

ABSTRACT
Carfilzomib (Kyprolis®), a second generation proteasome inhibitor, is FDA approved for single-agent use among relapsed/refractory multiple myeloma (MM). To enhance the therapeutic efficacy of carfilzomib, we sought to combine carfilzomib with other novel agents. TG02, a multi-kinase inhibitor, targets JAK2 and CDK9. The rationale for co-treatment with carfilzomib and TG02 is that both independently target Mcl-1 and most myeloma cells are dependent on this anti-apoptotic protein for survival. We observed at least additive effects using the combination treatment in MM cell lines and patient samples. To determine how the bone marrow environment affects the efficacy of the combination we conducted co-culture experiments with Hs-5 stromal cells. We also examined the mechanism of increased apoptosis by determining the affect on expression of the Bcl-2 family of proteins. We found that carfilzomib increases NOXA mRNA expression, as expected, and TG02 treatment caused a decrease in Mcl-1 protein but not mRNA levels. Consistent with this possibility, we find silencing CDK9 does not change carfilzomib sensitivity in the same manner as addition of TG02. Since changes in Mcl-1 protein occur in the presence of a proteasome inhibitor we hypothesize that regulation of Mcl-1 translation is the most likely mechanism. Taken together our data suggest that dual inhibition of Mcl-1 via decreased expression and the induction of its antagonist NOXA by the combination of carfilzomib and TG02 is active in myeloma and warrants further testing preclinically and in clinical trials. Moreover, regulation of Mcl-1 by TG02 is more complex than initially appreciated.
KEYWORDS: Apoptosis, Bcl-2, plasma cell dyscrasia
Abbreviations
- Cz
carfilzomib
- HMCL
human multiple myeloma cell lines
- MM
multiple myeloma
Introduction
Multiple Myeloma (MM) is a hematologic malignancy of bone marrow plasma cells. It is estimated that in the United States in 2015, 26,850 new cases of MM (16,090 in men and 12,760 in women) would be diagnosed, with an estimated 11,240 deaths.1 After the introduction of the concept of proteasome inhibition and immunomodulation among myeloma therapeutic options, the survival of myeloma patients has significantly improved. Newer treatment strategies aiming at novel mechanisms or improving the efficacy of currently available treatments by combining agents to enhance synergy are warranted to ensure continuing improvement in survival among MM patients.
Currently, carfilzomib achieved an FDA approval to treat relapsed and refractory multiple myeloma patients who have received at least 2 prior therapies, including treatment with bortezomib and an immunomodulation therapy.2 Carfilzomib is an irreversible proteasome inhibitor that targets the chymotrypsin-like activity of the proteasome.3 Although carfilzomib is active in this disease, responses to proteosome inhibitors can be improved with combination therapies.4,5
Plasma cells are terminally differentiated B cells that are responsible for antibody production.6 Due to the high rates of antibody production, plasma cells also have increased rates of protein turnover. The proteasome is responsible for the selective degradation of misfolded and high-turnover proteins.7,8 Therefore, both normal and malignant plasma cells are highly susceptible to proteasome inhibition.9 Apoptosis is induced when the proteasome is inhibited in part because of an accumulation of misfolded proteins which, leads to endoplasmic reticulum stress and the unfolded protein response.7,10-12
Apoptosis is a signaling cascade of events that leads to the controlled death of a cell, and is regulated by the Bcl-2 family of proteins.13 The Bcl-2 family of proteins consist of both pro- and anti-apoptotic proteins. The anti-apoptotic proteins, including Bcl-2, Bcl-xL and Mcl-1 work to inhibit the pro-apoptotic proteins. Upon apoptotic stimuli, pro-apoptotic proteins, Bax and Bak, will mulitmerize and induce mitochondrial outer membrane permeabilization (MOMP).14 This process allows cytochrome c to be released from the mitochondria and the subsequent activation of the caspase cascade. When caspases are activated they will cleave vital cellular substrates leading to the death of a cell.15
One hallmark of cancer is the ability to resist apoptotic signals.16 In order for MM cells to resist apoptosis they are dependent on the anti-apoptotic protein Mcl-1 for survival.17-19 Mcl-1 is an oncogene that promotes tumorigenesis by binding to pro-apoptotic proteins Bak and Bim, thus preventing the release of cytochrome c from the mitochondria and subsequent caspase activation.20 Given that most MM cells are dependent on Mcl-1 for survival it would be beneficial to inhibit Mcl-1 to increase the apoptotic response in these cells. NOXA is a BH3-only member of the Bcl-2 family of proteins.21 NOXA is an inhibitor of Mcl-1. When NOXA binds to Mcl-1, the pro-apoptotic protein Bim is released resulting in MOMP and ultimately cell death.22,23 Carfilzomib induces not only the unfolded protein response but also the expression of NOXA.24
Due to the established safety profile of carfilzomib, it is now being tested in combination therapies.5,25-27 Treating MM with a combination treatment is regarded as beneficial for many reasons, one being that multiple myeloma has clonal heterogeneity. Using a combination of drugs would allow for more cells in the heterogeneous population to be targeted.28 Combination treatments have been shown to synergistically increase apoptosis and demonstrate longer survival than single-agent treatments in patients.5,28-30
Since MM cells are dependent on Mcl-1, we sought to combine carfilzomib with other Mcl-1 targeting drugs. TG02 is a pyrimidine-based multi-kinase inhibitor that has been shown to target CDKs, JAK2 and FLT3.31-33 More interestingly, TG02 has been shown to increase Bax activation and decrease expression levels of anti-apoptotic protein Mcl-1.33 Therefore, the combination of carfilzomib and TG02 would allow us to target Mcl-1 via 2 different mechanisms. TG02 targets proteins that are involved with transcriptional regulation, such as CDK9. Therefore, proteins with a short half-life will be selectively depleted from the cell relative to long-lived proteins. Mcl-1 is a short half-life protein, which is an explanation as to why TG02 treatment can decrease Mcl-1 protein expression. We hypothesized that using the combination of carfilzomib and TG02 would allow dual inhibition of Mcl-1, the key anti-apoptotic protein essential for survival of MM cells.
Results
Four different human MM cell lines (HMCL), MM.1s, RPMI-8226, H929 and U266, were initially treated for 24 hours with increasing concentrations of carfilzomib or TG02. MM.1s were the most sensitive to carfilzomib treatment (IC50 6.2 nM) and H929 were the most resistant (IC50 31.6 nM) (Fig. 1A). MM.1s were also the most sensitive to TG02 treatment (IC50 101.1 nM) (Fig. 1B). Co-treatment with carfilzomib and TG02 for 24 hours resulted in at least additive cell death in all 4 lines (Fig. 1C). To better mimic the pharmacokinetics of carfilzomib in the clinic,34 pulse dosing of carfilzomib was also performed. When cells were pulsed for 1 hour with carfilzomib and TG02, then treated with only TG02 for an additional 23 hours, similar patterns of activity were observed compared with continuous dosing (Fig. 1D). However, higher concentrations of carfilzomib were required to reach an IC50. Activity of the combination was also observed in freshly isolated samples from relapsed/refractory MM patients (Fig. 1E). Cells were either unfractionated from the buffy coat (MM53 and MM55) or CD138+ plasma cells were isolated from bone marrow aspirates and treated (MM54) and myeloma cell death was measured by annexin V staining. The combination treatment in all 3 patient samples resulted in at least additive effects.
Figure 1.

Continuous carfilzomib and TG02 co-treatment results in at least additive cell death. Cells were plated at a density of 0.25 × 106 cells/mL and treated with the indicated concentrations of (A) carfilzomib, (B) TG02 or the (C) combination for 24 hours. (D) Cells were co-treated with carfilzomib (IC10 and IC50 concentrations) and TG02 for 1 hour, washed and then treated with TG02 for 23 hours. Cell death was determined via Annexin–V–FITC/PI flow cytometry. Data are presented as mean +/− SEM of at least 3 independent experiments. IC50 and IC10 significance are indicated above and below the mean values, respectively. (E) Ficoll isolated buffy coat from a myeloma patient BM aspirate was collected and washed with PBS. Plasma cells were either treated in the presence of buffy coat cells (MM53 and MM55) or CD138+ plasma cells were isolated and treated (MM54). Twenty-four hour apoptosis was determined by staining with anti-CD38, anti-CD45, and Annexin V-FITC. *p > 0.05, **p > 0.01, ***p > 0.001.
We next determined how the bone marrow environment influences the activity of this combination. HMCL were co-cultured with either Hs-5 stromal cells or the conditioned medium from these cells (Fig. 2). The Hs-5 stromal cells were able to protect against combination treatment induced apoptosis in MM.1s, RPMI-8226, and U266 cell lines. The protective effects of stromal cells appears to primarily impact carfilzomib, as the changes in cell death are greater with carfilzomib alone then with TG02 alone. Surprisingly, addition of Hs-5 cells or conditioned medium resulted in increased cell death in MM.1s when cells were treated with TG02 alone. This suggests that the Hs-5 protection that is effecting carfilzomib induced cell death is not having an effect on TG02 treatment. The addition of conditioned medium was only able to protect against combination treatment induced apoptosis in RPMI-8226 cells. Despite protection by the Hs-5 stromal cells and the conditioned medium, cell death was greater in the combination treatment in all 3 cell lines compared to single agent treatment.
Figure 2.
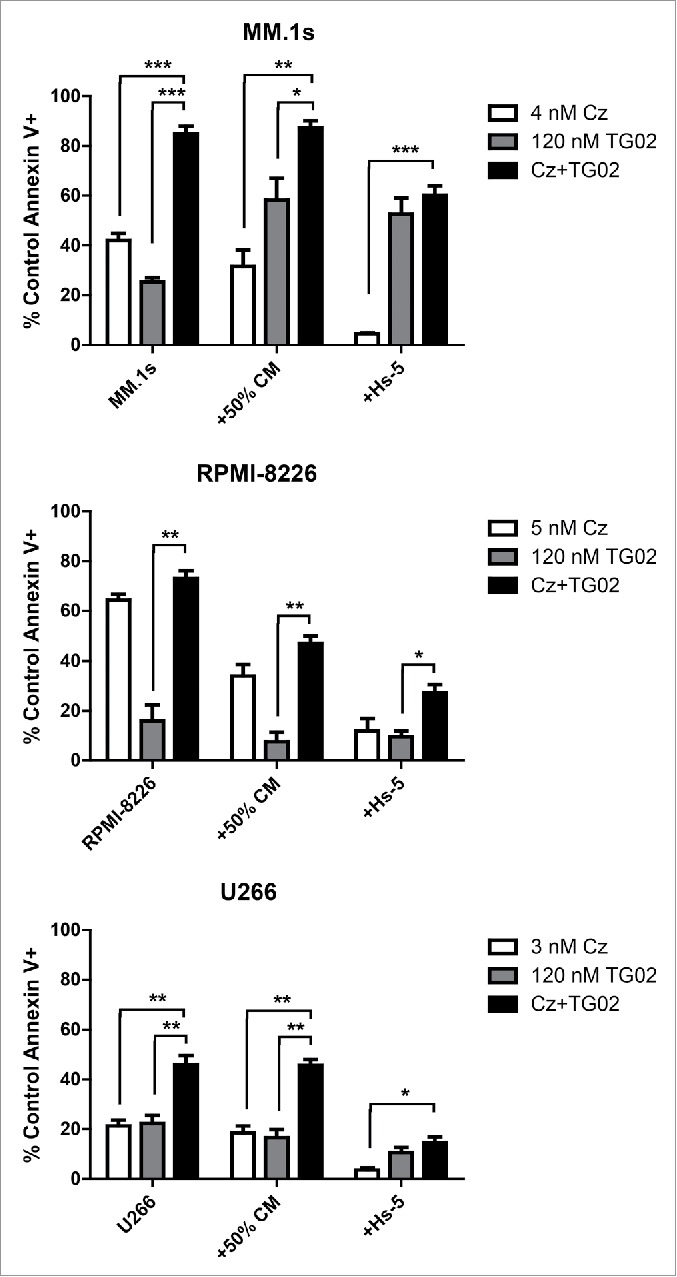
Co-culture with Hs-5 cells protected against combination treatment in 3 cell lines, while addition of Hs-5 conditioned medium was protective in only RPMI-8226. Hs-5 cells were plated at a density of 0.05 × 106 cells/mL and allowed to grow for 48 h. MM cells were added to the Hs-5 cells after 48 h, 30 min prior to drug treatment. Conditioned medium was collected and filtered then diluted to 50% and added to MM cells. 24 h cell death was assessed via Annexin-V-FITC/PI (control and conditioned medium treated cells) or Annexin-V-FITC/anti-CD38-PE (Hs-5 co-cultured cells) flow cytometry. Data in are presented as mean +/− SEM of at least 3 independent experiments.
We next determined the molecular basis for the increased apoptosis of this combination. Since the rationale for this combination was based on altering the expression of members of the Bcl-2 family, we initially focused on changes in the expression of both pro- and anti-apoptotic Bcl-2 family members. HMCL were treated with carfilzomib, TG02 or the combination for 6 hours. Cells were isolated for RT-qPCR to determine mRNA levels of the Bcl-2 family. Consistent with our hypothesis, treatment with carfilzomib caused an increase in NOXA, an inhibitor of Mcl-1, at the mRNA level in RPMI-8226 and H929 cells (Fig. 3A). When comparing mRNA levels of the other pro-apoptotic proteins, treatment with carfilzomib caused a decrease in Bmf expression in all cell lines except MM.1s. Bmf mRNA was significantly increased in all 4 cell lines with TG02 treatment, while Bid and Bim mRNA expression was significantly decreased (Fig. 3A).
Figure 3.

Treatment with carfilzomib causes an increase in NOXA mRNA and TG02 causes a decrease in Mcl-1 protein. Cells were plated at a density of 0.25 × 106 cells/mL and treated with carfilzomib, TG02 or the combination for 6 hours. RT-qPCR results for (A) pro-apoptotic and (B) anti-apoptotic mRNA expression levels compared to control were determined and presented as mean +/− SEM of at least 3 independent experiments. * p > 0.05, ** p > 0.01, *** p > 0.001. (C) Protein lysates were obtained after 6 hour carfilzomib, TG02 or the combination treatments (IC50) using RIPA buffer plus protease inhibitor cocktail. . 20 μg of protein were subjected to Western blot analysis with the antibodies shown. Numbers shown represent the quantified relative expression of Mcl-1 normalized to GAPDH.
The mRNA levels of the anti-apoptotic proteins were also determined using quantative real time PCR. Bcl-xL was increased with TG02 treatment in all 4 cell lines and Bcl-2 and Mcl-1 mRNA was decreased in 3 of the cell lines tested with carfilzomib treatment. We hypothesized that treatment with TG02 would result in a decrease in Mcl-1 expression, however, mRNA levels did not change following TG02 treatment (Fig. 3B).
To further investigate the effect of this combination on Bcl-2 expression we determined the protein expression for the Bcl-2 family of proteins after single and combination drug treatments. HMCL were treated with carfilzomib, TG02 or the combination for 6 hours. Cells were isolated for western blot analysis for the Bcl-2 family of proteins. Surprisingly, there was no increase in Bcl-xL or Bim protein following TG02 treatment, despite an increase in mRNA levels (Fig. 3C). Although there was no change in the Mcl-1 mRNA expression following TG02 treatment, TG02 did cause a decrease in Mcl-1 protein expression (Fig. 3C). Since the loss of Mcl-1 at the protein level occurs in the presence of carfilzomib, a proteasome inhibitor, the effect of TG02 is unlikely to be due to increased protein degradation. Therefore, we hypothesized that the decrease in Mcl-1 protein following TG02 treatment was due to translational regulation. TG02 has been shown to inhibit cyclin dependent kinase 9 (CDK9).32 To determine if TG02 was acting to inhibit CDK9, thus causing a decrease in Mcl-1 protein expression, we transiently knocked down CDK9 and treated the cells with carfilzomib, TG02 or the combination. The knockdown was verified using protein gel blot analysis (Fig. 4A). CDK9 silencing caused a decrease in Mcl-1 protein levels in MM.1s, although there was no difference in carfilzomib induced death was observed when comparing the siControl and siCDK9 transfected cells (Fig. 4B). To determine if TG02 is working via CDK9 to inhibit transcription of Mcl-1 we determined the phosphorylation status of the large subunit of RNA polymerase II, Rpb1. Knockdown of CDK9 had a minimal effect on Rpb1 phosphorylation. Treatment with TG02 did cause a decrease in Rpb1 phosphorylation, thus TG02 is working to inhibit transcription but not via inhibition of CDK9. Therefore, TG02 is likely to be working through a different mechanism than CDK9 inhibition in these cells.
Figure 4.
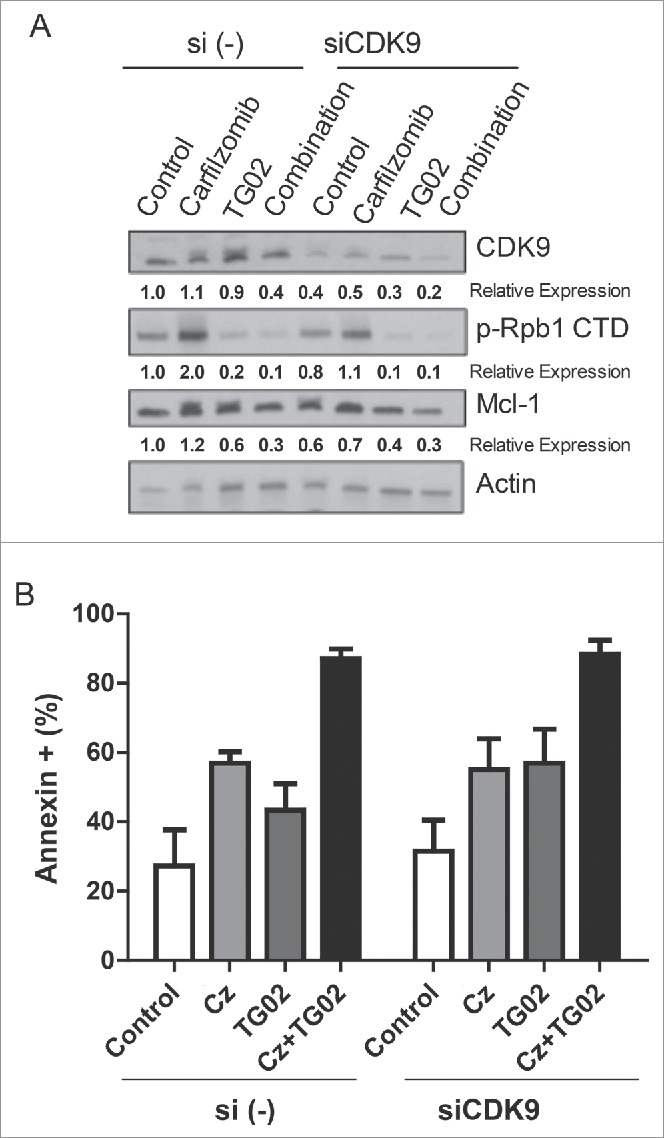
The decreased in Mcl-1 protein expression is not due to CDK9 inhibition. MM.1s cells were treated with siCDK9 or si (control) for 24 hours and then treated with carfilzomib, TG02 or the combination for an additional 24 hours. (A) Knockdown of siCDK9 was determined using protein gel blot and densitometry analysis. Relative expression, normalized to actin, of the indicated proteins are shown below the corresponding bands. (B) Cell death was determined via Annexin-V-FITC/PtdIns flow cytometry. Data are presented as mean +/− SEM of at least 3 independent experiments.
In order to further validate that Mcl-1 is the key player in the combination treatment induced apoptosis, we created RPMI-8226 cells that over expressed the anti-apoptotic proteins, Mcl-1, Bcl-2 and Bcl-xL. These cells were treated for 24 hours with increasing concentrations of carfilzomib or TG02 as well as the combination. Consistent with the possibility of the decrease in Mcl-1 protein being due to a regulation of translation mechanism, overexpression of Mcl-1 in RPMI-8226 cells confers significantly less protection than over expression of Bcl-2 or Bcl-xLwhen cells were treated with carfilzomib alone (Fig. 5A). 24 hour combination treatments with increasing doses of TG02 and 2 concentrations of carfilzomib had a similar result. The overexpression of Mcl-1 confers significantly less protection (Fig. 5C). This verifies that Mcl-1 protein is the key player in the combination treatment induced apoptosis.
Figure 5.

Overexpression of Mcl-1 in RPMI-8226 cells confers significantly less protection than over expression of Bcl-2 or Bcl-xL.RPMI-8226 cells overexpressing the anti-apoptotic proteins shown were treated with increasing doses of (A) carfilzomib (B) TG02 or (C) the combination for 24 hours. Cell death was determined via Annexin-V-FITC/PI flow cytometry. Data are presented as mean +/− SEM of at least 3 independent experiments. Statistical analysis for RPMI-8226 Mcl-1 over expressing cells only shown. *p > 0.05, **p > 0.01, ***p > 0.001.
Discussion
Multiple myeloma cells are dependent on Mcl-1 for survival.17-19 Therefore, we sought to induce dual inhibition of Mcl-1 by combining a proteasome inhibitor, carfilzomib, with a multi-kinase inhibitor, TG02. Individually these drugs could induce apoptosis in multiple myeloma cell lines, but in combination they had at least additive effects. This effect was also seen in the presence of stromal cells, mimicking the extracellular environment, and in relapsed/refractory multiple myeloma patient samples. This demonstrates that carfilzomib and TG02 work as a combination therapy and further testing preclinically and in clinical trials is warranted.
We next sought to determine the molecular mechanism by which the combination was working. We hypothesized that dual inhibition of Mcl-1 would cause increased apoptosis in multiple myeloma cells. It is known that carfilzomib induces the expression of NOXA and that NOXA acts to inhibit Mcl-1. Consistent with our hypothesis, NOXA had increased mRNA expression in RPMI-8226 and H929 cells when treated with carfilzomib. Carfilzomib also caused a decrease in Mcl-1 mRNA expression in 3 out of 4 cell lines, but the Mcl-1 protein expression in these cells lines increased. This is consistent with previous studies demonstrating Mcl-1 regulation by ubiquitination and proteasomal degradation.35,36
CDK9 is known to associate with a protein complex, TAK/P-TEFb, which functions as an elongation factor for RNA polymerase II. CDK9 phosphorylates RNA polymerase II on Serine 2, thereby activating it. Inhibition of CDK9 has been shown to cause a decrease in Mcl-1 expression attributed to the heightened dependence of labile protein on de novo transcription.37 TG02 has been found to inhibit CDK9 in a number of malignant cell types,32,33 causing a decrease in Mcl-1 protein expression. Therefore, we hypothesized that TG02 is inhibiting CDK9, thus causing a decrease in Mcl-1 protein expression. TG02 caused a decrease in Mcl-1 protein expression, as hypothesized, however only U266 cells demonstrated a decrease in Mcl-1 mRNA expression. Since it seemed unlikely that TG02 was working via CDK9, we formally tested the role of CDK9 in Mcl-1 expression and how CDK9 inhibition influenced carfilzomib-induced death. Transiently knocking down CDK9 did not have an effect on carfilzomib induced cell death in the same manner as TG02. Paradoxically, silencing CDK9 did affect TG02 activity. Treatment with TG02 did cause a decrease in Rpb1 phosphorylation, therefore, TG02 is working to inhibit transcription but not via CDK9.
The decrease in Mcl-1 protein expression is not a result of proteasomal degradation as the decreased in Mcl-1 protein was also seen in the combination with carfilzomib. However, we have not formally ruled out a role for selective autophagy of Mcl-1. Together these results point to a mechanism of translational regulation of Mcl-1 by TG02. Inhibition of Mcl-1 translation has been previously demonstrated as a means to induce cell death in tumor cells.38-40
To verify that Mcl-1 is the key target in the combination treatment we created cells that over express the anti-apoptotic proteins. Overexpression of Mcl-1 in RPMI-8226 cells confers significantly less protection compared to over expression of Bcl-xL. This further validates that Mcl-1 is the key target for this combination treatment.
We have demonstrated that the combination of carfilzomib and TG02 has at least additive effects in multiple myeloma cell lines and relapsed/refractory patient samples. We have shown that the additive effects of this combination are due to the dual inhibition of Mcl-1. Carfilzomib causes an increase in NOXA and TG02 causes a decreased in Mcl-1 protein but not mRNA. The mechanism by which TG02 causes a decrease in Mcl-1 requires additional investigation. Overall our results demonstrate a need for further testing of this combination preclinically. However, based on these and other promising preclinical studies with bortezomib 41 a phase I trial to test this combination for the treatment of multiple myeloma has been initiated (NCT01204164). Initial results from this trial were recently presented and demonstrated that this combination has a similar safety profile as TG02 alone and has activity in carfilzomib refractory patients.43
Materials and methods
Cell lines
U266, H929 and RPMI-8226 were obtained from ATCC. The MM.1s cell line was obtained from Dr. Steven Rosen (Northwestern University). Cells were cultured as previously described.42
Reagents
Propidium iodide (PI) was purchased from Sigma-Aldrich (P4170); Annexin-V–fluorescein isothiocyanate (FITC) was purchased from Biovision (1001–1000). Carfilzomib was generously provided by Onyx Pharaceuticals as part of their PRISM-NTP program. TG02 was generously provided by Tragara Pharmaceuticals.
Flow cytometric assay
Cells were collected at the various time points and treatments. 0.25–0.5 million cells were washed with phosphate buffered saline (PBS) and resuspended in 0.5 ml of FACS buffer (1% BSA in PBS containing 0.01% sodium azide) containing annexin V-FITC (BioVision 1001–1000) and propidium iodide (2 μg/ml Sigma). Cells were then measured with a BD FACSCanto II as described previously.20 Data were analyzed using FlowJo (TreeStar) software.
Pulse dosing experiment
Cell lines indicated were co-treated with carfilzomib (IC10 and IC50 concentrations) and TG02 for one hour, washed and then treated with TG02 for an additional 23 hours. Cell death was determined via Annexin-V-FITC/PtdIns flow cytometry.
Patient samples
Ficoll isolated buffy coat from a MM patient BM aspirate was collected and washed with PBS. Plasma cells were either treated in the presence of buffy coat cells or CD138+ plasma cells were isolated as previously described42 and treated. Twenty-four hour apoptosis was determined by staining with anti-CD38 (BD Biosciences 340927), anti-CD45 (BD Biosciences 348795), and Annexin V-FITC(BioVision 1001–1000). Samples were acquired with an IRB approved protocol.
Co-culture with Hs-5 stromal cells
Hs-5 cells were plated at a density of 0.05 × 106 cells/mL and allowed to grow for 48 h. MM cells were added to the Hs-5 cells after 48 h at a density of 0.25 × 106 cells/mL 30 min prior to drug treatment. Conditioned medium was collected and filtered then diluted to 50% and added to MM cells. 24 h cell death was assessed via Annexin-V-FITC/PI (control and conditioned medium treated cells) or Annexin-V-FITC/anti-CD38-PE (BD Biosciences 347687) (Hs-5 co-cultured cells) flow cytometry. Data in are presented as mean +/− SEM of at least 3 independent experiments.
Real time PCR
cDNA was prepared from RNA harvested at specified time points using the ABI high capacity cDNA kit (Applied Biosystems 4368814). Real-time PCR was performed using TaqMan gene expression master mix (ABI 4368814) with an ABI 9600 Fast thermocycler as described previously.42 The following probes were used: Bim (bcl2l11) Mm00437796_m1, Bid (BID) Hs00609632_m1, NOXA (PMAIP1) Hs00560402_m1, Bmf (BMF) Hs00372937_m1, Mcl-1 (mcl1) Mm00725832_s1, Bcl-2 (bcl2) Mm00477631_m1, Bcl-xL (bcl2l1) Mm00437783_m1, and GAPDH 4352932–0912031 were purchased from Applied Biosystems.
Immunoblotting
Cell lysate preparation and western blots were performed as described previously.20 Antibodies used were as follows: rabbit anti-BID pAb (Cell Signaling Technology 2002); rabbit anti–Puma pAb (Cell Signaling 12450); rabbit anti-Bim pAb (Chemicon AB17003); rabbit anti–Mcl-1 pAb (Stressgen PAB13415); rabbit anti–Bcl-xL pAb (13.6)26; mouse anti–Bcl-2 mAb (sc-509, Santa Cruz Biotechnology); rabbit anti-CDK9 mAb (Cell Signaling Technology C12F7); rabbit anti-phsopho Rpb1-CTD (Cell Signaling Technolog 4735) and the mouse anti-GAPDH mAb (Millipore AB2302). The ECL rabbit IgG, horseradish peroxidase-linked whole Ab (from donkey;GE Healthcare), and the anti–mouse IgG1-horseradish peroxidase conjugate (Santa Cruz sc-51625) were used as secondary antibody for Western blot.
siRNA
Silencing studies using small interfering RNAs (siRNAs) were obtained from Dharmacon RNA Technologies, selecting the ON-TARGETplus SMARTpool duplexes as the RNAi-specific technology platform. siRNA against human CDK9 and the siCON-TROL nontargeting siRNA[siRNA(−)] were used (L-003243-00-0005). siRNAs were electroporated into cells by nucleofection (Amaxa) following the manufacturer's instructions as previously described.20
Disclosures
- FB
Employee of Tragara Pharmaceuticals
- AN
Advisory board member for Onyx and Spectrum Pharmaceuticals
- JK
Consultant for Onyx and Millennium Pharmaceuticals, Novartis and Celgene. Research funding from Merck, Novartis and Onyx Pharmaceuticals
- SL
Consultant for Onyx and Millennium Pharmaceuticals, Bristol-Myers Squibb, Novartis, Janssen and Celegene
- LHB
Consultant for Onyx Pharmaceuticals and Novartis
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Shirin Kapur and Christopher Kirk for their advice regarding the use of carfilzomib. Support came from grants R01 CA127910 and 5 P30CA138292-07. LHB is supported by the TJ Martell Foundation and is a Georgia Research Alliance Cancer Scholar.
Contributions
KP designed, performed and analyzed experiments and wrote the manuscript. SMM designed and analyzed experiments and helped write the manuscript. SH and VAG designed and performed experiments. CS performed experiments and sample acquisition. FB analyzed data and helped to write the manuscript. AN helped write the manuscript and analyzed data. JK provided assistance in writing the manuscript and data analysis. SL analyzed data and helped write the manuscript. LHB conceived the idea, designed and analyzed experiments and wrote the manuscript.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2.Andreu-Vieyra C, Berenson JR. Carfilzomib in multiple myeloma. Exp Opin biol Ther 2014; 11:1685–1699; PMID: 25170966; http://dx.doi.org/22929803 10.1517/14712598.2014.953050v [DOI] [PubMed] [Google Scholar]
- 3.Zang Y, Thomas SM, Chan ET, Kirk CJ, Freilino ML, DeLancey HM, Grandis JR, Li C, Johnson DE. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin Cancer Res: Off J Am Assoc Cancer Res 2012; 18:5639-49; PMID:22929803; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lonial S, Boise LH. The future of drug development and therapy in myeloma. Semin Oncol 2013; 40:652-8; PMID:24135409; http://dx.doi.org/ 10.1053/j.seminoncol.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 5.Moreau P. The emerging role of carfilzomib combination therapy in the management of multiple myeloma. Exp Rev Hematol 2014; 7:265-90; PMID:24521249; http://dx.doi.org/ 10.1586/17474086.2014.873699 [DOI] [PubMed] [Google Scholar]
- 6.Tiburzy B, Kulkarni U, Hauser AE, Abram M, Manz RA. Plasma cells in immunopathology: concepts and therapeutic strategies. Semin Immunopathol 2014; 36:277-88; PMID:24740168; http://dx.doi.org/ 10.1007/s00281-014-0426-8 [DOI] [PubMed] [Google Scholar]
- 7.Delgado ME, Dyck L, Laussmann MA, Rehm M. Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis 2014; 5:e1011; PMID:24457955; http://dx.doi.org/ 10.1038/cddis.2013.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fennell DA, Chacko A, Mutti L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 2008; 27:1189-97; PMID:17828309; http://dx.doi.org/ 10.1038/sj.onc.1210744 [DOI] [PubMed] [Google Scholar]
- 9.Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood 2014; 124:1873-9; PMID:25097176; http://dx.doi.org/ 10.1182/blood-2014-05-578732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, et al.. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007; 110:3281-90; PMID:17591945; http://dx.doi.org/ 10.1182/blood-2007-01-065888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107:4907-16; PMID:16507771; http://dx.doi.org/ 10.1182/blood-2005-08-3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res 2007; 67:7395-405; PMID:17671210; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1015 [DOI] [PubMed] [Google Scholar]
- 13.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116:205-19; PMID:14744432; http://dx.doi.org/ 10.1016/S0092-8674(04)00046-7 [DOI] [PubMed] [Google Scholar]
- 14.Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science (New York, NY) 2006; 311:847-51; PMID:NOT_FOUND; http://dx.doi.org/ 10.1126/science.1115035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35:495-516; PMID:17562483; http://dx.doi.org/ 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 17.Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle (Georgetown, Tex) 2004; 3:1259-62; PMID:15467463; http://dx.doi.org/ 10.4161/cc.3.10.1196 [DOI] [PubMed] [Google Scholar]
- 18.Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002; 100:194-9; PMID:12070027; http://dx.doi.org/ 10.1182/blood.V100.1.194 [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002; 99:1885-93; PMID:11877256; http://dx.doi.org/ 10.1182/blood.V99.6.1885 [DOI] [PubMed] [Google Scholar]
- 20.Morales AA, Gutman D, Lee KP, Boise LH. BH3-only proteins Noxa, Bmf, and Bim are necessary for arsenic trioxide-induced cell death in myeloma. Blood 2008; 111:5152-62; PMID:18354037; http://dx.doi.org/ 10.1182/blood-2007-10-116889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science (New York, NY) 2000; 288:1053-8; PMID:10807576; http://dx.doi.org/15550399 10.1126/science.288.5468.1053 [DOI] [PubMed] [Google Scholar]
- 22.Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC, Hinds MG. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem 2005; 280:4738-44; PMID:15550399; http://dx.doi.org/ 10.1074/jbc.M411434200 [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17:393-403; PMID:15694340; http://dx.doi.org/ 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]
- 24.Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, et al.. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009; 114:3439-47; PMID:19671918; http://dx.doi.org/ 10.1182/blood-2009-05-223677 [DOI] [PubMed] [Google Scholar]
- 25.Gupta VA, Nooka AK, Lonial S, Boise LH. Clinical potential of carfilzomib in the treatment of relapsed and refractory multiple myeloma. Blood Lymphatic Cancer: Targets Ther 2013; 41-51; http://dx.doi.org/ 10.2147/BLCTT.431876 [DOI] [Google Scholar]
- 26.Shah JJ, Stadtmauer EA, Abonour R, Cohen AD, Bensinger WI, Gasparetto C, Kaufman JL, Lentzsch S, Vogl DT, Orlowski RZ, et al.. A multi-center phase I/II trial of carfilzomib and pomalidomide with dexamethasone (Car-Pom-d) in patients with relapsed/refractory multiple myeloma. ASH Annual Meeting Abstracts 2012; 120:74 [Google Scholar]
- 27.Kaufman JL, Zimmerman T, Rosenbaum CA, Nooka AK, Heffner LT, Harvey RD, Gleason C, Lewis C, Sharp C, Barron KW, et al.. Phase I study of the combination of carfilzomib and panobinostat for patients with relapsed and refractory myeloma: a multiple myeloma research consortium (MMRC) clinical trial. ASH Annual Meeting Abstracts 2014; 124:32 [Google Scholar]
- 28.Lonial S, Kaufman JL. The era of combination therapy in myeloma. J Clin Oncol: Off J Am Soc Clin Oncol 2012; 30:2434-6; PMID:22585685; http://dx.doi.org/ 10.1200/JCO.2011.40.6967 [DOI] [PubMed] [Google Scholar]
- 29.Lonial S, Mitsiades CS, Richardson PG. Treatment options for relapsed and refractory multiple myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res 2011; 17:1264-77; PMID:21411442; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1805 [DOI] [PubMed] [Google Scholar]
- 30.Orlowski RZ, Nagler A, Sonneveld P, Blade J, Hajek R, Spencer A, San Miguel J, Robak T, Dmoszynska A, Horvath N, et al.. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol: Off J Am Soc Clin Oncol 2007; 25:3892-901; PMID:17679727; http://dx.doi.org/ 10.1200/JCO.2006.10.5460 [DOI] [PubMed] [Google Scholar]
- 31.Pasha MK, Jayaraman R, Reddy VP, Yeo P, Goh E, Williams A, Goh KC, Kantharaj E. Preclinical metabolism and pharmacokinetics of SB1317 (TG02), a potent CDK/JAK2/FLT3 inhibitor. Drug Metab Lett 2012; 6:33-42; PMID:22372550; http://dx.doi.org/ 10.2174/187231212800229336 [DOI] [PubMed] [Google Scholar]
- 32.Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, Tan YC, Hu C, Jayaraman R, William AD, et al.. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012; 26:236-43; PMID:21860433; http://dx.doi.org/ 10.1038/leu.2011.218 [DOI] [PubMed] [Google Scholar]
- 33.Pallis M, Abdul-Aziz A, Burrows F, Seedhouse C, Grundy M, Russell N. The multi-kinase inhibitor TG02 overcomes signalling activation by survival factors to deplete MCL1 and XIAP and induce cell death in primary acute myeloid leukaemia cells. Brit J Haematol 2012; 159:191-203; PMID:22934750; http://dx.doi.org/ 10.1111/bjh.12018 [DOI] [PubMed] [Google Scholar]
- 34.Alsina M, Trudel S, Furman RR, Rosen PJ, O'Connor OA, Comenzo RL, Wong A, Kunkel LA, Molineaux CJ, Goy A. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin Cancer Res: Off J Am Assoc Cancer Res 2012; 18:4830-40; PMID:22761464; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-3007 [DOI] [PubMed] [Google Scholar]
- 35.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121:1085-95; PMID:15989957; http://dx.doi.org/ 10.1016/j.cell.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 36.Opferman JT. Unraveling MCL-1 degradation. Cell Death Diff 2006; 13:1260-2; PMID:16710358; http://dx.doi.org/ 10.1038/sj.cdd.4401978 [DOI] [PubMed] [Google Scholar]
- 37.Walsby E, Pratt G, Shao H, Abbas AY, Fischer PM, Bradshaw TD, Brennan P, Fegan C, Wang S, Pepper C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget 2014; 5:375-85; PMID:24495868; http://dx.doi.org/ 10.18632/oncotarget.1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong SE, Kim EK, Jin HO, Kim HA, Lee JK, Koh JS, Seol H, Kim JI, Park IC, Noh WC. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell Biol Toxicol 2013; 29:273-82; PMID:23942996; http://dx.doi.org/ 10.1007/s10565-013-9253-2 [DOI] [PubMed] [Google Scholar]
- 39.Kim SM, Yun MR, Hong YK, Solca F, Kim JH, Kim HJ, Cho BC. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol Cancer Therap 2013; 12:2145-56; PMID:23883584; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-1188 [DOI] [PubMed] [Google Scholar]
- 40.Zhou T, Li G, Cao B, Liu L, Cheng Q, Kong H, Shan C, Huang X, Chen J, Gao N. Downregulation of Mcl-1 through inhibition of translation contributes to benzyl isothiocyanate-induced cell cycle arrest and apoptosis in human leukemia cells. Cell Death Dis 2013; 4:e515; PMID:23449451; http://dx.doi.org/ 10.1038/cddis.2013.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Fernandez S, Ortiz-Ruiz MJ, Parrott T, Zaknoen S, Ocio EM, San Miguel J, Burrows FJ, Esparis-Ogando A, Pandiella A. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clin Cancer Res: Off J Am Assoc Cancer Res 2013; 19:2677-87; PMID:23532886; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2118 [DOI] [PubMed] [Google Scholar]
- 42.Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, Kaufman JL, Lee KP, Lonial S, Boise LH. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 2011; 118:1329-39; PMID:21659544; http://dx.doi.org/ 10.1182/blood-2011-01-327197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofmeister CC, Berdeja JG, Vesole DH, Suvannasankha A, Parrott T, Abonour R. TG02, an Oral CDK9-inhibitor, in combination with carfilzomib demonstrated objective responses in carfilzomib refractory multiple myeloma patients. Blood 2015; 126:3052 [Google Scholar]