsequent intravasation into the vasculature that ultimately favors their dissemination [58].
Mechanoregulation of invadosome and lamella assembly
So how could ECM stiffness foster invadosome and lamella assembly to drive cell invasion and migration? Cells sense mechanical stimuli from their microenvironment and transduce these cues to modify their behaviour through mechanotransduction (Fig. 3, Key Figure). There are several mechanosensors expressed by cells of which integrins are perhaps the best studied. Integrins are key regulators of cell invasion and migration and the current perspective is that they do so by modulating invadosome and lamella formation. For instance, dendritic cells that lack β2 integrin [59], and Src-transformed fibroblasts that have β1 integrin depleted [60], fail to assemble podosomes and are unable to degrade the underlying ECM. Similarly, keratinocytes with genetic deletion of α3 integrin fail to form stable lamella [61], whereas MDA-MB-231, MIP-101, and CCL-228 cancer cells treated with a function blocking antibody to α6β4 integrin neither assemble filopodia nor lamellipodia and cannot migrate [62].
Figure 3.
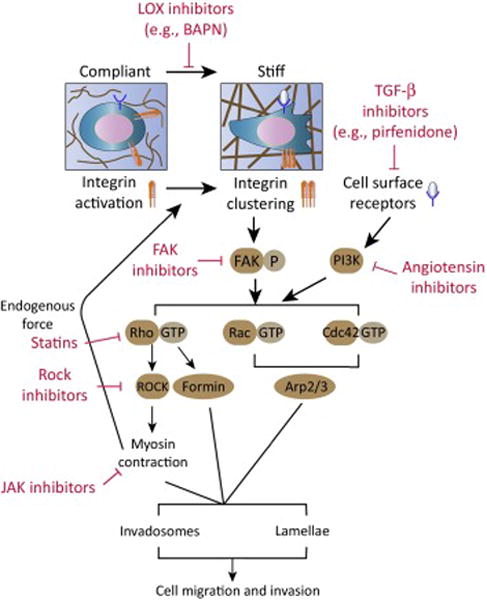
Key Figure. Biomechanical regulation of fibrotic disease across scales. ECM stiffening activates mechanotransduction signaling pathways that drive cell invasion and migration by promoting invadosome and lamella formation. Increased collagen crosslinking stiffens the ECM and drives integrin clustering, which subsequently results in focal adhesion kinase (FAK) activation and formation of FAK-Src complex. Stiff ECM can also promote PI3K signaling by potentiating signals transmitted from cell surface receptors, such as growth factor receptors. FAK-Src complex and PI3K can activate several pathways that lead to invadosome and lamella assembly via Rho GTPases. Activated Rac and Cdc42 GTPases can promote Arp2/3-nucleated dendritic actin network formation whereas RhoA GTPase activates formin-mediated linear F-actin assembly. Additionally, RhoA activates ROCK, which in turn enhances myosin contractibility to promote invadosome and lamella dynamics and reinforce integrin clustering. Currently available and pre-clinical drugs with potential utility in blocking mechanosignaling pathways are also listed (red font).
Pertinent to the central role for integrins in invadosome- and lamella-mediated invasion and migration, integrins are exquisitely sensitive to the stiffness of the ECM, such that a stiff ECM enhances integrin activation and focal adhesion (see Glossary) protein recruitment to drive adhesion plaque assembly [63, 64]. Indeed, focal adhesions are composed of a suite of integrin-associated adhesion plaque and signaling molecules including vinculin, paxillin and Rho GTPases, which regulate cytoskeletal organization and foster the assembly of invadosomes and lamella (see review [65–67]). A stiff ECM drives integrin clustering, enhances talin and vinculin recruitment and promotes the autophosphorylation of FAK at Tyr397 that fosters the assembly of a FAK-Src complex. The assembled FAK-Src complex, in turn, can activate Rac and Cdc42 GEFs (e.g. β-PIX and DOCK/ELMO complex) [68] that thereafter stimulate Rac and Cdc42 GTPase activity, which are required for lamella and invadosome assembly and dynamics (Fig. 3) [65, 69]. These integrin adhesion-associated GEFs also activate RhoA GTPases that stimulate the actomyosin contraction that is required for the proteolytic activity of the invadosomes and the formation of focal adhesions at the lamella [70, 71]. In this manner, integrins activate mechanosignaling to enhance the formation of invadosome and lamella that are necessary for cell invasion and migration.
In most adherent cells, invadosomes and lamella formation can be stimulated by external soluble factors. For instance, in primary endothelial cells, VEGF and TGF-β induce podosome formation [72, 73], whereas in VSMCs, PDGF drives podosome development [74, 75]. Lamella formation can also be stimulated by external stimuli such as EGF, PDGF, and TGF-β in diverse cell types [76–78]. Importantly, integrins can synergize with both growth factor receptors and G protein coupled receptors to enhance cell invasion and migration, and these interactions may potentiate invadosome and lamella assembly by amplifying Src and PI3K activity [15, 79, 80]. Given that a fibrotic ECM can harbour elevated exogenous factors and a stiff ECM enhances integrin adhesion assembly, it is perhaps not surprising that ECM stiffness also potentiates growth factor and G protein coupled receptor activity and signaling [15, 53, 81]. Thus, MECs plated on stiff “tumor-like” basement membrane-conjugated PA gel show a significant amplification in ERK and PI3K activation in response to EGF, and ErbB2-induced mammary tumor cell invasion into a stiffened collagenous-rich ECM requires integrin-dependent signaling [15]. Substrate stiffness can also enhance the autophosphorylation of the PDGFR, whose activity drives cell migration and promotes lamella formation in vascular smooth muscle cells [76, 80]. The in vivo relevance of an association between ECM stiffness, mechanosignaling, and cell invasion and migration was vividly illustrated by studies conducted in transgenic mice expressing oncogenic ErbB2 in the mammary epithelium, which showed that preventing collagen crosslinking and fibrosis-mediated stiffening of the ECM stroma with a LOX inhibitor reduced PI3K activity and impeded breast cancer invasion and migration [15]. Studies addressing the impact of ECM stiffness on tumor metastasis using mice expressing the oncogenic PyMT antigen (isolated from polyoma virus under the regulation of the MMTV promoter from mouse mammary tumor virus) and lacking the type II TGF-β receptor in the mammary epithelium, revealed that reducing collagen crosslinking not only decreased tissue fibrosis and stiffening, but also inhibited lung metastasis. Further analysis revealed that the decrease in metastasis in this mouse model was associated with reduced circulating tumor cells, likely reflecting lowered cell migration and extravasation [24]. Nevertheless, definitive in vivo evidence causally linking ECM stiffness to cell invasion and migration through integrin and GTPase-mediated modulation of invadosomes and lamella has yet to be demonstrated.
Therapeutic interventions targeting mechanosignaling
Overwhelming evidence indicate that ECM stiffening is a common feature of fibrotic diseases and suggest that this mechanophenotype contributes critically to the enhanced invasion and migration of exogenous and resident cells that contribute to the disease pathology associated with these conditions. Accordingly, treatments that can directly ameliorate tissue fibrosis should reduce ECM rigidity and potentially normalize tissue behavior. To this end, activated TGF-β undoubtedly stimulates tissue fibrosis and, accordingly, several anti-TGF-β pathway inhibitors have been developed and clinically tested [82]. Nevertheless, while experimental models employing anti-TGF-β therapies have been quite promising, a number of undesirable side effects have been highlighted in patients including skin rashes/lesions and gingival bleeding that have precluded their wide scale adoption [83]. More recently, Pirfenidone, which also reduces TGF-β activity as well as TNF-α, was developed as an FDA-approved anti-fibrotic treatment for idiopathic pulmonary fibrosis with fewer side effects [84, 85], although its clinical efficacy and applicability for other fibrotic diseases has yet to be elucidated.
Given the links between tissue fibrosis, ECM remodeling and stiffening, an alternative strategy has been to repress the activity of enzymes involved in cross-linking and processing of structural, fibrillar collagens that increase the tensile properties of the tissue. The LOX family members, prolyl 4-hydroxylase and the lysyl hydroxylase family have emerged as exciting new candidates with anti-fibrotic and anti-tension efficacy [86–88]. These collagen crosslinkers are elevated in a myriad of diseases including liver fibrosis, cardiovascular lesions, and cancers of the breast, colon, gut, kidney and lungs [86, 89]. These inhibitors have generated a tremendous amount of excitement, particularly since several compelling articles have been published in the past decade that attest to the abilities of the pharmaceutical inhibitor BAPN, which blocks the catalytic activity of LOX, and function-blocking antibodies generated against LOX and LOXL2, to profoundly reduce tissue fibrosis and repress disease progression, aggression and metastasis in experimental models of breast, colon and lung cancers, as well as cardiovascular disease [15, 90]. Despite the excitement associated with these agents, patients with Ras-driven tumors, including those of the pancreas and small cell lung carcinoma, may not be ideal candidates for these treatments because the pro-peptide cleaved from these enzymes has demonstrated anti-Ras activity [91]. Since the anti-fibrotic effects may be tempered with the release of Ras inhibition, this would likely result in restored proliferation of Ras-driven cancer cells [92]. Indeed, recent clinical studies assessing the efficacy of simtuzumab, an inhibitor of LOXL2, in combination with chemotherapy demonstrated no significant increase in progression-free survival in patients with previously untreated advanced pancreatic cancer (NCT01472198). Alternatively, inhibition of prolyl 4-hydroxylase, an essential enzyme in collagen biosynthesis and triple helix stabilization, reduces fibrillar collagen secretion and may thus prove therapeutically beneficial for inhibiting the development of fibrosis in disease [93].
As the increased cell migration and invasion induced in response to elevated ECM stiffness is linked to enhanced actomyosin contractility, one attractive and tractable approach has been to bypass the stroma and inhibit the elevated cellular tension or the cellular mechanotransduction machinery directly. Indeed, targeting of FAK using small molecule-based inhibitors has been tested in pre-clinical and clinical studies, demonstrating efficacy in various fibrotic diseases including mesothelioma [94]. Additionally, many fibrotic tissues show RhoA GTPase and ROCK activity towards which highly specific ROCK inhibitors have been developed [95]. Unfortunately, while highly effective in culture and in vivo, inhibition of ROCK may prove challenging as one profound side effect of systemic inhibition is the lowering of patient blood pressure [95]. Re-purposing of readily available, affordable drugs with known safety records is another treatment approach. Lipophilic statins, which are used widely to treat cardiovascular diseases and high cholesterol, have been proposed to treat patients with fibrosis-associated diseases. Several preclinical studies reported that invasion players such as RhoA and C proteins are dependent targets of statins, with statins inhibiting cellular adhesion, migration, and chemotaxis [96, 97]. Similarly, the oft-prescribed angiotensin inhibitors, used to treat patients with high blood pressure, increased metastatic renal cell carcinoma patient survival, and reduced tumor fibrosis and enhanced chemotherapeutic efficacy in experimental models of cancer, possibly by inhibiting growth factor-stimulated PI3K signaling [98–100]. Similarly, various fibrotic pathologies are characterized by excessive activation of the Janus family of cytoplasmic tyrosine kinases (JAK) and their associated signal transducer and activator of transcription factors (STAT) [101]. Given evidence that JAK signaling can drive actomyosin contractility in parallel with ROCK, therapeutic inhibition of the JAK/STAT3 pathway offers considerable benefit, and this idea is emphasized by successful clinical applications of JAK-specific small molecule inhibitors for the treatment of inflammatory disorders and multiple cancers [102–104]. Importantly, while affordable and readily available, re-purposed drugs are not without side effects and still require safety and efficacy testing for each new disease modality.
Concluding remarks
While anti-tension treatments and re-purposed therapies are attractive approaches with which to treat mechano-fibrotic diseases, these options should not be thought of as a final solution. Ideally, through the identification of the proverbial “Achilles heel” linking ECM stiffness to the aberrant invasive and migratory phenotype that drives the pathology associated with many chronic fibrotic conditions, scientists can begin to design specific strategies to combat various fibrotic conditions. As discussed, one common feature of pathological fibrosis, whether it be cancer or otherwise, is the promotion of cell invasion and migration by elevated tissue stiffness, which likely arises via the promotion of invadosome and lamella assembly. Accordingly, studies aimed at clarifying this interplay may identify novel and specific mechanics regulated molecular candidates for drug targeting (see Outstanding questions). Indeed, with the advent of sophisticated 3D tissue-like model systems [30, 34], state-of-the-art intravital imaging technology in transgenic mouse models (FRET and biosensor based), and genetic and drug screens, we are well-positioned to clarify the interplay between fibrosis, ECM stiffness and cellular invasion, and to identify novel and specific molecular candidates for drug targeting.
Outstanding questions.
Does pathological stiffening in diseased tissues drive cell migration and invasion in vivo?
Although multiple lines of evidence demonstrate that a stiff ECM serves as an environmental cue to promote cell migration and invasion in vitro, a causal relationship between ECM stiffening and cell migration and invasion in vivo remains to be investigated.
How can we selectively target cell migration/invasion at diseased sites specifically without affecting cell migration/invasion during normal processes, such as immunosurveillance?
Do cells switch migration/invasion mode (e.g. from a mesenchymal to an amoeboid mode of migration) when ECM stiffness is reduced in three-dimensional cultures or in vivo?
While the role of ECM stiffness in invadosome and lamella formation has been examined in 2D culture systems, what role does ECM stiffness have in regulating invadosome/lamella formation in 3D cultures? Does ECM stiffness drive invadosome/lamella formation in fibrotic tissue through the conventional pathways that have been identified using traditional stiff 2D culture surfaces?
Does mechanotransduction downstream of different mechanical cues, in different cell types and under various contexts share common mechanisms? As the mechanotransduction field is still in its infancy, only a handful of proteins/molecules have been implicated as comprising mechanotransduction machinery. A more comprehensive identification mechanosignaling pathways and machinery would allow for therapeutic targeting of pathways contributing to fibrosis and disease.
Trends box.
Fibrotic diseases frequently coincide with stiffened ECMs, and elevated migration and invasion of resident and exogenous cells.
A stiff matrix fosters the formation of actin-rich invadosomes and lamella.
Invadosomes and lamella are essential subcellular machinery to drive cell invasion and migration.
Reducing invadosome and lamella formation (and, thus, cellular invasion) through the therapeutic targeting of tissue tension and mechanosignaling could ameliorate disease pathology.
Acknowledgments
The authors apologize to all colleagues whose work cannot be cited owing to space limitations. We thank Janna K. Mouw for helpful editing of the manuscript. This work was supported by DOD grant BCRP BC122990, and NIH R01 grants CA192914, CA174929, CA08592, CA138818, and GM059907, U01 grant CA202241, U54 grant CA163155, and R33 grant CA183685 and AACR PANCAN A121989 to VMW.
Glossary
- Actin-related protein 2/3 (Arp2/3) complex
A multi-protein complex that nucleates the highly branched F-actin meshwork within the lamellipodia
- Extracellular matrix (ECM)
A multi-protein complex surrounding cells within tissue. The ECM provides biochemical and biomechanical cues to cells and modulates cellular behavior
- Filopodia
Finger-like protrusions comprised of parallel actin bundles in its core located at the leading edge of the migrating cells
- Focal adhesion
A macromolecular signaling complex that links the ECM to the intracellular actin cytoskeleton network through transmembrane protein integrin
- Invadosome
Cellular protrusion that mediates matrix metalloproteinase-dependent proteolytic degradation of the ECM
- Lamella
Actin-rich subcellular compartments, including lamellipodia and filopodia, that are located at the leading cell edge and drive membrane protrusions during cell migration
- Lamellipodia
Sheet-like protrusions comprised of highly branched Arp2/3 complex-nucleated actin network at the leading edge of migrating cells
- Lysyl oxidase (LOX)
An extracellular enzyme that catalyzes the covalent crosslinking between collagen molecules to stabilize the supramolecular collagen structure
- Mechanotransduction
Process by which cells sense and translate mechanical cues by converting them into intracellular biochemical signals to control cellular behaviors
- Metalloproteinases (MMP)
A family of membrane-bound or secreted proteolytic enzymes that degrade and remodel ECM proteins
- Neuronal Wiskott-Aldrich Syndrome protein (N-WASP)
A Rho GTPase-activated effector protein that stimulates Arp2/3-dependent actin polymerization
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mouw JK, et al. Extracellular matrix assembly: a multiscale deconstruction. Nature reviews Molecular cell biology. 2014;15:771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnans C, et al. Remodelling the extracellular matrix in development and disease. Nature reviews Molecular cell biology. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyd NF, et al. Mammographic density and breast cancer risk: current understanding and future prospects. Breast cancer research: BCR. 2011;13:223. doi: 10.1186/bcr2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Serag HB. Hepatocellular carcinoma. The New England journal of medicine. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 5.Weber KT, et al. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nature reviews Cardiology. 2013;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 6.Rockey DC, et al. Fibrosis–a common pathway to organ injury and failure. The New England journal of medicine. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, et al. Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:9424–9429. doi: 10.1073/pnas.1505700112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellicoro A, et al. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nature reviews Immunology. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 9.Ruffell B, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seneviratne A, et al. Biomechanical factors and macrophages in plaque stability. Cardiovascular research. 2013;99:284–293. doi: 10.1093/cvr/cvt097. [DOI] [PubMed] [Google Scholar]
- 11.Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal immunology. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linder S, et al. Degrading devices: invadosomes in proteolytic cell invasion. Annual review of cell and developmental biology. 2011;27:185–211. doi: 10.1146/annurev-cellbio-092910-154216. [DOI] [PubMed] [Google Scholar]
- 13.Petrie RJ, Yamada KM. At the leading edge of three-dimensional cell migration. Journal of cell science. 2012;125:5917–5926. doi: 10.1242/jcs.093732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kothapalli D, et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell reports. 2012;2:1259–1271. doi: 10.1016/j.celrep.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acerbi I, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative biology: quantitative biosciences from nano to macro. 2015 doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Molecular cancer therapeutics. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 18.Barron DA, Rowley DR. The reactive stroma microenvironment and prostate cancer progression. Endocrine-related cancer. 2012;19:R187–204. doi: 10.1530/ERC-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katsuda S, et al. Collagens in human atherosclerosis. Immunohistochemical analysis using collagen type-specific antibodies. Arteriosclerosis and thrombosis: a journal of vascular biology/American Heart Association. 1992;12:494–502. doi: 10.1161/01.atv.12.4.494. [DOI] [PubMed] [Google Scholar]
- 20.Le TT, et al. Label-free molecular imaging of atherosclerotic lesions using multimodal nonlinear optical microscopy. Journal of biomedical optics. 2007;12:054007. doi: 10.1117/1.2795437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alcolado R, et al. Pathogenesis of liver fibrosis. Clinical science. 1997;92:103–112. doi: 10.1042/cs0920103. [DOI] [PubMed] [Google Scholar]
- 22.Frantz C, et al. The extracellular matrix at a glance. Journal of cell science. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller BW, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO molecular medicine. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickup MW, et al. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer research. 2013;73:5336–5346. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pelham RJ, Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isenberg BC, et al. Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength. Biophysical journal. 2009;97:1313–1322. doi: 10.1016/j.bpj.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prager-Khoutorsky M, et al. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nature cell biology. 2011;13:1457–1465. doi: 10.1038/ncb2370. [DOI] [PubMed] [Google Scholar]
- 28.Lang NR, et al. Biphasic response of cell invasion to matrix stiffness in three-dimensional biopolymer networks. Acta biomaterialia. 2015;13:61–67. doi: 10.1016/j.actbio.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cassereau L, et al. A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. Journal of biotechnology. 2015;193:66–69. doi: 10.1016/j.jbiotec.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubashkin MG, et al. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer research. 2014;74:4597–4611. doi: 10.1158/0008-5472.CAN-13-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haage A, Schneider IC. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:3589–3599. doi: 10.1096/fj.13-245613. [DOI] [PubMed] [Google Scholar]
- 33.Wolf K, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. The Journal of cell biology. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miroshnikova YA, et al. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Physical biology. 2011;8:026013. doi: 10.1088/1478-3975/8/2/026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plotnikov SV, et al. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell. 2012;151:1513–1527. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gray DS, et al. Repositioning of cells by mechanotaxis on surfaces with micropatterned Young’s modulus. Journal of biomedical materials research Part A. 2003;66:605–614. doi: 10.1002/jbm.a.10585. [DOI] [PubMed] [Google Scholar]
- 37.Vincent LG, et al. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnology journal. 2013;8:472–484. doi: 10.1002/biot.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albiges-Rizo C, et al. Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. Journal of cell science. 2009;122:3037–3049. doi: 10.1242/jcs.052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagedorn EJ, et al. The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. The Journal of cell biology. 2013;201:903–913. doi: 10.1083/jcb.201301091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamaguchi H, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. The Journal of cell biology. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X, et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. The Journal of cell biology. 2012;199:527–544. doi: 10.1083/jcb.201203025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gligorijevic B, et al. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. Journal of cell science. 2012;125:724–734. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leong HS, et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell reports. 2014;8:1558–1570. doi: 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 44.Euteneuer U, Schliwa M. Persistent, directional motility of cells and cytoplasmic fragments in the absence of microtubules. Nature. 1984;310:58–61. doi: 10.1038/310058a0. [DOI] [PubMed] [Google Scholar]
- 45.Johnson HE, et al. F-actin bundles direct the initiation and orientation of lamellipodia through adhesion-based signaling. The Journal of cell biology. 2015;208:443–455. doi: 10.1083/jcb.201406102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu C, et al. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collin O, et al. Spatiotemporal dynamics of actin-rich adhesion microdomains: influence of substrate flexibility. Journal of cell science. 2006;119:1914–1925. doi: 10.1242/jcs.02838. [DOI] [PubMed] [Google Scholar]
- 48.Alexander NR, et al. Extracellular matrix rigidity promotes invadopodia activity. Current biology: CB. 2008;18:1295–1299. doi: 10.1016/j.cub.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Labernadie A, et al. Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nature communications. 2014;5:5343. doi: 10.1038/ncomms6343. [DOI] [PubMed] [Google Scholar]
- 50.Parekh A, et al. Sensing and modulation of invadopodia across a wide range of rigidities. Biophysical journal. 2011;100:573–582. doi: 10.1016/j.bpj.2010.12.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carman CV, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collin O, et al. Self-organized podosomes are dynamic mechanosensors. Current biology: CB. 2008;18:1288–1294. doi: 10.1016/j.cub.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 54.Yeung T, et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell motility and the cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 55.Giannone G, et al. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116:431–443. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 56.Giannone G, et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128:561–575. doi: 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong S, et al. Fibroblasts probe substrate rigidity with filopodia extensions before occupying an area. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17176–17181. doi: 10.1073/pnas.1412285111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patsialou A, et al. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital. 2013;2:e25294. doi: 10.4161/intv.25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gawden-Bone C, et al. A crucial role for beta2 integrins in podosome formation, dynamics and Toll-like-receptor-signaled disassembly in dendritic cells. Journal of cell science. 2014;127:4213–4224. doi: 10.1242/jcs.151167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Destaing O, et al. beta1A integrin is a master regulator of invadosome organization and function. Molecular biology of the cell. 2010;21:4108–4119. doi: 10.1091/mbc.E10-07-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choma DP, et al. Integrin alpha3beta1 directs the stabilization of a polarized lamellipodium in epithelial cells through activation of Rac1. Journal of cell science. 2004;117:3947–3959. doi: 10.1242/jcs.01251. [DOI] [PubMed] [Google Scholar]
- 62.Rabinovitz I, Mercurio AM. The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. The Journal of cell biology. 1997;139:1873–1884. doi: 10.1083/jcb.139.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dumbauld DW, et al. How vinculin regulates force transmission. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9788–9793. doi: 10.1073/pnas.1216209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lawson C, et al. FAK promotes recruitment of talin to nascent adhesions to control cell motility. The Journal of cell biology. 2012;196:223–232. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spuul P, et al. Importance of RhoGTPases in formation, characteristics, and functions of invadosomes. Small GTPases. 2014;5:e28195. doi: 10.4161/sgtp.28713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sit ST, Manser E. Rho GTPases and their role in organizing the actin cytoskeleton. Journal of cell science. 2011;124:679–683. doi: 10.1242/jcs.064964. [DOI] [PubMed] [Google Scholar]
- 67.Hoshino D, et al. Signaling inputs to invadopodia and podosomes. Journal of cell science. 2013;126:2979–2989. doi: 10.1242/jcs.079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. Journal of cell science. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- 69.Ridley AJ, et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 70.Jerrell RJ, Parekh A. Cellular traction stresses mediate extracellular matrix degradation by invadopodia. Acta biomaterialia. 2014;10:1886–1896. doi: 10.1016/j.actbio.2013.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parsons JT, et al. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nature reviews Molecular cell biology. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Osiak AE, et al. Subconfluent endothelial cells form podosomes downstream of cytokine and RhoGTPase signaling. Experimental cell research. 2005;307:342–353. doi: 10.1016/j.yexcr.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 73.Varon C, et al. Transforming growth factor beta induces rosettes of podosomes in primary aortic endothelial cells. Molecular and cellular biology. 2006;26:3582–3594. doi: 10.1128/MCB.26.9.3582-3594.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quintavalle M, et al. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. The Journal of cell biology. 2010;189:13–22. doi: 10.1083/jcb.200912096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgstaller G, Gimona M. Podosome-mediated matrix resorption and cell motility in vascular smooth muscle cells. American journal of physiology Heart and circulatory physiology. 2005;288:H3001–3005. doi: 10.1152/ajpheart.01002.2004. [DOI] [PubMed] [Google Scholar]
- 76.Brown XQ, et al. Effect of substrate stiffness and PDGF on the behavior of vascular smooth muscle cells: implications for atherosclerosis. Journal of cellular physiology. 2010;225:115–122. doi: 10.1002/jcp.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edlund S, et al. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Molecular biology of the cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harms BD, et al. Directional persistence of EGF-induced cell migration is associated with stabilization of lamellipodial protrusions. Biophysical journal. 2005;88:1479–1488. doi: 10.1529/biophysj.104.047365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baker AM, et al. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene. 2013;32:1863–1868. doi: 10.1038/onc.2012.202. [DOI] [PubMed] [Google Scholar]
- 80.Huynh J, et al. Substrate Stiffness Regulates PDGF-Induced Circular Dorsal Ruffle Formation Through MLCK. Cellular and molecular bioengineering. 2013;6 doi: 10.1007/s12195-013-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schrader J, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nature reviews Drug discovery. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Connolly EC, et al. Complexities of TGF-beta targeted cancer therapy. International journal of biological sciences. 2012;8:964–978. doi: 10.7150/ijbs.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.King TE, Jr, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. The New England journal of medicine. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 85.Spond J, et al. Inhibition of experimental acute pulmonary inflammation by pirfenidone. Pulmonary pharmacology & therapeutics. 2003;16:207–214. doi: 10.1016/S1094-5539(03)00026-9. [DOI] [PubMed] [Google Scholar]
- 86.Kagan HM. Intra- and extracellular enzymes of collagen biosynthesis as biological and chemical targets in the control of fibrosis. Acta tropica. 2000;77:147–152. doi: 10.1016/s0001-706x(00)00128-5. [DOI] [PubMed] [Google Scholar]
- 87.Luo Y, et al. A novel profibrotic mechanism mediated by TGFbeta-stimulated collagen prolyl hydroxylase expression in fibrotic lung mesenchymal cells. The Journal of pathology. 2015;236:384–394. doi: 10.1002/path.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xiao Q, Ge G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer microenvironment: official journal of the International Cancer Microenvironment Society. 2012;5:261–273. doi: 10.1007/s12307-012-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer metastasis reviews. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cox TR, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015;522:106–110. doi: 10.1038/nature14492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 91.Contente S, et al. Expression of gene rrg is associated with reversion of NIH 3T3 transformed by LTR-c-H-ras. Science. 1990;249:796–798. doi: 10.1126/science.1697103. [DOI] [PubMed] [Google Scholar]
- 92.Min C, et al. The tumor suppressor activity of the lysyl oxidase propeptide reverses the invasive phenotype of Her-2/neu-driven breast cancer. Cancer research. 2007;67:1105–1112. doi: 10.1158/0008-5472.CAN-06-3867. [DOI] [PubMed] [Google Scholar]
- 93.Saika S, et al. Effect of fibrostatin C, an inhibitor of prolyl 4-hydroxylase, on collagen secretion by human Tenon’s capsule fibroblasts in vitro. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 1996;234(Suppl 1):S214–222. doi: 10.1007/BF02343075. [DOI] [PubMed] [Google Scholar]
- 94.Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Frontiers in bioscience. 2014;19:687–706. doi: 10.2741/4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olson MF. Applications for ROCK kinase inhibition. Current opinion in cell biology. 2008;20:242–248. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kleikers PW, et al. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. Journal of molecular medicine. 2012;90:1391–1406. doi: 10.1007/s00109-012-0963-3. [DOI] [PubMed] [Google Scholar]
- 97.Maher BM, et al. Statins alter neutrophil migration by modulating cellular Rho activity–a potential mechanism for statins-mediated pleotropic effects? Journal of leukocyte biology. 2009;85:186–193. doi: 10.1189/jlb.0608382. [DOI] [PubMed] [Google Scholar]
- 98.Chauhan VP, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nature communications. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Keizman D, et al. Angiotensin system inhibitors and outcome of sunitinib treatment in patients with metastatic renal cell carcinoma: a retrospective examination. European journal of cancer. 2011;47:1955–1961. doi: 10.1016/j.ejca.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao Y, et al. Ang II-AT1R increases cell migration through PI3K/AKT and NF-kappaB pathways in breast cancer. Journal of cellular physiology. 2014;229:1855–1862. doi: 10.1002/jcp.24639. [DOI] [PubMed] [Google Scholar]
- 101.Mair M, et al. JAK-STAT signaling in hepatic fibrosis. Frontiers in bioscience. 2011;16:2794–2811. doi: 10.2741/3886. [DOI] [PubMed] [Google Scholar]
- 102.Gritsina G, et al. Targeted Blockade of JAK/STAT3 Signaling Inhibits Ovarian Carcinoma Growth. Molecular cancer therapeutics. 2015;14:1035–1047. doi: 10.1158/1535-7163.MCT-14-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Quintas-Cardama A, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sanz-Moreno V, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer cell. 2011;20:229–245. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]