

Abstract
Transmissible spongiform encephathalopathies or prion diseases are a group of neurological disorders characterized by neuronal loss, spongiform degeneration, and activation of astrocytes or microglia. These diseases affect humans and animals with an extremely high prevalence in some species such as deer and elk in North America. Although rare in humans, they result in a devastatingly swift neurological progression with dementia and ataxia. Patients usually die within a year of diagnosis. Prion diseases are familial, sporadic, iatrogenic, or transmissible. Human prion diseases include Kuru, sporadic, iatrogenic, and familial forms of Creutzfeldt–Jakob disease, variant Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker disease, and fatal familial insomnia. The causative agent is a misfolded version of the physiological prion protein called PrPSc in the brain. There are a number of therapeutic options currently under investigation. A number of small molecules have had some success in delaying disease progression in animal models and mixed results in clinical trials, including pentosan polysulfate, quinacrine, and amphotericin B. More promisingly, immunotherapy has reported success in vitro and in vivo in animal studies and clinical trials. The three main branches of immunotherapy research are focus on antibody vaccines, dendritic cell vaccines, and adoptive transfer of physiological prion protein-specific CD4+ T-lymphocytes. Vaccines utilizing antibodies generally target disease-specific epitopes that are only exposed in the misfolded PrPSc conformation. Vaccines utilizing antigen-loaded dendritic cell have the ability to bypass immune tolerance and prime CD4+ cells to initiate an immune response. Adoptive transfer of CD4+ T-cells is another promising target as this cell type can orchestrate the adaptive immune response. Although more research into mechanisms and safety is required, these immunotherapies offer novel therapeutic targets for prion diseases.
Keywords: neurodegeneration, immunization, spongiform encephalopathies, PrPC, PrPSc, tans-missible spongiform encephalopathies Creutzfeldt-Jacob disease, dendritice cells, Alzheimer disease
Video abstract
Introduction
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are progressive, fatal neurodegenerative disorders characterized by neuronal loss, spongiform degeneration, and activation of astrocytes or microglia. Prion diseases are rare in the population but result in a devastating neurodegenerative condition. They are believed to result from the transformation of the normal prion protein (PrPc) into aberrantly folded isoforms that are resistant to breakdown by proteases (PrPres). These aberrant prion proteins then accumulate and propagate in both the central nervous system (CNS) and the peripheral nervous system where they cause neurodegeneration, leading to dementia and rapid death.
Prevalence of prion diseases
Animal prion diseases include chronic wasting disease (CWD) in cervids (deer and elk), scrapie in sheep and goats, and bovine spongiform encephalopathy (BSE) in cattle. Scrapie was the first TSE to be described. CWD is extremely contagious and prevalent in both the wild and captive population, with prevalence as high as 30% and 100%, respectively.1,2 CWD has been detected in free-ranging cervids in 20 US states and two Canadian provinces.3 Of all the prion diseases, only BSE has been proven to be zoonotic.4
The highest incidence of a human prion disease was the famous 1950s outbreak of Kuru in the Fore tribe of Papua New Guinea, where the incidence was estimated to be as high as 20%.5 The human prion diseases include the sporadic, iatrogenic, and inherited forms of Creutzfeldt–Jakob disease (CJD), variant CJD (vCJD), Gerstmann–Sträussler–Scheinker (GSS) disease, and fatal familial insomnia (FFI). There are currently no proven therapeutic agents. Currently, the most prevalent human prion disease is sporadic CJD (sCJD), which is very rarely due to mutations in the prion gene.6 The most recent data from the Centers for Disease Control and Prevention reveal that there have been 478 deaths attributed to sCJD in the US alone or close to five people in 1 million in 2013. Worldwide trends estimate the prevalence of sCJD at 1–1.5 deaths per 1 million population per year.3 Clinical diagnosis of sCJD is made using a combination of neurological assessment, magnetic resonance imaging, electroencephalogram, and measurement of 14-3-3 or tau protein levels in cerebrospinal fluid, or brain biopsy. The 14-3-3 proteins are a family of conserved regulatory molecules and are increased in the cerebrospinal fluid.7 In the UK, familial prion diseases account for 5%–15% of TSEs.8 These data include familial CJD, which has a prevalence of ∼1 in a million people per year; GSS disease, which affects less than five in 100 million people/year; and FFI, which has only been diagnosed in ∼50 families.9–11
In the 1980s, the outbreak of BSE led to a new form of CJD. This new form was termed variant CJD or vCJD and was linked to the consumption of BSE-contaminated meat. The majority of TSE cases are believed to be transmitted by the oral route from recycled animal products in feed or environmental contamination. This has largely been minimized by removing animal protein from ruminant feeds. Remaining cases of BSE are thought to be atypical BSE, a spontaneous form of the prion disease.
Genetics
Mutations in the prion protein gene (PRNP) account for 10%–15% of all TSEs. Familial prion diseases resulting from mutations in the PRNP gene include GSS disease and FFI. Inherited prion disease in humans is associated with the mutation of T183A and other mutations.12,13 It is thought to act by disrupting glycosylation,12 promoting structural instability and misfolding,14,15 and accelerating conversion from α-helical to a β-sheet-rich conformation.12 Data on genetic TSEs were collected by the European CJD project from registries in Australia, Austria, Canada, France, Germany, Italy, the Netherlands, Slovakia, Spain, Switzerland, and the UK.16 The frequency of point and insertional mutations of the PRNP gene varies significantly between countries, with the most common mutation being E200K.
Codon 129 in the PRNP gene is the site for a common methionine (M)/valine (V) polymorphism. Within the Caucasian population, it is estimated that 52% of individuals are M homozygous (MM), 36% are heterozygous (MV), and 12% are V homozygous (VV).17 Gambetti et al18 observed that the phenotype of the prion disease, whether familial, sporadic, or infectious, was different depending on the affected individual’s genotype at codon 129. Therefore, it is postulated that codon 129 may act as a modifier of the prion disease phenotype in humans. This group also observed that human prion diseases are associated with two types of scrapie prion protein, and it is the type of scrapie protein (type 1 or 2) and the sequence variation at codon 129 that determines the disease phenotype.
The prion protein
The prevailing hypothesis of prion disease is that the normal physiological form of prion protein (PrPc) converts to the diseased form (PrPSc) due to a mutation in the PRNP gene or contact with another PrPSc protein. To add further complexity, within the PrPSc proteins, there are PrPSensitive (PrPsen) and PrPResistant (PrPres) forms that differ in their sensitivities to proteinase K digestion. The majority of PrPSc-infected tissue contains the PrPres form; however, this is not an absolute requirement.19 The PrPsen configuration has also been detected in infected tissue, suggesting that there could be a number of different conformations contributing to prion disease.20 A popular hypothesis is that the PrPc form offers neuroprotection, and so conversion to the PrPSc form attenuates this protection or possibly involves a gain in neurotoxicity. This theory is based on the observation that only oligomers, and not monomers, are infectious (Figure 1).21
Figure 1.
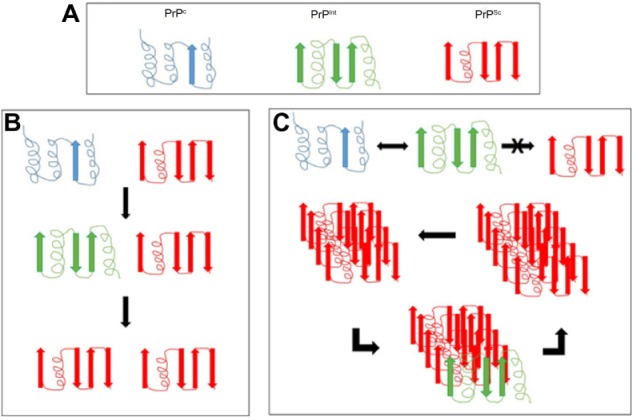
Proposed mechanisms of conversion of the prion protein.
Notes: (A) The physiological prion protein PrPc is composed mainly of alpha helices. It can convert to an intermediate form PrPInt or a pathological form PrPSc, composed mainly of beta sheets. There are many different intermediate forms, but only one is represented here for clarity. (B) Template-directed refolding proposes that PrPc can convert to PrPInt and then to PrPSc in the presence of a single PrPSc protein. (C) Nucleated polymerization proposes that a PrPc to PrPInt change is reversible; however, the PrPInt to PrPSc change can only occur in the presence of PrPSc oligomers.
Mechanisms of conversion
The molecular mechanisms of conversion of PrPc to PrPSc are still being determined. There are currently two plausible theories. The first is template-directed refolding (Figure 1B), which suggests that PrPSc acts as a substrate for the reaction converting PrPc to PrPSc. The newly formed PrPSc triggers a cascade of misfolding of neighboring PrPc proteins, resulting in self-propagation. The PrPSc is believed to lower the energy barrier of the reaction. The second theory is nucleated polymerization (Figure 1C), which suggests that this conversion is a thermodynamically controlled, noncatalytic process. This theory suggests that it is a nucleated polymerization reaction that is reversible and that PrPc is converted to PrPSc only in the presence of PrPSc aggregates or oligomers, which increases stability (Figure 1).21
Possible therapeutic targets
Theoretically, there are a number of potential targets available for research into a vaccine or cure for prion diseases. These include blocking conversion of PrPc into PrPSc, accelerating the clearance of amyloid PrP deposits in peripheral tissues and brain, stopping prion progression in secondary lymphoid organs, reducing brain inflammation, and promoting neuronal healing.
Current therapeutic approaches rely on the assumption that PrP proteins convert to PrPres proteins and subsequently accumulate in the CNS and peripheral nervous system. Based on this, recent targets include direct inhibition of this conversion, degradation of PrPres, altering PrPc expression and/or cell surface localization, and interference with vital accessory molecules such as glycosaminoglycans and fragment antigen binding.
Small molecules
There is interest in small molecules as therapeutic agents for prion disease.22 Due to space constraints, we will discuss only those that have been applied to human patients. The high-affinity prion receptor laminin (LPR/LR) is a potential candidate for therapy. Evidence suggests that LPR/LR acts as a receptor for both PrPc and PrPres isoforms23 and may aid PrPres binding and cellular internalization.24 Gauczynski et al23 reported that polysulfated glycans can inhibit PrPSc synthesis via inhibition of LPR/LR-dependent binding of prions to target cells. Leucht et al24 used antisense RNA to knock down the LPR/LR. More research into ligands targeting this receptor is required.
Pentosan polysulfate
Pentosan polysulfate (PPS) is a large polyglycoside molecule and showed early promise as a prophylactic against TSEs. Biologically, it is thought that PPS acts competitively with endogenous heparin sulfate proteoglycans as a coreceptor for PrP on the cell surface.25 PPS does not cross the blood–brain barrier after oral or parenteral administration.26 PPS was given into the cerebral ventricles of mice after prion infection and was associated with prolonged survival in all cases in a dose-dependent manner.27
There have been a limited number of case studies of patients with forms of CJD being treated with PPS. A 22-year-old man with vCJD was given continuous intraventricular PPS (32 µg/kg/d) commencing 19 months after the onset of symptoms for a total of 31 months. This experience suggests that PPS was safe and well tolerated and prolonged life expectancy when compared with natural history studies to a total of 52 months.28 An observational study in the UK assessed the effect of continuous intraventricular infusion of PPS in seven patients at different centers across the UK. The results showed that PPS was well tolerated over a large dose range (11–110 µg/kg/d) by all patients. Major complications of intraventricular catherization occurred in four of seven patients. Survival of all the seven patients exceeded mean survival of untreated patients.29
One issue with PPS treatment is that no safe and effective dose has been established for use in humans, and therefore, the dosing varies widely. It has been reported that doses ranging between 11 µg/kg/d, established from animal studies, and 110 µg/kg/d have been administered to patients with no overt toxicity or increase in seizures or hemorrhagic complications. However, it is difficult to extrapolate from this information, as only a small number of patients have been studied.
Quinacrine
Quinacrine, a derivative of acridine, is reported to inhibit PrPres formation in neuroblastoma cells infected with scrapie.30 The therapeutic mechanism is thought to involve the additional nitrogen side chain on its quinoline ring.31 Quinacrine is an antimalarial drug; therefore, its biological effects and complications have been extensively tested, providing an obvious choice for clinical trials. However, a number of clinical trials involving small numbers of patients with CJD have shown either limited or no beneficial effect using quinacrine treatment.32,33
Amphotericin B
Similarly, the macrolide polyene antibiotic amphotericin B showed some promise in early in vivo animal studies,34,35 but these results did not translate to the bedside due to a small clinical trial failing to observe any therapeutic improvement in two patients with CJD.36
Immunotherapies
Fifteen years ago, evidence that antibodies could block the propagation of prion diseases in vitro in infected cell lines37,38 and in vivo in scrapie-bearing mice39,40 was greeted with much enthusiasm. This vaccine theory of neurodegeneration is further supported by immunization studies using antibodies to Aβ and α- synuclein (α-syn) in transgenic mouse models of Alzheimer’s disease (AD)41 and Parkinson’s disease (PD).42
Antibody-based immunotherapies
Targeting disease-specific epitopes
Active and passive immunization strategies have been shown to be effective in the clearance of misfolded Aβ protein in transgenic mouse models of AD. Their efficacy and efficiency in human disease are currently being determined and are discussed later in this review.
Passive immunization
Madampage et al43 have developed a potential vaccine targeting a PrP epitope exposed after protein misfolding that produces PrPSc-specific antibody responses (Figure 2). This polyclonal antibody (SN6b) was reported to bind to a bovine prion protein with mutation of T194A [bPrP(T194A)]. This protein is a homolog of the human T183A mutation of PrPc that is known to cause early onset familial dementia.44 Binding of SN6b to bPrP(T194A) was confirmed by nanopore analysis, enzyme-linked immunosorbent assay, and immunoprecipitation of recombinant and cellular expressed forms of bPrP(T194A). Results showed consistent and sustained serum PrPSc-specific IgG levels with no adverse effects to the health of the animal. This group then vaccinated prion-susceptible Tga20 mice with the SN6b antigen and found no evidence of clinical disease up to 255 days postvaccination and no signs of proteinase K-resistant PrP.45 Homozygous Tga20 mice carry 60 copies of the PRNP gene and express tenfold higher PrP than wild-type (wt) mice, making them highly susceptible to prion disease.46 Therefore, the SN6b antibody holds great promise for vaccinating cervid populations, such as deer and elk, that are susceptible to CWD in the US and Canada.
Figure 2.

Immunology of antibody vaccines.
Notes: The PrPSc/antibody complex is too large to intercalate. (A) A mutation in the PRNP gene disrupts two hydrogen bonds between the second β-sheet and second α-helix exposing the SNP6 binding site and enabling the more flexible protein to interact with the receptor. (B) The PrPSc/antibody complex it too large to intercalate with the receptor.
Abbreviation: SNP6, single-nucleotide polymorphism 6.
Active immunization
Current immunization protocols obviously need to be improved to be useful in treating human prion diseases. Initially, vaccinations need to elicit a stronger and more specific anti-PrP immune response. One way to achieve this is with the use of an adjuvant such as deoxycytidyl-deoxyguanosine (CpG) oligodeoxynucleotides, which has been shown to stimulate T- and B-cell repertoires against PrP.47 A number of studies have utilized highly immunogenic viral and bacterial vectors encoding the PRNP sequence.48,49 Current successful vaccination strategies in mice have delivered the vaccine concomitantly or shortly after infection with prion proteins. As human prion diseases can be asymptomatic for years after infection, this strategy relies on earlier detection of prion diseases. Active immunization strategies using dendritic cells (DCs) and CD4+ T-cells are discussed later in this review.
Antiprion gene-based antibody approaches
More recently, gene-based antibody approaches have been trialed in cell and animal studies of neurodegenerative diseases such as AD, Huntington’s disease (HD), PD, and prion diseases. Intrabodies are recombinant antibody fragments that are engineered to block or modify the function of target proteins. Intrabodies show particular promise in prion diseases, as they can be specifically selected against conformational PrPC epitopes.50
Challenges for disease-specific epitope immunotherapy
The main challenge in developing effective immunotherapies for prion diseases is self-tolerance. As the PrPSc protein is a modified version of PrPc, the body’s immune system does not recognize it as foreign and therefore does not mount a humoral response. Wt mice challenged with prion proteins do not develop a characteristic immune response.51 PrPSc is hydrophobic, which suggests that it is resistant to proteolytic degradation by antigen-presenting cells (APCs) resulting in impaired presentation to T-cells.52 To counteract this problem of self-tolerance, a number of approaches have been made using adjuvants and immunization regimens.
PrP is known to be highly tolerogenic, and therefore, the T- and B-lymphocytes that escape tolerance and are thus available for vaccination appear to be both quantitatively and qualitatively impaired. Due to immune selection, the lymphocytes with high self-reactivity are selected against. This means that the lymphocytes that recognize PrP with high specificity and affinity, and would therefore be the most useful therapeutically, are destroyed by the immune system. This may explain why it is difficult to generate a robust immune response against PrP using current vaccine strategies.
A further challenge to the development of an effective vaccine is the struggle to deliver antibodies into the CNS, as they generally cannot cross the blood–brain barrier. Current in vivo studies in mice have overcome this limitation by delivering antibodies through intraventricular infusion via an osmotic pump.53 This delivery method has been shown to be successful in delaying disease onset and even attenuating disease progression when treatment was commenced up to 120 days postinoculation. A number of groups have developed single-chain variable fragment (Fv) antibodies that can penetrate the brain more readily owing to their low molecular weight.54,55 A recent advancement in this field has been the insertion of single-chain Fv antibody genes directed against the 37/67 kDa laminin receptor (LRP/LR), a known prion receptor, into specific viral vectors, for example, adeno-associated vectors, to increase brain penetrability of antibodies in scrapie-infected mice.56 The results demonstrated that disease progression was slowed but ultimately all mice succumbed to the neurological lesions.
Challenges to the use of antibody vaccines
A significant challenge to developing immunotherapy for prion diseases is the ability of the treatment to distinguish between self and non-self. Classically, T- or B-lymphocytes, which have receptors for recognizing self-proteins, are destroyed or prevented from initiating an immune response.57 Therefore, a central problem in developing effective therapy is overcoming tolerance. This can be achieved in a number of ways. First, the conformational changes in the affected protein reveal disease-specific epitopes (DSEs) that are hidden in the conventional folding of the protein. These DSEs can be targeted with antibodies in an attempt to neutralize them. The second major challenge is access to the CNS via the blood–brain barrier.
A prion vaccine could be extremely useful in at-risk populations or individuals; however, the rate of prion disease in humans is not large enough to justify vaccination in whole populations. Vaccines could be given to prion researchers, those with a genetic propensity toward the disease, or those individuals suspected of consuming prion-infected meat or other sources of contamination. A prion vaccination could also be useful for for pre-manifest individuals with genetic mutations for familial prion disease. One substantial limitation to the development of an effective vaccine is to target the misfolded proteins and preserve the function of the healthy tissue. Classically, there is a clear difference between self and non-self; however, the distinction in prion diseases is less clear. In prion diseases, “self” is the correctly folded protein, while “non-self” is the misfolded protein. A logical target for vaccine candidates are DSEs. DSEs elicit strong immune responses that are highly specific to the misfolded protein. Vaccines based on DSEs have already been targeted in human diseases such as prion diseases58,59 and AD.60 Further research is required to determine the specificity of DSE-induced antibody responses to clarify that they do not recognize correctly folded proteins.
There is a concern that targeting the ubiquitous cell surface PrPc protein may have negative consequences. Systemic autoreactive PrPc antibodies could interfere with the function of PrPc, which could lead to inappropriate cell signal activation or stimulate suppressor T-lymphocytes.61 There is evidence that antibodies that crosslink PrPc antibodies in the brain could induce apoptosis;62 however, a more recent study by Klohn et al63 contradicts this by reporting no observable apoptosis using a range of antibodies, thus questioning the neurotoxicity of these antibodies. In addition, antibody binding to PrPc causes alterations in signal transduction in human neurons.64 Therefore, the goal is to elicit a PrPSc-specific effective immune response that will not develop autoreactive PrPc antibodies and therefore limit adverse effects. One way to bypass this issue of self-tolerance is to utilize cell-based vaccines such as DC or CD4+ cell therapy.
Cell-based immunotherapies
DC vaccines
In vitro studies have demonstrated that DCs can be specifically targeted for use as a vaccine.65,66 DCs are taken from a patient; differentiated and expanded from bone marrow, peripheral, or cord blood precursors; loaded with antigen; and then reinfused into the same patient. The clear advantage is that the antigen can target the correct APC population that has already matured. This technique can avoid issues with antigen being delivered to irrelevant DC or APC populations that could result in anergic T-cells or T-cells skewed toward an inappropriate inflammatory response such as T helper (TH) 17 cells.
Vaccination with antigen-pulsed or antigen-transduced DCs to bypass self-tolerance is based on the knowledge that antigen presentation to DCs is an absolute requirement for a successful immune response (Figure 3). The current dogma suggests that DCs can activate naive T-cells in order to initiate a primary immune response. Successful activation of a naive T-cell is a three-step process. First, an antigen-specific signal is required between the T-cell receptor and the antigen–major histocompatibility complex (MHC) of the DCs. Second, costimulatory molecules such as CD80, CD86, and CD40 on the surface of the APCs act to amplify the signal. Third, APCs secrete a number of cytokines to direct the T-cell milieu (Figure 3). Studies utilizing antigen-loaded DCs in cancer models have had some success with generating CD8+ cytotoxic T-cells that are able to kill cells expressing tumor-specific antigens.67
Figure 3.
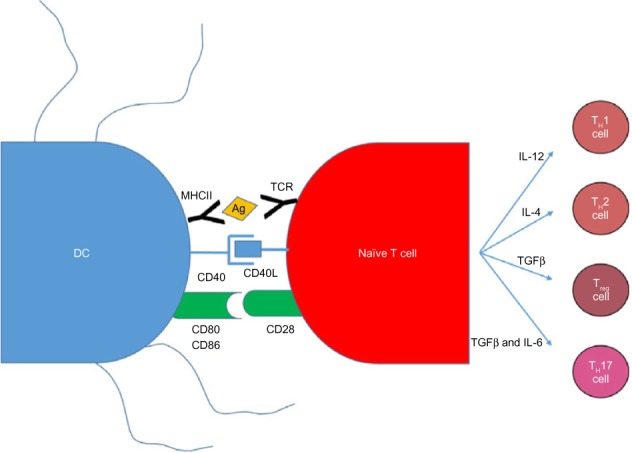
DC and naive T-cell interactions produce an adaptive immune response.
Notes: There are three signals required to initiate an adaptive immune response. The first is the presentation of antigen by the DCs to the T-cell via the MHC II–TCR interaction. Second, costimulatory molecules on the DCs such as CD40, CD80, and CD86 bind to their respective receptors on the T-cell to amplify the signal. Third, cytokines produced by the now activated T-cell skew the T-cell response toward a TH1, TH2, Treg, or TH17 response.
Abbreviations: DC, dendritic cell; IL-4, interleukin 4; IL-6, interleukin 6; IL-12, interleukin 12; MHCII, major histocompatibility complex class II; TCR, T-cell receptor; TGFβ, transforming growth factor beta; Treg, T regulatory cells; TH1, T helper 1; TH2, T helper 2; TH17, T helper 17.
There have been a number of small trials reporting some success with antigen-loaded DC therapy in a range of cancers. In a small study of 12 patients with advanced colon or nonsmall-cell lung cancer, immunization with DCs loaded with carcinoembyrogenic antigen produced carcinoembyrogenic antigen-specific CD8+ cytotoxic T-cells.68 Furthermore, in vivo treatment with the hematopoietic growth factor Flt3 ligand expanded DCs 20-fold. Two patients demonstrated dramatic tumor regression, one showed a mixed response, and two had stable disease. In another trial, 16 patients with advanced melanoma were immunized with peptide- or lysate-pulsed DCs in the presence of granulocyte–macrophage colony-stimulating factor and interleukin-4 (IL-4).69 All the patients tolerated the vaccine well, with no signs of developing autoimmunity. The results showed that vaccination induced a delayed-type hypersensitivity reaction to peptide-pulsed DCs in eleven patients in addition to recruitment of peptide-specific cytotoxic T-lymphocytes. This study demonstrated that antigen-specific immunity could be successfully induced during DC vaccination.
The modest success of DC vaccination in cancer was then translated into prion diseases. A study published 10 years ago revealed that two MHC class II-restricted 30-mer peptides of PrP (PrP98–127 and PrP158–187), when combined with an adjuvant, could strongly stimulate CD4+ T-cells in PrP-deficient (PrP−/−) but not Wt mice.70 Bachy et al71 extended this research to demonstrate that DC pulsed twice with these peptides could overcome tolerance in Wt mice by generating CD4+ T-cells with the ability to produce IL-4 and interferon-γ. Interestingly, the PrP158–187 peptide produced an IL-4 and interferon-γ response, whereas the PrP98–127 peptide produced only an IL-4 response. Using the PrP98–127 peptide to pulse DC, Bachy et al71 were able to demonstrate modest success in delaying disease progression of scrapie (strain 139A) in mice. Of ten mice infected, two never succumbed to disease and the remaining eight showed prolonged survival compared with controls.
Adoptive transfer of CD4+ T-cells
Another logical choice for designing a specific immunotherapy for prion disease is the CD4+ lymphocyte, as it plays a crucial role in orchestrating the adaptive immune response. This cell type can generate a wide range of immunogenic responses as they can differentiate into a plethora of cell types, including TH1, TH2, TH17, or T regulatory cells (Figure 4).72–74 It is the DC that controls the CD4+ cell lineage and can be modified by altering DC maturation conditions or by reprogramming the CD4+ T-cell.75
Figure 4.

The potential roles of CD4+ cells in prion disease.
Notes: CD4+ cells can be skewed into a TH2 or Treg phenotype in prion disease. TH2 cells may aid in the differentiation of antibody-producing plasma cells from B-cells. TH2 cells could increase the number and efficiency of secreted therapeutic antibodies by inducing an Ig class switch that could aid treatment of prion or neurological diseases. They may also be beneficial in advanced or familial prion disease by secreting proinflammatory cytokines that may attract macrophages to the brain in an attempt to clear prion deposits. Treg cells can secrete the cytokines IL-10 or TGFβ to decrease inflammation when necessary.
Abbreviations: Ig, immunoglobulin; IgA, immunoglobulin A; IgG, immunoglobulin G; IL-4, interleukin 4; IL-10, interleukin 10; IL-13, interleukin 13; Treg, T regulatory cells; TH2, T helper 2; TGFβ, transforming growth factor beta.
There are a number of theories regarding the potential role of CD4+ cells in prion diseases. First, in their TH2 role, they may aid in the differentiation of antibody-producing plasma cells from B-cells. TH2 cells could increase the number and efficiency of secreted therapeutic antibodies by inducing an immunoglobulin switch. CD4+ cells could also be beneficial in advanced disease or in familial or sporadic forms of prion disease by secreting proinflammatory cytokines and chemokines (Figure 4). These proinflammatory cytokines and chemokines could potentially attract macrophages to the brain and activate microglial cells to clear prion deposits. T regulatory cells could decrease inflammation through the release of IL-10 or transforming growth factor beta (Figure 4). To attempt to test some of these hypotheses, Gourdain et al76 extracted CD4+ cells from PrP-deficient mice and injected them into histocompatible mice sensitive to scrapie. They discovered that the CD4+ cells were still able to proliferate and produce cytokines up to 3 months after transfer, suggesting that these cells were somewhat resistant to tolerance. The transferred CD4+ cells did require regular antigen injections to remain functional. Additionally, these transferred CD4+ cells were able to protect donor mice by significantly attenuating the progression of prions in secondary lymphoid organs and were able to delay disease progression. Interestingly, PrPc-specific circulating antibodies were low, suggesting that the functional response was not mediated by antibodies.
This team also generated a transgenic mouse that expresses the β-chain of a T-cell receptor recognizing the PrP epitope presented by the MHC class II complex. Iken et al77 demonstrated that transgene-bearing T-cells offer protection from scrapie infection through a cell-mediated pathway involving IL-4 production. These results suggest that cell-mediated immunity may not be as harmful in prion diseases as once believed and may even be beneficial if adequately controlled. Adoptive transfer of PrP-sensitized CD4+ cells could promote humoral, cell-mediated, or regulatory responses, coordinate adaptive and innate immunity, and have long-lasting effects.
Challenges to cell-based immunotherapies
One substantial challenge in developing an effective cell-based strategy is in diagnosing the disease before significant brain damage occurs. Most of the current immunotherapies under development in animals and humans are only beneficial if administered in conjunction with or shortly after infection with a prion protein. A second limitation is the lack of access of immune-based therapies into the CNS. The third limitation is tolerance to self-PrP. More research is needed into earlier detection of prion diseases if an effective immunotherapy is to be developed that can act on the disease to delay or even reverse the pathological process.
Relationships to other neurodegenerative conditions
Protein misfolding is not unique to prion diseases. In humans, AD, PD, HD, and amyotrophic lateral sclerosis (ALS) have all been related to protein misfolding and propagation – the so called prion hypothesis of neurodegenerative disorders (Table 1). One major distinguishing feature of prion diseases is that they can be transmitted; however, recent evidence suggests that AD, PD, HD, and ALS, while not transmissible between individuals, can also self-propagate, as has recently shown for multiple system atrophy – an α-synucleinopathy.78–80 Therefore, research into a prion vaccine might be helpful in developing treatments that could halt the progression of other neurodegenerative disorders.
Table 1.
Comparison of prion-like mechanisms and neurotoxicity in neurodegenerative diseases
| Disease | Misfolded protein | Studies revealing evidence for self-propagation | Neuronal loss | Fatal disease |
|---|---|---|---|---|
| Prion diseases | PrPSc | 19,20 | Yes | Yes |
| AD | Aβ | 81–84 | Yes | Yes |
| Tau | 85 | Yes | Yes | |
| PD | α-syn | 88 | Yes | Yes |
| Multiple system atrophy | α-syn | 78 | Yes | Yes |
| ALS | SOD1 | 93 | Yes | Yes |
| TDP-43 | 96 | Yes | Yes | |
| HD | PolyQ repeat | 95 | Yes | Yes |
Notes: Evidence of self-propagation of the causative agent in a number of neurodegenerative diseases. The majority of evidence comes from studies where brain homogenates from sick donor mice were injected either intracerebrally or intraperitoneally into recipient mice. Donor mice were analyzed for disease symptoms, accumulation, and self-propagation of the misfolded protein and, in some cases, travel of the misfolded protein from the injection site to other areas of the brain.
Abbreviations: AD, Alzheimer’s disease; PD, Parkinson’s disease; ALS, amyotrophic lateral sclerosis; HD, Huntington’s disease; SOD1, superoxide dismutase 1; PolyQ, polyglutamine.
It was originally thought that misfolded protein aggregates were a consequence rather than a causative agent of neurodegeneration in AD, PD, HD, and ALS. Amyloidogenic proteins in these diseases–Aβ42 and tau for AD, α-syn for PD, polyglutamine (PolyQ) repeat expansions in HD, super-oxide dismutase 1 (SOD1) and TDP-43 for ALS–can induce misfolding in their healthy structural counterparts (Table 1). Thus, the misfolded proteins are involved in disease initiation and progression.
One obvious distinction between prion diseases and AD, PD, HD, and ALS is that prion diseases can be both infectious (ie, from an exogenous source) and self-propagating (able to convert neighboring proteins to the pathologic form). Conversely, it is extremely unlikely that there is an exogenous infectious agent involved in these neurodegenerative conditions; however, there is evidence that they are self-propagating.
Prion and Alzheimer’s link
AD is characterized by Aβ plaques in the brain and neurofibrillary tangles of hyperphosphorylated tau protein. The first evidence of a prion-like mechanism in AD came from primate studies 20 years ago. Human brain tissue from patients with AD was injected into primate cerebrums, and results showed transmissibility of Aβ plaques.81 These data were strengthened by transgenic mouse models of AD, demonstrating that Aβ plaques could be generated in transgenic mice injected intracerebrally with brain tissue from patients with AD. These data proved that a component of the AD brain, although not necessarily Aβ plaques themselves, could initiate plaque formation.82 Furthermore, pretreatment of the donor brain extracts with an antibody to neutralize amyloid abrogated the development of plaques in recipient mice, lending strong support to the theory that β-amyloid is a toxic agent with the ability to self-propagate.83 In another study, an Aβ-rich brain homogenate was injected intraperitoneally into amyloid precursor protein transgenic mice, which resulted in widespread cerebral β-amyloidosis,84 suggesting that peripheral or systemic Aβ can travel to the brain. Interestingly, it is not only the Aβ protein that can potentially be self-propagating: brain extracts containing misfolded tau, when injected into the brains of tau-transgenic mice caused tau protein misfolding, which spread to neighboring tissues.85
Parkinson’s disease
A characteristic of PD is the presence of Lewy body inclusions in the brain, which are largely made up of the protein α-syn. The oligomeric isoform of α-syn is thought to be the causative agent in PD, as elimination of this isoform can modify disease progression.86 Further evidence to support the role of α-syn is that familial forms of PD often display mutations in the α-syn gene, and overexpression of Wt α-syn results in PD-like toxicity.87 CNS injection of α-syn seeds or brain homogenates from mice showing α-syn pathology promoted the induction and aggregation of α-syn, resulting in selective loss of dopaminergic neurons and motor function deterioration.88 These studies suggest that α-syn may have a similar mechanism of action as prions in self-propagation and induction of disease, as has recently been shown for another α-synucleinopathy – multiple system atrophy.78
Similar to the CD4+ adoptive transfer studies of prion immunotherapy mentioned previously, adoptive transfer of nitrated α-syn T-cells polarized to a TH1 phenotype increased neuronal loss in vivo, whereas CD4+ cells polarized to a TH2 phenotype reversed this effect.89
Amyotrophic lateral sclerosis
ALS is the most common type of motor neuron disease. It leads to paralysis and death in 1–5 years after onset.90 As with prion diseases, there are familial and sporadic forms of ALS. The familial form accounts for ∼10% of cases, while the sporadic form accounts for the remaining 90% – although both show similar clinical signs.91 A common cause of familial ALS is misfolded and mutated Cu/Zn SOD1.92 Misfolded SOD1 has been reported to induce misfolding of natively structured Wt SOD1 via protein–protein interaction.93 The role of the misfolded SOD1 in sporadic ALS still remains unclear. TDP-43, an RNA/DNA-binding protein, also has the ability to misfold and is implicated in ALS pathology as well as a number of other neurodegenerative diseases. However, there is contradictory evidence whether TDP-43 exhibits prion-like behaviors such as seeding and self-propagation.94
Huntington’s disease
HD is characterized by misfolded Huntington protein caused by variable CAG repeat length expansions resulting in PolyQ repeats. There is some emerging evidence for prion-like mechanisms. Aggregates of pathogenic PolyQ expansion peptides taken up into cultured cells have the ability to recruit soluble physiological PolyQ expansions into the aggregate core.95 The pathological importance of this finding has yet to be determined.
Future prospects
To date, there is no effective treatment or cure for prion diseases. Current treatment options only relieve symptoms but have no effect on disease pathology. Both passive and active immunization strategies have shown different levels of success in mouse models of prion and other neurological diseases. However, the serious side effects associated with active immunization, such as autoimmune meningoencephalitis, raise doubts about its safety for human trials. Thus, most researchers favor passive immunization strategies. Unfortunately, there is no evidence that passive immunization with antibodies can prolong survival in mice when administered after prion accumulation in the brain or after clinical symptoms have developed. Passive immunization appears to only be successful in prolonging survival if administered before or immediately after contact with PrPSc. Earlier diagnosis of prion diseases would enable passive immunization to commence earlier and have a better chance of delaying disease progression and prolonging life. One major obstacle to earlier diagnosis is that, aside from folding, PrPc is identical to PrPSc. While prophylactic treatment to whole populations may not be feasible, passive immunization could be targeted to at-risk groups such as individuals with known mutations of the PRNP gene or those at risk from exposure.
Conclusion
Prion diseases are devastating in their neurological effects and poor survival. Although rare in the population, research is being conducted into earlier diagnosis and more targeted therapies in an effort to improve prognosis.
Both DC vaccination and CD4+ adoptive therapy require more research before they can be transferred into large-scale clinical trials. Further information is required to determine the most effective type of adaptive immune response to delay or even reverse disease progression, for example, cell-mediated, humoral, innate, inflammatory, regulatory, or a combination of these approaches. This approach could then be adapted to the patients’ disease state. Current research into passive immunization strategies appears to offer promise for the future, with fewer serious side effects when compared to active immunization. However, its usefulness is dependent on earlier detection. Lessons learnt from these immunotherapies for prion diseases could then be used to develop treatments for other neurodegenerative diseases.
Acknowledgments
This work was funded by Neurodegenerative Disorders Research Pty Ltd.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Williams ES. Chronic wasting disease. Vet Pathol. 2005;42(5):530–549. doi: 10.1354/vp.42-5-530. [DOI] [PubMed] [Google Scholar]
- 2.Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochim Biophys Acta. 2007;1772(6):610–618. doi: 10.1016/j.bbadis.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cdc.gov [homepage on the internet] Atlanta: Centers for Disease Control and Prevention; [Accessed November 15, 2015]. [updated 2015 Nov 10]. Available from: http://www.cdc.gov/ [Google Scholar]
- 4.Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389(6650):448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 5.Gajdusek DC. Kuru: an appraisal of five years of investigation. Eugen Q. 1962;9(1):69–74. doi: 10.1080/19485565.1962.9987505. [DOI] [PubMed] [Google Scholar]
- 6.Panegyres PK, Goh J, Goldblatt J. Codon 200 mutation of the prion gene: genotype-phenotype correlations. J Neurol. 2012;259(12):2579–2584. doi: 10.1007/s00415-012-6539-x. [DOI] [PubMed] [Google Scholar]
- 7.Zerr I, Pocchiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55(6):811–815. doi: 10.1212/wnl.55.6.811. [DOI] [PubMed] [Google Scholar]
- 8.Windl O, Dempster M, Estibeiro JP, et al. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet. 1996;98(3):259–264. doi: 10.1007/s004390050204. [DOI] [PubMed] [Google Scholar]
- 9.Glatzel M, Ott PM, Linder T, et al. Human prion diseases: epidemiology and integrated risk assessment. Lancet Neurol. 2003;2(12):757–763. doi: 10.1016/s1474-4422(03)00588-x. [DOI] [PubMed] [Google Scholar]
- 10.Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989;338(6213):342–345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 11.Beekes M. Prions and prion diseases. FEBS J. 2007;274(3):575. doi: 10.1111/j.1742-4658.2006.05629.x. [DOI] [PubMed] [Google Scholar]
- 12.Nitrini R, Rosemberg S, Passos-Bueno MR, et al. A familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol. 1997;42(2):138–146. doi: 10.1002/ana.410420203. [DOI] [PubMed] [Google Scholar]
- 13.Finckh U, Müller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66(1):110–117. doi: 10.1086/302702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiachopoulos S, Bracher A, Winklhofer KF, Tatzelt J. Pathogenic mutations located in the hydrophobic core of the prion protein interfere with folding and attachment of the glycosylphosphatidylinositol anchor. J Biol Chem. 2005;280(10):9320–9329. doi: 10.1074/jbc.M412525200. [DOI] [PubMed] [Google Scholar]
- 15.van der Kamp MW, Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol. 2010;404(4):732–748. doi: 10.1016/j.jmb.2010.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovacs GG, Puopolo M, Ladogana A, et al. EUROCJD Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118(2):166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- 17.Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991;337(8755):1441–1442. doi: 10.1016/0140-6736(91)93128-v. [DOI] [PubMed] [Google Scholar]
- 18.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and clarification. Br Med Bull. 2003;66(1):213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 19.Lasmezas CI, Deslys JP, Robain O, et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science. 1997;275(5298):402–405. doi: 10.1126/science.275.5298.402. [DOI] [PubMed] [Google Scholar]
- 20.Caughey B, Kocisko DA, Raymond GJ, Lansbury PT., Jr Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem Biol. 1995;2(12):807–817. doi: 10.1016/1074-5521(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 21.Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009;89(4):1105–1152. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- 22.Panegyres PK, Armari E. Therapies for human prion diseases. Am J Neurodegener Dis. 2013;2(3):176–186. [PMC free article] [PubMed] [Google Scholar]
- 23.Gauczynski S, Nikles D, El-Gogo S, et al. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycans. J Infect Dis. 2006;194(5):702–709. doi: 10.1086/505914. [DOI] [PubMed] [Google Scholar]
- 24.Leucht C, Vana K, Renner-Muller I, et al. Knock-down of the 37-kDa laminin receptor in mouse brain by transgenic expression of specific antisense LRP RNA. Transgenic Res. 2004;13(1):81–85. doi: 10.1023/b:trag.0000017177.35197.89. [DOI] [PubMed] [Google Scholar]
- 25.Larramendy-Gozalo C, Barret A, Daudigeous E, et al. Comparison of CR36, a new heparin mimetic, and pentosan polysulfate in the treatment of therapies for human prion diseases. Am J Neurodegener Dis. 2013;2(3):176–186. [Google Scholar]
- 26.MacGregor IV, Dawes J, Paton L, Pepper DS, Prowse CV, Smith M. Metabolism of sodium pentosan polysulphate in man. Catabolism of iodinated derivatives. Thromb Haemost. 1984;51(3):321–325. [PubMed] [Google Scholar]
- 27.Farquhar C, Dickinson A, Bruce M. Prophylactic potential of pentosan polysulphate in transmissible spongiform encephalopathies. Lancet. 1999;353(9147):117. doi: 10.1016/S0140-6736(98)05395-1. [DOI] [PubMed] [Google Scholar]
- 28.Parry A, Baker I, Stacey R, Wimalaratna S. Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intra-ventricular pentosan polysulphate. J Neurol Neurosurg Psychiatry. 2007;78(7):733–734. doi: 10.1136/jnnp.2006.104505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bone L, Belton L, Walker AS, Darbyshire J. Intraventricular pentosan polysulphate in human prion diseases: an observational study in the UK. Eur J Neurol. 2008;15(5):458–464. doi: 10.1111/j.1468-1331.2008.02108.x. [DOI] [PubMed] [Google Scholar]
- 30.Doh-ura K, Ishikawa K, Murakami-Kubo I, et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78(10):4999–5006. doi: 10.1128/JVI.78.10.4999-5006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami-Kubo I, Doh-Ura K, Ishikawa K, et al. Quinoline derivatives are therapeutic candidates for transmissible spongiform encephalopathies. J Virol. 2004;78(3):1281–1288. doi: 10.1128/JVI.78.3.1281-1288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haïk S, Brandel JP, Salomon D, et al. Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology. 2004;63(12):2413–2415. doi: 10.1212/01.wnl.0000148596.15681.4d. [DOI] [PubMed] [Google Scholar]
- 33.Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8(4):334–344. doi: 10.1016/S1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pocchiari M, Schmittinger S, Masullo C. Amphotericin B delays the incubation period of scrapie in intracerebrally inoculated hamsters. J Gen Virol. 1987;68(1):219–223. doi: 10.1099/0022-1317-68-1-219. [DOI] [PubMed] [Google Scholar]
- 35.Demaimay R, Adjou KT, Beringue V, et al. Late treatment with polyene antibiotics can prolong the survival time of scrapie-infected animals. J Virol. 1997;71(12):9685–9689. doi: 10.1128/jvi.71.12.9685-9689.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masullo C, Macchi G, Xi YG, Pocchiari M. Failure to ameliorate Creutzfeldt-Jakob disease with amphotericin B therapy. J Infect Dis. 1992;165(4):784–785. doi: 10.1093/infdis/165.4.784. [DOI] [PubMed] [Google Scholar]
- 37.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A. 2001;98(16):9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412(6848):739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 39.Heppner FL, Musahl C, Arrighi I, et al. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;294(5540):178–182. doi: 10.1126/science.1063093. [DOI] [PubMed] [Google Scholar]
- 40.White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422(6927):80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- 41.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 42.Masliah E, Rockenstein E, Adame A, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46(6):857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Madampage CA, Määttänen P, Marciniuk K, et al. Binding of bovine T194A PrPC by PrPSc-specific antibodies: potential implications for immunotherapy of familial prion diseases. Prion. 2013;7(4):301–311. doi: 10.4161/pri.25148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol. 2004;108(6):476–484. doi: 10.1007/s00401-004-0913-4. [DOI] [PubMed] [Google Scholar]
- 45.Määttänen P, Taschuk R, Marciniuk K, et al. PrP(Sc)-specific antibodies do not induce prion disease or misfolding of PrPc in highly susceptible Tga20 mice. Prion. 2013;7(5):434–439. doi: 10.4161/pri.26639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischer M, Rülicke T, Raeber A, et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15(6):1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 47.Rosset MB, Ballerini C, Gregoire S, Metharom P, Carnaud C, Aucouturier P. Breaking immune tolerance to the prion protein using prion protein peptides plus oligodeoxynucleotide-CpG in mice. J Immunol. 2004;172(9):5168–5174. doi: 10.4049/jimmunol.172.9.5168. [DOI] [PubMed] [Google Scholar]
- 48.Goni F, Prelli F, Schreiber F, et al. High titers of mucosal and systemic anti-PrP antibodies abrogate oral prion infection in mucosal-vaccinated mice. Neuroscience. 2008;153(3):679–686. doi: 10.1016/j.neuroscience.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nikles D, Bach P, Boller K, et al. Circumventing tolerance to the prion protein (PrP): vaccination with PrP-displaying retrovirus particles induces humoral immune responses against the native form of cellular PrP. J Virol. 2005;79(7):4033–4042. doi: 10.1128/JVI.79.7.4033-4042.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cardinale A, Biocca S. Gene-based antibody strategies for prion diseases. Int J Cell Biol. 2013;2013:710406. doi: 10.1155/2013/710406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porter DD, Porter HG, Cox NA. Failure to demonstrate a humoral immune response to scrapie infection in mice. J Immunol. 1973;111(5):1407–1410. [PubMed] [Google Scholar]
- 52.Spinner DS, Kascsak RB, LaFauci G, et al. CpG oligodeoxynucleotide-enhanced humoral immune response and production of antibodies to prion protein [PrP.sup.Sc] in mice immunized with 139A scrapie-associated fibrils. J Leukoc Biol. 2007;81(6):1374–1385. doi: 10.1189/jlb.1106665. [DOI] [PubMed] [Google Scholar]
- 53.Song CH, Furuoka H, Kim CL, et al. Effect of intraventricular infusion of anti-prion protein monoclonal antibodies on disease progression in prion-infected mice. J Gen Virol. 2008;89(6):1533–1544. doi: 10.1099/vir.0.83578-0. [DOI] [PubMed] [Google Scholar]
- 54.Luginbuhl B, Kanyo Z, Jones RM, et al. Directed evolution of an anti-prion protein scFv fragment to an affinity of 1 pM and its structural interpretation. J Mol Biol. 2006;363(1):75–97. doi: 10.1016/j.jmb.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 55.Padiolleau-Lefevre S, Alexandrenne C, Dkhissi F, et al. Expression and detection strategies for an scFv fragment retaining the same high affinity than Fab and whole antibody: implications for therapeutic use in prion diseases. Mol Immunol. 2007;44(8):1888–1896. doi: 10.1016/j.molimm.2006.09.035. [DOI] [PubMed] [Google Scholar]
- 56.Zuber C, Mitteregger G, Schuhmann N, et al. Delivery of singlechain antibodies (scFvs) directed against the 37/67 kDa laminin receptor into mice via recombinant adeno-associated viral vectors for prion disease gene therapy. J Gen Virol. 2008;89(8):2055–2061. doi: 10.1099/vir.0.83670-0. [DOI] [PubMed] [Google Scholar]
- 57.Zinkernagel RM, Hengartner H. Regulation of the immune response by antigen. Science. 2001;293(5528):251–253. doi: 10.1126/science.1063005. [DOI] [PubMed] [Google Scholar]
- 58.Flego M, Ascione A, Zamboni S, Dupuis ML, Imperiale V, Cianfriglia M. Generation of human scFvs antibodies recognizing a prion protein epitope expressed on the surface of human lymphoblastoid cells. BMC Biotechnol. 2007;7(1):38–45. doi: 10.1186/1472-6750-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marciniuk K, Määttänen P, Taschuk R, et al. Development of a multi-valent, PrPSc-specific prion vaccine through rational optimization of three disease-specific epitopes. Vaccine. 2014;32(17):1988–1997. doi: 10.1016/j.vaccine.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 60.Lobello K, Ryan JM, Liu E, Rippon G, Black R. Targeting beta amyloid: a clinical review of immunotherapeutic approaches in Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:628070. doi: 10.1155/2012/628070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mouillet-Richard S, Ermonval M, Chebassier C, et al. Signal transduction through prion protein. Science. 2000;289(5486):1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 62.Solforosi L, Criado JR, McGavern DB, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303(5663):1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- 63.Klohn P-C, Farmer M, Linehan JM, et al. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science. 2012;335(6064):52. doi: 10.1126/science.1215579. [DOI] [PubMed] [Google Scholar]
- 64.Arsenault RJ, Li Y, Potter A, Griebel PJ, Kusalik A, Napper S. Induction of ligand-specific PrP (C) signaling in human neuronal cells. Prion. 2012;6(5):477–488. doi: 10.4161/pri.21914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360(6401):258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 66.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179(4):1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 68.Fong L, Hou Y, Rivas A, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A. 2001;98(15):8809–8814. doi: 10.1073/pnas.141226398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 70.Gregoire S, Logre C, Metharom P, et al. Identification of two immunogenic domains of the prion protein – PrP – which activate class II-restricted T cells and elicit antibody responses against the native molecule. J Leukoc Biol. 2004;76(1):125–134. doi: 10.1189/jlb.1203656. [DOI] [PubMed] [Google Scholar]
- 71.Bachy V, Ballerini C, Gourdain P, et al. Mouse vaccination with dendritic cells loaded with prion protein peptides overcomes tolerance and delays scrapie. J Gen Virol. 2010;91(3):809–820. doi: 10.1099/vir.0.013417-0. [DOI] [PubMed] [Google Scholar]
- 72.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 73.Stockinger B, Veldhoen M, Martin B. TH17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19(6):353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 74.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11(1):7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 75.Bluestone JA, Mackay CR, O’Shea JJ, Stockinger B. The functional plasticity of T cell subsets. Nat Rev Immunol. 2009;9(11):811–816. doi: 10.1038/nri2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gourdain P, Gregoire S, Iken S, et al. Adoptive transfer of T lymphocytes sensitized against the prion protein attenuates prion invasion in scrapie-infected mice. J Immunol. 2009;183(10):6619–6628. doi: 10.4049/jimmunol.0804385. [DOI] [PubMed] [Google Scholar]
- 77.Iken S, Bachy V, Gourdain P, et al. Th2 polarised PrP-specific transgenic T-cells confer partial protection against murine scrapie. PLoS Pathog. 2011;7(9):e1002216. doi: 10.1371/journal.ppat.1002216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112(38):E5308–E5317. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209(5):889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336(6088):1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ. Induction of [beta](A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate – comparison with transmission of spongiform encephalopathy. Mol Neurobiol. 1994;8(1):25–39. doi: 10.1007/BF02778005. [DOI] [PubMed] [Google Scholar]
- 82.Kane MD, Lipinski WJ, Callahan MJ, et al. Evidence for seeding of [beta]-amyloid by intracerebral infusion of Alzheimer brain extracts in [beta]-amyloid precursor protein-transgenic mice. J Neurosci. 2000;20(10):3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral [beta]-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 84.Eisele YS, Obermuller U, Heilbronner G, et al. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science. 2010;330(6006):980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eriksen JL, Dawson TM, Dickson DW, Petrucelli L. Caught in the act: [alpha]-synuclein is the culprit in Parkinson’s disease. Neuron. 2003;40(3):453–456. doi: 10.1016/s0896-6273(03)00684-6. [DOI] [PubMed] [Google Scholar]
- 87.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 88.Luk KC, Kehm VM, Carroll J, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in non-transgenic mice. Science. 2012;338(6109):949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate TH17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184(5):2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2(2):806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 91.Bruijin LI, Miller TM, Cleveland DW. Unravelling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 92.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn super-oxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 93.Grad LI, Guest WC, Yanai A, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Nat Acad Sci U S A. 2011;108(39):16398–16403. doi: 10.1073/pnas.1102645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smethurst P, Sidle KCL, Hardy J. Review: prion-like mechanisms of transactive response DNA binding protein of 43kDa (TDP-43) in amyotrophic lateral sclerosis (ALS) Neuropathol Appl Neurobiol. 2015;41(5):578–597. doi: 10.1111/nan.12206. [DOI] [PubMed] [Google Scholar]
- 95.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;2(2):219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N. A seeding reaction recapitulates intracellular formation of sarkosyl-insoluble TAR DNA binding protein-43 inclusions. J Biol Chem. 2011;286(16):18664–18672. doi: 10.1074/jbc.M111.231209. [DOI] [PMC free article] [PubMed] [Google Scholar]