

Abstract
Many viral pathogens encode the motor proteins named RNA helicases which display various functions in genome replication. General strategies to design specific and selective drugs targeting helicase for the treatment of viral infections could act via one or more of the following mechanisms: inhibition of the NTPase activity, by interferences with ATP binding and therefore by limiting the energy required for the unwinding and translocation, or by allosteric mechanism and therefore by stabilizing the conformation of the enzyme in low helicase activity state; inhibition of nucleic acids binding to the helicase; inhibition of coupling of ATP hydrolysis to unwinding; inhibition of unwinding by sterically blocking helicase translocation. Recently, by in vitro screening studies, it has been reported that several benzotriazole, imidazole, imidazodiazepine, phenothiazine, quinoline, anthracycline, triphenylmethane, tropolone, pyrrole, acridone, small peptide, and Bananin derivatives are endowed with helicase inhibition of pathogen viruses belonging to Flaviviridae, Coronaviridae, and Picornaviridae families.
1. Introduction
To convert a closed double-stranded DNA or RNA helix into two open single strands, so that other protein machinery can manipulate the polynucleotides, the cells require helicases. They are motor proteins that use energy derived from ATP hydrolysis [1–4]. Several DNA and RNA helicases have been isolated from all kingdoms of life, from virus to man [5–8]. Detailed structural information, biological mechanisms, and clear outlook on inhibitors of therapeutic relevance as antiviral agents are recently provided by Xi et al. [9], Kwong et al. [10], and overall Frick et al. [11, 12].
Several ssRNA+ (positive sense single-stranded RNA) helicases have been studied in detail including those from Dengue fever virus (DFV), West Nile virus (WNV), and Japanese encephalitis virus (JEV). More in general, a recent article on anti-Flaviviridae chemotherapy has been published by Ghosh and Basu [13], who expand the original information regarding the role of helicases in Flaviviridae previously reported by Borowski [14].
This enzyme is a promising target to develop new therapies and preventative agents, since ssRNA+ viruses belonging to families like Flaviviridae, Coronaviridae, and Picornaviridae cause clinically significant diseases both in humans and animals, determining life lost, economical loss, and higher productivity costs. Examples are the bovine viral diarrhea virus (BVDV), a serious welfare problem that significantly damages the farm business, and the Hepatitis C virus [HCV], that is now a global public health issue, being a major cause of human hepatitis [15]. Actually, with the exception of YFV, no vaccine exists against the various Flaviviridae members therefore, new therapies and preventative agents are strongly needed.
Viruses belonging to Picornaviridae family cause a variety of illnesses, including meningitis, cold, heart infection, conjunctivitis, and hepatitis [16]. This family includes nine genera, some of which comprise major human pathogens, namely, Enterovirus (including Poliovirus, Coxsackievirus, Echovirus), Rhinovirus (approximately 105 serotypes), and Hepatovirus (Hepatitis A virus). At present, no specific antiviral therapy is available for the treatment of Picornaviridae infections.
Finally, Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), an enveloped virus, has recently infected thousand of humans, with about 800 deaths, and no vaccine or specific antiviral therapy is known against this virus.
No retroviruses or ssRNA− viruses have been reported to encode the synthesis of a helicase; they might simply utilize helicases encoded by the host cell instead of their own proteins, as recently shown for HIV replication, which requires the human DDX3 DEAD-box RNA helicase [17, 18].
In ssRNA+ viruses, the RNA helicases are implicated in several functions including RNA genome replication, ribosome biogenesis, messengers RNA transcription, pre-mRNA splicing, RNA maturation, RNA export and degradation, as well as RNA translation [19, 20].
Basing on certain signature motifs in the amino acid sequence, Gorbalenya and Koonin have shown that all helicases can be classified in several genetic families [21]. All but two of the helicase families can be grouped into one of three larger “superfamilies,” designed as superfamily 1 (SF1), superfamily 2 (SF2) [22], and superfamily 3 (SF3) [23].
Of the remaining 2 families, one is similar to the DnaB helicase of E. coli [5] and the other resembles the E. coli Rho helicase that is used in transcriptional termination [24]. Only the DnaB-like family, sometimes called family 4 (F4) or superfamily 4 (SF4), contains viral proteins [25].
All helicases bind NTP using two structurally common amino acidic sequences named motif I and motif II, described by Walker et al. [25] and Subramanya et al. [26]. Motif I (also known as Walker A motif/boxes A) is a phosphate-binding P-loop that also interact with the ribose, while motif II (also known as Walker B/boxes B) is a Mg2+ co-factor binding loop. The ATP-binding site of helicase is completed by an arginine “finger” and a catalytic base, which accepts a proton from the attacking water molecule. In related proteins, this catalytic base has been demonstrated to be a conserved glutamate near the Walker B motif [27, 28]. Arginine amino acids often interact with the beta and gamma phosphates of the bound ATP, stabilizing the transition state [29, 30], Figure 1.
Figure 1.

Mechanism of helicase-catalyzed ATP hydrolysis. Helicases coordinate an ATP, Mg2+ and a water molecule using a conserved Lys and Asp in the Walker A and B motifs on one RecA-like domain and an Arg on an adjacent RecA-like domain. A Glu likely acts as a catalytic base by accepting a proton from the attacking water molecule [11].
All helicases can also be classified according to their movement relative to the nucleic acid strand to which they are primarily associated or to their quaternary structures.
Thus, a helicase can be classified basing on each of the three above schemes. For example, the helicase encoded by HCV (Hepatitis C Virus) is an SF2, nonring, 3′–5′ RNA helicase. Human papillomavirus helicase is an SF3, ring, 3′–5′ DNA helicase.
The functional importance of helicases means that inhibitors or modulators for these enzymes are potentially important as therapeutic agents. Over the past decade, significant progress has been made in the development of highly selective inhibitors as antiviral and anticancer drugs for clinical uses. Developing nontoxic helicase inhibitors as antiviral drugs is considerably more difficult than developing drugs designed to inhibit other viral enzymes. In fact, in contrast with proteases and polymerases, the helicases-dependent reactions are still not fully elucidated. Furthermore, the helicase ATP-binding site is conserved not only in all the classes of helicases, but also in other proteins necessary for the cellular lifecycle, such as small GTPases, kinases, the AAA+ family (ATPases associated with various cellular activities), and even the mitochondrial ATP synthase (F1 ATPase). Thus, compounds that inhibit helicases via their ATP-binding sites could have toxic effects on the host cells.
2. Viral RNA Helicases As Antiviral Drug Targets
Many viral pathogens encode RNA helicases which have been demonstrated essential for viral replication and pathogenesis [31–33]. Between them are
emerging or re-emerging viruses with pandemic potential, such as SARS-Cov (Severe Acute Respiratory Syndrome-Coronavirus), Dengue, West Nile, and Japanese encephalitis viruses,
viruses that have a stable spread worldwide, such as HCV (Hepatitis C Virus),
viruses that do not have a large spread, but can generate serious health problems because of lack or limited availability of effective drugs, such as CVB (Human Coxsackie B Virus).
General strategies to design specific and selective drugs for the treatment of viral infections targeting helicase could act via one or more of the following mechanisms:
inhibition of the NTPase activity by interferences with ATP binding and therefore by limiting the energy required for the unwinding and translocation,
inhibition of the NTPase activity by allosteric mechanism and therefore by stabilizing the conformation of the enzyme in low helicase activity state,
inhibition of nucleic acids binding to the helicase,
inhibition of coupling of ATP hydrolysis to unwinding,
inhibition of unwinding by sterically blocking helicase translocation,
development of small molecule antagonists against essential protein-protein interactions involving helicases.
Some characteristics of helicase families of pathogen viruses belonging to Flaviviridae, Coronaviridae, and Picornaviridae families are reported in Table 1 [9, 10, 34].
Table 1.
Viral helicases of same ssRNA+ Viruses (belonging to Flaviviridae, Coronaviridae, and Picornaviridae families) [9, 10, 33].
| Family | Species | Helicase family | Helicase name | In vitro activity |
|---|---|---|---|---|
| Flaviviridae | Yellow fever virus | SF2 | NS3 | RNA stimulated NTPase |
| West Nile virus | SF2 | NS3 | RTPase 3′–5′ helicase | |
| Dengue fever virus | SF2 | NS3 | 3′–5′RNA helicase RTPase | |
| Japanese encephalitis virus | SF2 | NS3 | 3′–5′RNA helicase | |
| Bovine viral diarrhea virus | SF2 | NS3 | 3′–5′RNA helicase | |
| Hepatitis C virus | SF2 | NS3 | 3′–5′RNA /DNA helicase | |
| Hepatitis G virus | SF2 | NS3 | 3′–5′RNA /DNA helicase | |
| Hepatitis A virus | SF3 | 2C | NTPase | |
| Coronaviridae | Human coronavirus 229E | SF1 | Nsp 13 | 3′–5′RNA/DNA helicase RTPase |
| SARS Coronavirus | SF1 | Nsp 13 | 3′–5′RNA/DNA helicase RTPase | |
| Picornaviridae | Poliovirus | SF3 | 2C | NTPase |
| Rhinovirus | SF3 | 2C | NTPase |
3. Flaviviridae
The Flaviviridae is a large family of related positive-strand RNA viruses that currently consists of three genera: Flavivirus, Pestivirus (from the Latin pestis, plague), and Hepacivirus (from the Greek hepatos, liver). In addition, the family includes two groups of viruses, GBV-A and GBV-C, that are currently unassigned to a specific genus and await formal classification [35]. Within this family are comprised viruses that cause significant diseases in human and animal populations. From Flavivirus genus is Dengue virus (DENV) with its associated dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), Japanese encephalitis virus (JEV), West Nile virus (WNV), Yellow Fever virus (YFV), and tick-borne encephalitis virus (TBEV). The Pestiviruses are animal pathogens of major economic importance for the livestock industry, like bovine viral diarrhea virus (BVDV), border disease virus (BDV) of sheep, and classical swine fever virus (CSFV). The Hepacivirus genus includes only the hepatitis C virus (HCV), an important human pathogen.
HCV, identified in 1989 [36], is a major cause of human hepatitis, globally, and infects about 3% of the world's population [37]. Hepacivirus is spread primarily by direct contact with human blood; hence, the major causes of infection are use of unscreened blood transfusions and reuse of needles and syringes that have not been adequately sterilised. The World Health Organization (WHO) estimates that over 170 million people worldwide are presently infected with this virus [38]. Most infections become persistent and about 60% of cases progress towards chronic liver disease, that can lead to development of cirrhosis, hepatocellular carcinoma, and liver failure [39, 40].
Pegylated interferon in combination with ribavirin is used in the clinic for hepatitis due to HCV. Unfortunately, this therapy requires lengthy periods of administration and is often associated with severe and adverse events. Furthermore, this drug has limited efficacy and the sustained virological response rate is of 40–50% in genotype HCV-1 infected patients, and of 80% in those infected with genotypes 2 and 3 [41, 42].
This emphasizes that new therapies are clearly needed, since for the treatment of this infection, and generally for diseases caused by viruses belonging to the Flaviviridae family, therapeutic strategies really effective and selective are not available.
All of the 12 HCV genotypes, which have nucleotide sequences that differ by as much as 30%, produce a single polyprotein of about 3,000 amino acids, which is subsequently processed by viral and host proteases into four structural proteins and six nonstructural proteins (altogether 10 mature proteins). As summarized in Figure 2, the structural proteins (S proteins: core, E1, E2, and p7) generate the viral capsid and envelope proteins and are cleaved by host-signal peptidases, while the six nonstructural proteins (NS proteins: NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are responsible for genome replication and are largely generated by HCV-encoded protease [43].
Figure 2.

Simplified representation of structure of Hepacivirus and Flaviviruses polyprotein.
HCV Helicase is part of the bi-functional NS3 protein, carrying three different enzymatic activities: helicase, NTPase, and serine protease activities.
NS3 helicase is essential for viral replication, and this makes it one of the most promising target for the antiviral therapy.
The known HCV helicase inhibitors can be classified on the base of their mechanism of action, into the first four groups of those above cited:
inhibitors of NTPase activity by interference with NTP binding,
inhibitors of NTPase activity by allosteric mechanism,
competitive inhibitors of RNA binding,
inhibitors of the coupling of NTP hydrolysis at the unwinding reaction.
3.1. Inhibition of NTPase Activity by Interference with NTP Binding
The hydrolysis of ATP supplies the energy that allows the helicase to adopt various nucleotide ligation states that allosterically cause conformational changes in the nucleic acid binding site to drive the movement of the helicase along the length of the nucleic acid chain [19]. So, competitive NTPase inhibitors may lead to decreased ATPase activity and therefore to reduction of the unwinding rate.
Consequently, non-(or slowly) hydrolysable ATP-analogs seemed to be effective tools for inhibiting the helicase activity, like adenosine-5′γ-thiotriphosphate (ATP-γ-S), which is used to determine a low level of unwinding of HCV dsRNA [44, 45]. However, ribavirin 5′-triphosphate (RTP), that inhibits the HCV NTPase/helicase by a competitive mechanism in regard to ATP [46], and ribavirin 5′-diphosphate (RDP), both reported in Figure 3, even showing IC50 values in the micromolar range, demonstrates to determine only a weakly enzymatic inhibition [34]. The same behavior has been put in evidence for paclitaxel, compound structurally nonrelated to NTP. This derivative is able to block the NTP-binding site (IC50 = 22 μM) and to inhibit the ATPase activity (IC50 = 17 μM) in a competitive way, but is not able to inhibit the helicase activity at concentration lower than 1 mM [14]
Figure 3.

Structure of three competitive HCV helicase inhibitors ribavirin 5′-triphosphate (1), ribavirin 5′-diphosphate (2), and paclitaxel (3).
The partial unwinding activity mediated by these competitive NTPase inhibitors is common to all members of the class, and the concentrations needed for the helicase inhibition usually exceed the IC50 value by 3–5 times. At these concentrations, the NTPase activity reached 10–35% of the control [46–48]. The basis for the phenomenon remains unclear.
On the other hand, most potent benzotriazole helicase inhibitors were identified during the course of a random screening study [49, 50]. In particular, 4, 5, 6, 7-tetrabromobenzotriazole (TBBT) (4), known as a potent and highly selective inhibitor of protein kinase 2, and 5,6-dichloro-1-(β-D-ribofuranosyl) benzotriazole (DRBT) (5) displayed IC50 values of 20 and 1.5 μM, respectively (Figure 4).
Figure 4.
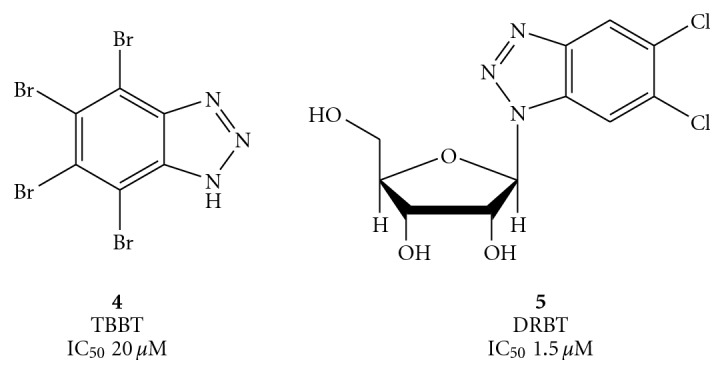
Structure of the halogenated benzotriazoles TBBT (4) and DRBT (5).
On the contrary, the corresponding imidazole derivative of DRBT, the 5, 6-dichloro-1-(β-D-ribofuranosyl) benzimidazole (DRBI), against NTPase/helicase of a large number of members of the Flaviviridae family (HCV, WNV, DENV, and JEV) resulted to be completely inactive.
To explain this finding, Bretner et al. synthesized and studied a new series of substituted (alkyl, hydroxy alkyl, chloro alkyl, ribofuranose) 1H-benzimidazole and 1H-benzotriazole derivatives shown in Figures 5 and 6 [50, 51].
Figure 5.

Synthesis of TBBT (4) and its N-alkyl derivatives.
Figure 6.

Synthesis of 4, 5, 6, 7-tetrabromo 1H-benzimidazole.
TBBT (more less DRBT) resulted effective in HCV subgenomic replicon system in a comparable way to the inhibition reported in the enzymatic essays, showing a property that has been detected only for a handful group of HCV inhibitors [52].
It has been reported that the starting compounds 1H-benzotriazole (6) and 1H-benzimidazole (17), screened for their effect against the HCV-helicase, showed
very low activity (IC50 200 μM and 500 μM, respectively) when measured with a DNA substrate,
no activity when measured either with an RNA substrate or against the flavivirus enzymes of WNV, DENV, and JEV (IC50> 500 μM).
On the contrary, the whole halogenation of 1H-benzotriazole (6) with bromine atoms, to afford the above cited 4, caused either a 10-fold or 9-fold more effective inhibition of the HCV helicase when determined with a DNA substrate or an RNA substrate, respectively, and of 25-fold in the case of the JEV enzyme (IC50 20 μM).
The corresponding bromination of 1H-benzimidazole (17) afforded the derivative (18), which resulted to be less effective than 4 and 2–2.5 times more potent than parent 17 against HCV helicase.
When 1- or 2-alkyl benzotriazoles were screened for their effect on the HCV-helicase activity using the DNA substrate, the 2-alkylated derivatives (10–12) resulted to be significantly more potent inhibitors of the enzyme (2- to 7- times) than the respective 1-alkylated analogues (7–9).
On the other hand, enhancement of the activity was observed when the aliphatic chain was elongated in both 1-alkylated benzotriazoles (7–9) and 1-alkylated benzimidazoles (19–21) than the respective 2-alkylated analogues. In the case of the benzimidazole derivatives (19–21), however, the inhibitory activity was very low and ranged between 250 and 500 μM. Furthermore, the HCV helicase activity of the alkylated benzimidazoles tested using the RNA substrate, as well as using other viral NTPase/helicases, displayed no inhibitory activity.
This behaviour suggests that these inhibitors do not act by blocking the NTP binding sites of the enzymes and that occupation of an allosteric nucleoside binding site should be considered, as previously suggested by Porter [53].
Furthermore, in this study the authors observed that replacement of the alkyl side-chain by a substituent endowed with higher hydrophilicity (hydroxyethyl derivatives 13 and 14 in Figure 5) or with higher hydrophobicity (chloroethyl derivatives 15 and 16 in Figure 5) dramatically decreases the activity of the tetrabromobenzotriazoles. Consequently, it seems that a small hydrophobic alkyl moiety (methyl or ethyl) at position 2- of the tetrabromobenzotriazole could play a crucial role in the inhibition of the HCV NTPase/helicase.
Introduction of a ribofuranosyl ring in both benzotriazole and tetrabromobenzotriazole improves the water solubility but leads to a decrease of the inhibitory activity against HCV and all the enzymes tested. The same substituent in the position 1 of the 5,6-dichlorobenzotriazole DRBT (5) was, as above reported, very effective in inhibiting the HCV and WNV helicases (IC50 1.5 μM and 3.0 μM, respectively) but ineffective against JEV helicase [49]. On the contrary, replacement of chlorine atoms of the benzotriazole ring with either bromine atoms or methyl groups (compounds 28–30, Figure 7) showed lower activity compared to DRBT.
Figure 7.

Synthesis of compounds 5, 27–30.
In an extension of this study, an additional class of nucleoside analogues known as ring-expanded nucleosides (REN or “fat”) involving 6-aminoimidazo [4,5-e] [1, 3] diazepine-4,8-dione ring were reported to be active against the helicase unwinding reaction [54]. A number of RENs, such as compounds 31 and 32 of Figure 8, displayed IC50 values in the micromolar range. In view of the observed tight complex between some nucleosides and RNA and/or DNA substrates of a helicase, the mechanism of REN action might involve binding to the minor or major groove of the helical nucleic acid substrate.
Figure 8.
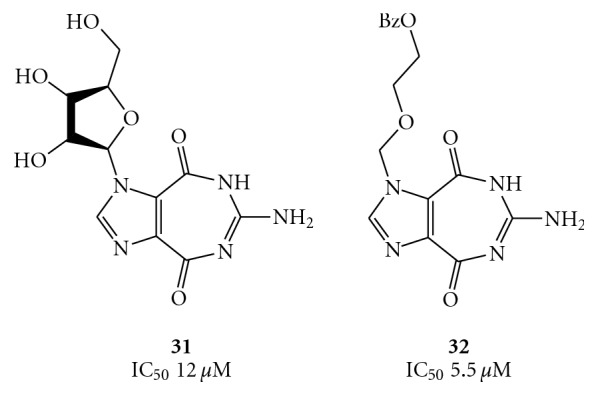
Structures of the ring expanded nucleosides 31 and 32.
The fat nucleosides 31, 32, and TBBT (4) and nogalamycin (see compound 76) have been recently used to construct a pharmacophore model for designing new Japanese encephalitis virus NS3 helicase/NTPase inhibitors, using a refined structure of this enzyme [55].
On the other hand, the REN 5′-triphosphates, such as compounds 33 and 34 of Figure 9, did not influence the unwinding reaction while exerting their inhibitory effect (IC50 0.55 μM and 1.5 μM, respectively) on the ATPase activity of the enzyme. As reported in Figure 9, compounds 33 and 34, containing the 5 : 7-fused heterocyclic systems, imidazo [4,5-e] [1, 3] diazepine and imidazo [4,5-e] [1, 2, 4] triazepine, respectively, were synthesized from the corresponding nucleosides 36 and 37, employing the Ludwig's procedure [56]. The nucleosides 36 and 37, in turn, were synthesized by Vorbrüggen ribosylation [57–60] of the respective heterocycles 35 and 38 [61, 62].
Figure 9.

Synthesis of compounds 33 and 34.
Therefore, in exploring the potential anti-Flaviviridae activity of the ring system contained in 31, the same authors focused on different substituents (alkyl, arylalkyl, and aromatic groups) at position 6, along with variations of sugar moieties at position 1 (ribose, 2′-deoxyribose, or acyclic derivatives) as well as their attachment to the base (α or β configuration) [63].
The general method for the synthesis of the designed nucleosides (41–59) was involved, as reported in Figure 10, the Vorbrüggen ribosylation [53, 54] of dimethyl imidazole-4,5-dicarboxylate (39) [64, 65], followed by condensation of the resulting imidazole nucleoside (40) with the appropriately substituted guanidine derivatives.
Figure 10.

Synthesis of the compounds 41–59.
The modulation effect exerted by RENs can result in an inhibition or activation. In the first case, the mechanism may involve the interaction of RENs with a DNA or an RNA substrate through binding to the major or minor groove of the double-helix. In the case of activation, the mechanism may involve an allosteric binding site that can be occupied by nucleoside/nucleotide-type molecules including, but not limited to RENs. The occupation of this allosteric site on the enzyme is dependent upon the high level of ATP (NTP) concentration in the reaction mixture.
RENs obtained with the above procedures were screened for inhibition of NTPase/helicase of the WNV. One of the most promising among these early inhibitors is 1-(2′-O-methyl-β-D-ribofuranosyl)imidazo[4,5-d]pyridazine-4,7(5H,6H)-dione (HMC-HO4) (60), Figure 11, which produces a promising antiviral effect (EC50 = 25–30 μM) [66]. At all the concentrations of HMC-HO4, ATP hydrolysis is stimulated, suggesting that the inhibitor somehow uncouples the ATPase and helicase functions. In that regard, RENs may represent a starting point for the development of highly selective inhibitors of WNV NTPase/helicase.
Figure 11.
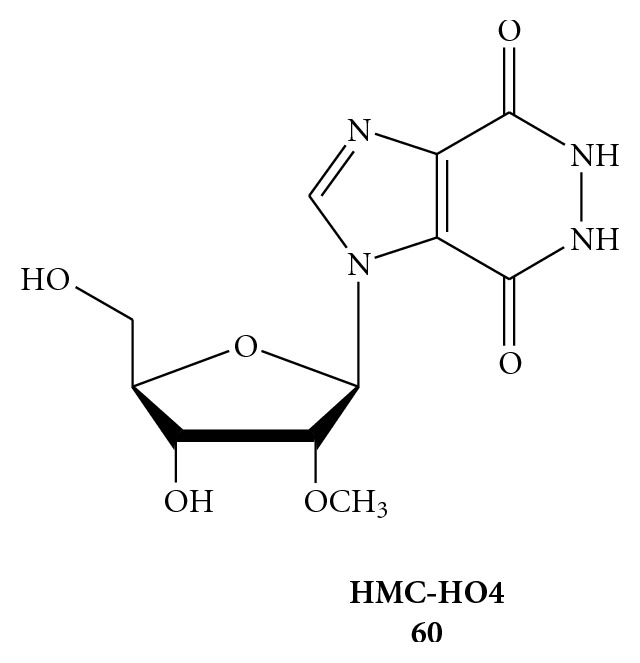
Chemical structure of 1-(2′-O-Methyl-β-D-ribofuranosyl)imidazo[4,5-d]pyridazine-4, 7(5H, 6H)-dione (HMC-HO4) (60).
An other recent starting point is represented by triphenylmethane derivatives, as reported from Chen et al. [67]. Compound (61) of Figure 12, where the triphenylmethane moiety is linked to a 2-(3-bromo-4-hydroxyphenyl)propane, was identified as a good inhibitor that suppresses HCV RNA replication in the HCV replicon cells through both the inhibition of ATP hydrolysis and the RNA substrate binding [67].
Figure 12.
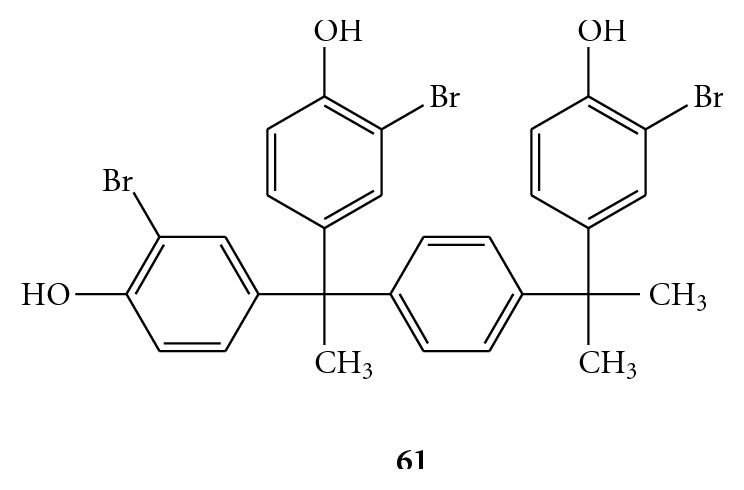
Chemical structure of 4, 4′-(1-(4-(2-(3-bromo-4-hydroxyphenyl)propan-2-yl)phenyl)ethane-1,1-diyl)bis(2-bromophenol) (61).
3.2. Inhibition of NTPase Activity by Allosteric Mechanisms
The partial inhibition mediated by the competitive NTPase inhibitors may be avoided by utilizing compounds chemically unrelated to NTP, which reduce the accessibility to the NTP-binding site in a noncompetitive manner [68]. An example is the calmodulin antagonist trifluoperazine (62, Figure 13). Although the molecule is known to interact with domain 1 of HCV helicase, it is uncertain if inhibition results from conformational changes or from blockage of the ATP-binding site [46].
Figure 13.
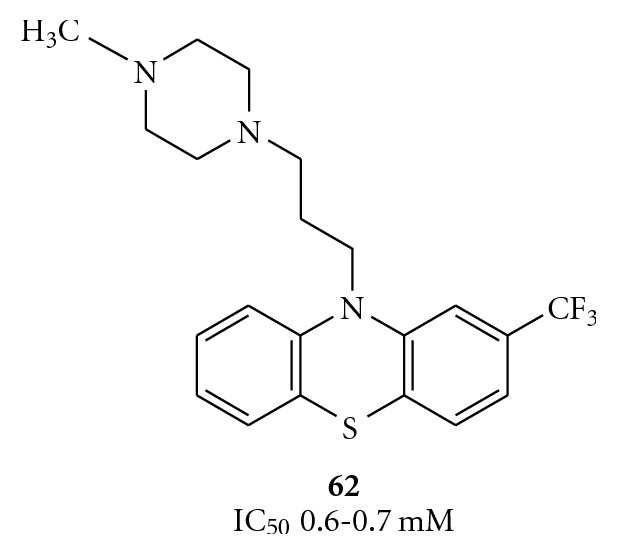
Structure of the calmodulin antagonist trifluoperazine (62).
Even some tropolones have been screened as inhibitors of HCV helicase-catalyzed DNA unwinding. Recently Bernatowicz et al. have described several derivatives bearing a side chain that connect the seven-member ring system to some N-heterocycles.
The most active compound, 3,5,7-tri[(40-methylpiperazin-10-yl)methyl]tropolone (63), inhibited RNA replication by 50% at 46.9 μM (EC50), showing an IC50 = 3.4 μM and a CC50> 1000 μM (SI > 21), whereas the most efficient was 3,5,7-tri[(30-methylpiperidin-10-yl)methyl]tropolone (64), with an EC50 of 35.6 μM, which unfortunately exhibited a lower SI (9.8) derived by a CC50 = 348 μM. These tropolone derivatives, reported in Figure 14, are the first antihelicase compounds that inhibit HCV replication with the ability to cause the appearance of resistant mutants, suggesting that inhibition of replication is the result of inhibition of the enzyme activity. They also inhibit replication of the HCV subgenomic replicon in cell cultures [69].
Figure 14.

Structure and synthesis of the compounds 63 and 64.
3.3. Competitive Inhibition of RNA Binding
Several polynucleotides displayed inhibitory HCV helicase activity. The inhibition is believed to result from the competition of the polynucleotides with DNA or RNA substrates, an effect that could be mimicked by synthetic macromolecules [46].
With the aim of discovering new anti-HCV agents, ViroPharma synthesized several benzimidazole derivatives, two of them (compounds 65 and 66, Figure 15) showing high activity against HCV helicase [70]. Although the exact mechanism of 65 and 66 is still not clear, they might compete with nucleic acids in the manner above mentioned. In particular, the benzene ring and the NH group could interact by hydrophobic interaction and hydrogen bound, respectively.
Figure 15.

The HCV helicase inhibitors reported by ViroPharma Inc.
In the attempt to extend the SAR analysis, some new dimers containing benzimidazole, benzoxazole, pyridinoxazole, and benzothiazole rings, attached to symmetrical linkers, were synthesized by Phoon et al., as summarized in Figure 16 [70, 71]. Preliminary studies of these compounds showed a significant decrease in potency when the benzimidazole moiety was replaced by the benzoxazole or benzothiazole rings (compounds 67). On the other hand, the aminobenzimidazole-diamides (68) and aminophenyl benzimidazole-diureas (69) derivatives displayed, at 25 μg/mL, 6–13, and 20–28 percent inhibitory activity, respectively.
Figure 16.

Structures of diamides (67), aminobenzimidazole-derived diamides (68), and two aminophenyl benzimidazole-derived diureas (69).
Likewise, the linker was also implicated in the inhibitory activity since replacement of the diamide linkage possessed by 65 with the diurea linkage (compounds 69) led to reduced potency. Thus, the SAR data indicate that the benzimidazole ring, the benzene group at the C2 position of the benzimidazole moiety, and the nature of the linker are essential for the activity [70].
The synthesis of these analogues is outlined in Figure 17. Aminophenols and thiophenols, or the corresponding pyridine derivatives, reacted easily with p-aminobenzoic acid in the presence of polyphosphoric acid to afford the corresponding oxazole and thiazole derivatives (72). Subsequent coupling of 72 and 2-aminobenzoimidazole with the opportune acid dichlorides furnished the products 67–69.
Figure 17.

Synthesis of the diamides (67), aminobenzimidazole-derived diureas (68), and aminophenyl benzimidazole-derived diamides (69).
Belon et al. recently described how a prototype in the symmetrical benzimidazolephenyl series, the N1,N4-bis[4-(1H-benzimidazol-2-yl)phenyl]benzene-1,4-dicarboxamide (BIP)2B, (69a, derivative of 69 with Y = phenyl, Figure 18), binds directly the HCV NS3 helicase in the same binding site for RNA in a competitive manner. Furthermore, they reported that 69a interacts with NS3 encoded non only by various HCV genotypes, but even by Dengue virus (DV), Japanese encephalitis virus (JEV) and, even if less tightly, the related human helicase [72].
Figure 18.

Symmetrical benzimidazolephenylcarboxamide (BIP)2B.
Also small peptides specifically inhibit HCV helicases, even in cells bearing HCV replicon. Between them, a peptide expressed in bacteria, composed of 14 amino acids (p14, RRGRTGRGRRGIYR), demonstrated to be the best enzyme inhibitor. P14 has the same amino-acidic sequence as that surrounding the putative HCV helicase arginine finger and inhibits the replication of HCV replicon in cells with an EC50 = 83 μM [73], while reduces the DNA unwinding with an IC50 of 0.2 μM [74].
A new selective inhibitor of the HCV helicase, QU663 (compound 73 of Figure 19), discovered by Maga and coworkers, showed a potent and selective inhibition with Ki of 0.75 μM [75]. The study of the inhibition mechanism has revealed that QU663 is a specific inhibitor of the strand-displacement activity, without affecting the ability of NS3 helicase to hydrolyse ATP. QU663 could function as a competitive inhibitor with respect to nucleic acid substrate by decreasing the affinity of the enzyme for the substrate. Molecular docking studies further support this explanation. Therefore, QU663 inhibits the unwinding activity of NS3 in a competitive manner with respect to the DNA substrate, making it a promising candidate for a novel class of anti-HCV drugs.
Figure 19.
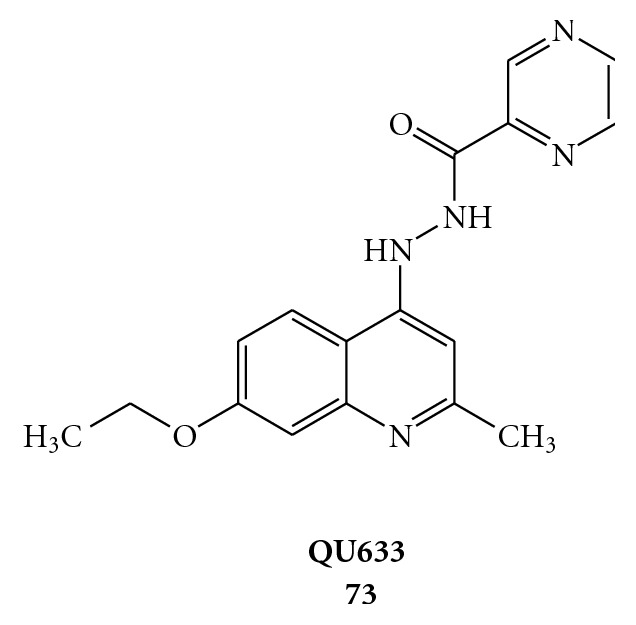
Molecular formula of (N′-(pyrazinecarbonyl)-N′′-(7-ethoxy-2-methylquinolin-4-yl)hydrazine) (QU633).
Recently, a new rational approach for the design of selective inhibitors of the HCV NS3 helicase brought the discovery of a novel HCV helicase inhibitor that potentially could compete for the nucleic acid binding site, occupying the NS3/RNA binding cler. In consequence of this de novo drug design, the predicted (E)-methyl 4-((5-(3-oxobut-1-enyl)-1H-pyrrole-2-carboxamido)methyl)benzoate (74, Figure 20) was synthesized and tested in the HCV replicon system. It inhibits HCV replicons with an EC50 of 9 μM, but showing a CC50 = 30 μM [76].
Figure 20.
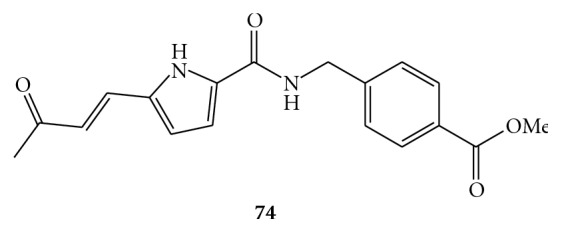
Molecular structure of compound 74.
3.4. Inhibition of the Unwinding through Intercalation of Polynucleotide Chain
DNA and RNA intercalating compounds are potential helicase inhibitors by increasing the energy required for duplex/intercalator complex unwinding [77–79].
In particular, two anthracycline derivatives, doxorubicin and nogalamycin (compounds 75 and 76, Figure 21), have been shown to be effective inhibitors of the unwinding reaction [77]. The limits in their application for the treatment of chronic viral infections is their high cytotoxicity and weak penetration into the cell. Thus, if intercalative modulation of the DNA or RNA substrates is to be considered as a possible antiviral therapy, less toxic and more selective derivatives must be identified.
Figure 21.

Structures of two DNA/RNA intercalators doxorubicin (75) and nogalamycin (76) that have displayed inhibition of the unwinding reaction catalyzed by HCV helicase.
As previously seen, the antibiotic nogalamycin (76), that interacts with allosteric binding site, has been recently used to obtain a structure-based pharmacophore model for JEV NS3 helicase/NTPase [55].
In the aim to find less toxic compounds, a large group of amidinoanthracyclines, with decreased acute toxicity and cardiotoxicity compared to the parent antibiotics, were screened against HCV helicase. From this studies emerged one of the most potent and selective inhibitors of helicase activity described in the literature. The derivative 77, showed in Figure 22, acts not only via intercalation into the double-stranded DNA substrate, but also impeding formation of the active helicase complex via competition with the enzyme for access to the substrate. Tested in the subgenomic HCV replicon system, 77 it showed an EC50 of 0.13 μM and a CC50 = 4.3 μM [80].
Figure 22.
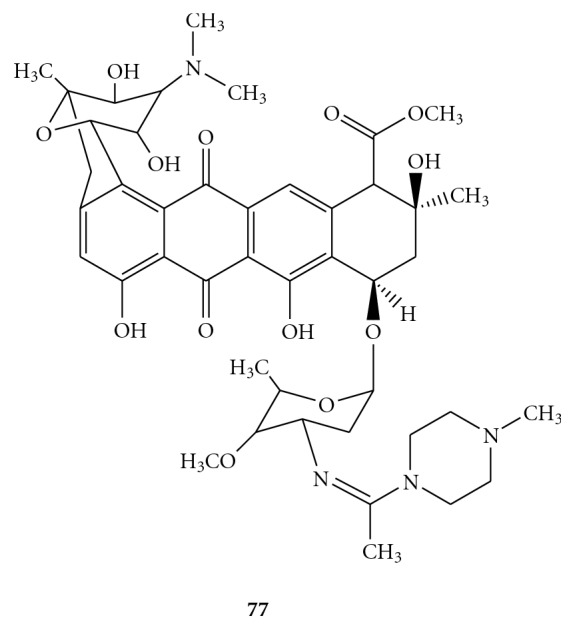
Structural formula of amidinoanthracycline derivative 77.
An other class of compounds that probably acts via intercalation into double-stranded nucleic acids with strong specificity for RNA are the acridone derivatives, but a direct interaction with the viral NS3 helicase cannot be excluded. A large group of acridones were tested from Stankiewicz-Drogon et al. using the direct fluorometric helicase activity assay to determine their inhibitory properties towards the NS3 helicase of HCV. From a preliminary study, N-(pyridin-4-yl)-amide (78) and N-(pyridin-2-yl)-amide (79) of acridone-4-carboxylic acid emerged to be efficient RNA replication inhibitors with a good specificity in subgenomic replicon system and low cytotoxicity (Figure 23) [81]. Even the thiazolpiperazinyl acridone derivative 80 demonstrated to act as a potent agent against HCV replicons (EC50 = 3 μM) and as a selective inhibitor of the HCV NS3 helicase, albeit with low potency (IC50 = 110 μM) [82]. Comparing acridone derivatives 78 and 79 with 80, we can see that the amide bonding formed after the derivatization of acridone-4-carboxylic acid with amines seemed to increase affinity and selectivity for the NS3 enzyme [81].
Figure 23.

Structures and activity of acridone-4-carboxylic acid derivatives 78 and 79 and of 7-amino-1,3,10-trimethoxy-6-(4-(thiazol-2-yl)piperazin-1-yl)acridin-9(10H)-one 80.
Finally, with the intent to improve the antiviral activity of acridones, Stankiewicz-Drogon et al. prepared a new class of compounds, namely, 5-methoxyacridone-4-carboxylic acids (MACA). From this group, compound 81 (Figure 24) came out not only as an efficient inhibitor of the NS3 helicase in the in vitro assay but also as a potent inhibitor of HCV replication endowed with low cytotoxicity for human hepatoma cells [83].
Figure 24.
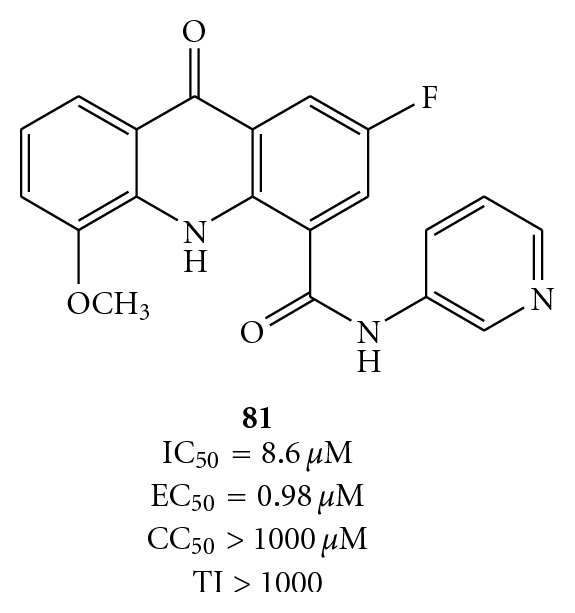
Structures of 2-fluoro-5-methoxy-9-oxo-N-(pyridin-3-yl)- -9, 10-dihydroacridine-4-carboxamide (81).
4. Coronaviridae
An enveloped single-stranded positive-sense RNA (ssRNA+) virus, SARS coronavirus (SARS-CoV), has been recently identified as the etiological agent of severe acute respiratory syndrome (SARS) in humans [84–88]. About ten thousand cases of SARS worldwide, including 800 deaths, were reported in 2003 (WHO data). Although this initial global outbreak, SARS appears to has been successfully contained, but it remains a serious concern because no vaccine or effective drug treatment is actually available. Recently, Tanner and coworkers have found that Bananin and three of their derivatives, Figure 25, function as non-competitive SARS-CoV helicase inhibitors (with IC50 values in the micromolar range) at a site different from the ATP and nucleic acid binding site, causing inhibition probably through an allosteric mechanism [89]. In foetal rhesus kidney-4cells infected with SARS-Cov, Bananin inhibited the viral replication (IC50 = 10 μM) with low host cellular toxicity (CC50 = 390 μM) [89].
Figure 25.

Molecular formula of Bananin (BAN) (82), Iodobananin (IBN) (83), Vanillinbananin (VBN) (84), Eubananin (EUB) (85).
Finally, in the last years various molecules have been detected showing an interesting and promising anti-Coronaviridae activity. Unfortunately, for many of them, was not identified a clear molecular target or mechanism of action. The fact remains that the eventual target could be the NS3 helicase. With this in mind, we report briefly the new classes of compounds that have emerged in recent published works. Among them glycopeptide antibiotics [90], which seem to interfere with the Coronavirus entry process but do not exclude an unknown cellular target; pyridine N-oxide derivatives [91]; plant lectins [92], which most probably interfere with the glycans on the spike protein during virus entry and virus release; phenanthroindolizines and phenanthroquinolizidines [93]; tetrahydroquinoline oxocarbazate derivatives as inhibitor of human cathepsin L and as entry blockers [94].
5. Picornaviridae
Picornaviridae family includes 9 genera, 3 of which are human pathogens: Enterovirus (containing poliovirus, enterovirus, coxsackievirus, echovirus), Rhinovirus (approximately 105 serotypes), and Hepatovirus (Hepatitis A virus). At present no specific antiviral therapy is available for the treatment of Picornaviridae infections. The viruses belonging to this family, all having a single-stranded positive-sense RNA (ssRNA+) genome, cause a dramatic variety of illnesses, including meningitis, colds, heart infection, conjunctivitis, and hepatitis.
Recently Carta and coworkers reported the synthesis and antiviral screening of a series of N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamides (86(1-yl), 87(2-yl) [95] and N,N′-bis-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkyldicarboxamides (88(1-yl), 89(2-yl)) [78] (see Figure 26).
Figure 26.

Molecular formula of novel N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamides (86–87) and N,N′-bis-[4-4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamides (88–89).
Compounds were evaluated in vitro for cytotoxicity and antiviral activity against a wide spectrum of ssRNA+ viruses, like Bovine Viral Diarrhea Virus (BVDV), Yellow Fever Virus (YFV), Coxsakie Virus B (CVB-2), Polio Virus (Sb-1), and Human Immunodeficiency Virus (HIV-1). Only CVB-2 and Sb-1 were inhibited by N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamide derivatives. In particular, two of them emerged for their selectivity: 87e, which was the most active against CVB-2 (EC50 = 10 μM and CC50 > 100 μM) and 86h, which was the most active against Sb-1 (EC50 = 30 μM and CC50 = 90 μM), Figure 27 [95]. N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamides (86a–e,g,h and 87a–g) were prepared by condensation of the amino derivatives 90, 91 with the appropriate anhydrides 92 under stirring at 100°C for 2 h, as shown in Figure 28. The N,N′-bis[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkyldicarboxamides (88a–d, g, h and 88a–d89a–g, i–k) were in turn prepared, as reported in Figure 29, by condensation of the amines 1(2)-(4-aminophenyl)benzotriazoles (90, 91) with the suitable diacyl dichlorides (93).
Figure 27.

Molecular formula of compounds 86h and 87e.
Figure 28.

Synthesis of N-[4-1H(2H)-benzotriaol-1(2)-yl)phenyl]alkylcarboxamides (86 and 87).
Figure 29.

Synthesis of N,N′-bis[4-1H(2H)-benzotriaol-1(2)- yl)phenyl]alkylcarboxamides 88a–d, g, h and 89a–g, i–k.
Among N,N′-bis-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkyldicarboxamides, the bis-5,6-dimethyl-derivatives (89d–f) exhibited good activity against Enteroviruses (EC50 were 7–11 μM against CVB-2 and 19–52 μM against Sb-1) and the bis-5,6-dichloro-benzotriazol-2-yl derivatives (80i–k) showed very selective activity against CVB-2 (EC50 = 4–11 μM) resulting to be completely inactive against all the other viruses screened [96].
N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamides (86 and 87) were evaluated in silico against the 3D model of the Sb-1 helicase, as exemplified by compounds 86h (a) and 89f (b) in Figure 30. The portion of the enzyme containing the binding site interacting with the inhibitors consists of two loops, part of two β-sheets, and part of three helices.
Figure 30.
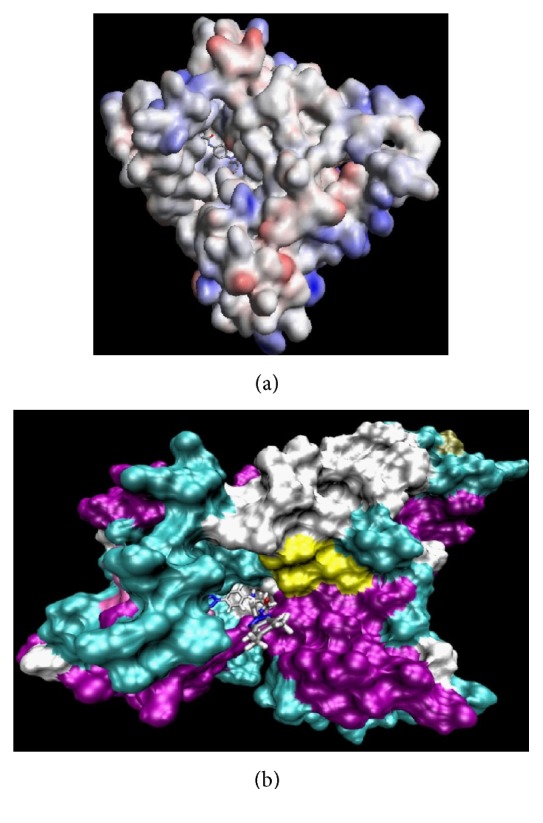
Binding of compounds 86h (a) and 89f (b) to the putative binding site on the surface of Polio (Sb-1) helicase.
It is important to notice that, with respect to the N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamide series, all the N,N′-bis[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkyldicarboxamide derivatives bind helicase Sb-1 in a different manner, as expected, due to their different shapes and dimensions. This is well quantified by the value of the solvent accessible volume of this new class of inhibitors, which on average has almost doubled compared to that of the previous molecular series (e.g., 1672 Å3 versus 891 Å3, resp.). Accordingly, it is impossible for the protein pocket to host N,N′-bis[4-(1H(2H)-benzotriazol-1(2)-yl) phenyl]alkyldicarboxamides with the same binding mode, and the results form the docking study reveal that only one of the two identical inhibitors moieties can be positioned well within the binding pocket. However, in correspondence of the most favored binding mode for the most active compounds, the formation of a new, small network of H-bonds between 89f and enzyme was observed. In particular, the analysis of the trajectories of the MD simulations for the 89f/helicase complex as an example indicates that there is a constant presence of an H-bond which involves the carbonyl oxygen atom of the Asn179 side chain and the triazole N(1) atom of the drug, characterized by an average dynamic length (ADL) of 3.0 Å. At the same time, it is possible to verify the formation of other two H-bond interactions, the former between the C=O backbone group of Ser221 and the NH group of the amidic moiety of 89f (ADL = 1.6 Å), and the latter between the carbonyl oxygen atom of the C=O group of the same amidic moiety of 89f and the side chain hydroxyl group of Ser221 (ADL = 2.6 Å).
Compounds 89i–k also exhibited selectivity against Coxsackie B2CVB-2. Unfortunately, homology standard techniques were not able to produce a reliable 3D model for the CVB-2 virus helicase, due to very low sequence identities found during alignment processes.
In the absence of a 3D model for the CVB-2 helicase, the activity of 89i–k can be explained adopting a 2D alignment analysis.
The putative binding site proposed by Carta and coworkers for Sb-1 is composed by 30 residues and, according to their 2D alignment, the binding site for CVB-2 differs for 7 residues only. Among these, Ser296 in Sb-1 is mutated to Arg237 in CBV-2. Following their analysis, and a preliminary visual inspection based on the swapping of Ser to Arg in the Polio helicase, they concluded that this is the most important residue in the case of compounds 89i–k, featuring chlorine atoms as substituents. In fact, the positively charged side chain of Arg237 is placed at an average distance of 3.5 Å from the Cl atoms, thus sensibly resulting is strong electrostatic interactions between the inhibitor and the protein. These speculations, which may account in part for the selectivity of these compounds with respect to CVB-2, clearly await further confirmation from the simulations performed on the corresponding protein 3D models.
References
- 1.Lohman T. M., Bjornson K. P. Mechanisms of helicase-catalyzed DNA unwinding. Annual Review of Biochemistry. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 2.Soultanas P., Wigley D. B. Unwinding the 'Gordian knot' of helicase action. Trends in Biochemical Sciences. 2001;26(1):47–54. doi: 10.1016/S0968-0004(00)01734-5. [DOI] [PubMed] [Google Scholar]
- 3.Singleton M. R., Wigley D. B. Modularity and specialization in superfamily 1 and 2 helicases. Journal of Bacteriology. 2002;184(7):1819–1826. doi: 10.1128/JB.184.7.1819-1826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin M. K., Patel S. S. Helicases as motor proteins. In: Schliwa M., editor. Molecular Motors. Weinheim, Germany: Wiley-VCH; 2002. pp. 179–198. [Google Scholar]
- 5.Ilyina T. V., Gorbalenya A. E., Koonin E. V. Organization and evolution of bacterial and bacteriophage primase-helicase systems. Journal of Molecular Evolution. 1992;34(4):351–357. doi: 10.1007/BF00160243. [DOI] [PubMed] [Google Scholar]
- 6.Lohman T. M. Escherichia coli DNA helicases: mechanisms of DNA unwinding. Molecular Microbiology. 1992;6(1):5–14. doi: 10.1111/j.1365-2958.1992.tb00831.x. [DOI] [PubMed] [Google Scholar]
- 7.Matson S. W., Bean D. W., George J. W. DNA helicases: enzymes with essential roles in all aspects of DNA metabolism. BioEssays. 1994;16(1):13–22. doi: 10.1002/bies.950160103. [DOI] [PubMed] [Google Scholar]
- 8.Lain S., Riechmann J. L., Garcia J. A. RNA helicase: a novel activity associated with a protein encoded by a positive strand RNA virus. Nucleic Acids Research. 1990;18(23):7003–7006. doi: 10.1093/nar/18.23.7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xi X. G. Helicases as antiviral and anticancer drug targets. Current Medicinal Chemistry. 2007;14(8):883–915. doi: 10.2174/092986707780362998. [DOI] [PubMed] [Google Scholar]
- 10.Kwong A. D., Rao B. G., Jeang K. T. Viral and cellular RNA helicases as antiviral targets. Nature Reviews Drug Discovery. 2005;4(10):845–853. doi: 10.1038/nrd1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frick D. N., Lam A. M. I. Understanding helicases as a means of virus control. Current Pharmaceutical Design. 2006;12(11):1315–1338. doi: 10.2174/138161206776361147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belon C. A., Frick D. N. Helicase inhibitors as specifically targeted antiviral therapy for hepatitis C. Future Virology. 2009;4(3):277–293. doi: 10.2217/fvl.09.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh D., Basu A. Present perspectives on flaviviral chemotherapy. Drug Discovery Today. 2008;13(13-14):619–624. doi: 10.1016/j.drudis.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Borowski P., Niebuhr A., Schmitz H., et al. NTPase/helicase of Flaviviridae: inhibitors and inhibition of the enzyme. Acta Biochimica Polonica. 2002;49(3):597–614. [PubMed] [Google Scholar]
- 15.Hayashi P. H., Di Bisceglie A. M. The progression of hepatitis B- and C-infections to chronic liver disease and hepatocellular carcinoma: epidemiology and pathogenesis. Medical Clinics of North America. 2005;89(2):371–389. doi: 10.1016/j.mcna.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Enterovirus surveillance—United States, 2002–2004. Morbidity and Mortality Weekly Report. 2006;55(6):153–156 . [PubMed] [Google Scholar]
- 17.Bennett R. J., Keck J. L., Wang J. C. Structure and function of RecQ DNA helicases. Critical Reviews in Biochemistry and Molecular Biology. 2004;4(2):79–97. doi: 10.1080/10409230490460756. [DOI] [PubMed] [Google Scholar]
- 18.Yedavalli V. S. R. K., Neuveut C., Chi YA. H., Kleiman L., Jeang K. T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119(3):381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 19.Lüking A., Stahl U., Schmidt U. The protein family of RNA helicases. Critical Reviews in Biochemistry and Molecular Biology. 1998;33(4):259–296. doi: 10.1080/10409239891204233. [DOI] [PubMed] [Google Scholar]
- 20.Cordin O., Banroques J., Tanner N. K., Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367(1-2):17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Gorbalenya A. E., Koonin E. V. Helicases: amino acid sequence comparisons and structure-function relationships. Current Opinion in Structural Biology. 1993;3(3):419–429. [Google Scholar]
- 22.Gorbalenya A. E., Koonin E. V., Donchenko A. P., Blinov V. M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Research. 1989;17(12):4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorbalenya A. E., Koonin E. V., Wolf Y. I. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Letters. 1990;262(1):145–148. doi: 10.1016/0014-5793(90)80175-I. [DOI] [PubMed] [Google Scholar]
- 24.Richardson J. P. Loading Rho to terminate transcription. Cell. 2003;114(2):157–159. doi: 10.1016/S0092-8674(03)00554-3. [DOI] [PubMed] [Google Scholar]
- 25.Walker J. E., Saraste M., Runswick M. J., Gay N. J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO Journal. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanya H. S., Bird L. E., Brannigan J. A., Wigley D. B. Crystal structure of a DExx box DNA helicase. Nature. 1996;384(6607):379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- 27.Orelle C., Dalmas O., Gros P., Di Pietro A., Jault J. M. The conserved glutamate residue adjacent to the Walker-B Motif is the catalytic base for ATP hydrolysis in the ATP-binding cassette transporter BmrA. Journal of Biological Chemistry. 2003;278(47):47002–47008. doi: 10.1074/jbc.M308268200. [DOI] [PubMed] [Google Scholar]
- 28.Goetzinger K. R., Rao V. B. Defining the ATPase center of bacteriophage T4 DNA packaging machine: requirement for a catalytic glutamate residue in the large terminase protein gp17. Journal of Molecular Biology. 2003;331(1):139–154. doi: 10.1016/S0022-2836(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 29.Ahmadian M. R., Stege P., Scheffzek K., Wittinghofer A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nature Structural Biology. 1997;4(9):686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 30.Nadanaciva S., Weber J., Wilke-Mounts S., Senior A. E. Importance of F-ATPase residue α-Arg-376 for catalytic transition state stabilization. Biochemistry. 1999;38(47):15493–15499. doi: 10.1021/bi9917683. [DOI] [PubMed] [Google Scholar]
- 31.Kleymann G., Fischer R., Betz U. A. K., et al. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nature Medicine. 2002;8(4):392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 32.Crumpacker C. S., Schaffer P. A. New anti-HSV therapeutics target the helicase-primase complex. Nature Medicine. 2002;8(4):327–328. doi: 10.1038/nm0402-327. [DOI] [PubMed] [Google Scholar]
- 33.Frick D. N. Helicases as antiviral drug targets. Drug News and Perspectives. 2003;16(6):355–362. doi: 10.1358/dnp.2003.16.6.829307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon C. P., Keller P. A. Control of hepatitis C: a medicinal chemistry perspective. Journal of Medicinal Chemistry. 2005;48(1):1–20. doi: 10.1021/jm0400101. [DOI] [PubMed] [Google Scholar]
- 35.Lindenbach Brett D., Thiel Heinz-Jurgen, Rice Charles M. Flaviviridae: the viruses and their replication. In: Knipe D. M., Howley P. M., editors. Fields Virology. 5th. Philadelphia, Pa, USA: Lippincott-Raven; 2007. [Google Scholar]
- 36.Choo Q. L., Kuo G., Weiner A. J., Overby L. R., Bradley D. W., Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 37.World Health Organization. Hepatitis C. Weekly Epidemiological Record. 1997;72:65–69. [Google Scholar]
- 38.Memon M. I., Memon M. A. Hepatitis C: an epidemiological review. Journal of Viral Hepatitis. 2002;9(2):84–100. doi: 10.1046/j.1365-2893.2002.00329.x. [DOI] [PubMed] [Google Scholar]
- 39.Echevarría-Mayo J. M. Etiology and pathogenesis of viral hepatitis. Enfermedades Infecciosas y Microbiologia Clinica. 2006;24(1):45–56. doi: 10.1157/13083375. [DOI] [PubMed] [Google Scholar]
- 40.Bosch F. X., Ribes J., Cléries R., Díaz M. Epidemiology of hepatocellular carcinoma. Clinics in Liver Disease. 2005;9(2):191–211. doi: 10.1016/j.cld.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 41.Cornberg M., Wedemeyer H., Manns M. P. Treatment of chronic hepatitis C with PEGylated interferon and ribavirin. Current Gastroenterology Reports. 2002;4(1):23–30. doi: 10.1007/s11894-002-0034-y. [DOI] [PubMed] [Google Scholar]
- 42.Tan S. L., Pause A., Shi Y., Sonenberg N. Hepatitis C therapeutics: current status and emerging strategies. Nature Reviews Drug Discovery. 2002;1(11):867–881. doi: 10.1038/nrd937. [DOI] [PubMed] [Google Scholar]
- 43.Hwang L. H., Hsieh C. L., Yen A., Chung YI. L., Chen D. S. Involvement of the 5' proximal coding sequences of hepatitis C virus with internal initiation of viral translation. Biochemical and Biophysical Research Communications. 1998;252(2):455–460. doi: 10.1006/bbrc.1998.9673. [DOI] [PubMed] [Google Scholar]
- 44.Shuman S. Vaccinia virus RNA helicase: an essential enzyme related to the DE-H family of RNA-dependent NTPases. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10935–10939. doi: 10.1073/pnas.89.22.10935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner J. D. O., Jankowsky E., Company M., Pyle A. M., Abelson J. N. The DEAH-box protein PRP22 is an ATPase that mediates ATP-dependent mRNA release from the spliceosome and unwinds RNA duplexes. EMBO Journal. 1998;17(10):2926–2937. doi: 10.1093/emboj/17.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borowski P., Mueller O., Niebuhr A., et al. ATP-binding domain of NTPase/helicase as a target for hepatitis C antiviral therapy. Acta Biochimica Polonica. 2000;47(1):173–180. [PubMed] [Google Scholar]
- 47.Borowski P., Kuehl R., Mueller O., Hwang L. H., Zur Wiesch J. S., Schmitz H. Biochemical properties of a minimal functional domain with ATP-binding activity of the NTPase/helicase of hepatitis C virus. European Journal of Biochemistry. 1999;266(3):715–723. doi: 10.1046/j.1432-1327.1999.00854.x. [DOI] [PubMed] [Google Scholar]
- 48.Borowski P., Lang M., Niebuhr A., et al. Inhibition of the helicase activity of HCV NTPase/helicase by 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide-5′-triphosphate (ribavirin-TP) Acta Biochimica Polonica. 2001;48(3):739–744. [PubMed] [Google Scholar]
- 49.Borowski P., Deinert J., Schalinski S., et al. Halogenated benzimidazoles and benzotriazoles as inhibitors of the NTPase/helicase activities of hepatitis C and related viruses. European Journal of Biochemistry. 2003;270(8):1645–1653. doi: 10.1046/j.1432-1033.2003.03540.x. [DOI] [PubMed] [Google Scholar]
- 50.Bretner M., Baier A., Kopańska K., et al. Synthesis and biological activity of 1H-benzotriazole and 1H-benzimidazole analogues—inhibitors of the NTPase/helicase of HCV and of some related Flaviviridae. Antiviral Chemistry and Chemotherapy. 2005;16(5):315–326. doi: 10.1177/095632020501600504. [DOI] [PubMed] [Google Scholar]
- 51.Bretner M., Najda A., Podwińska R., et al. Inhibitors of the NTPase/helicases of hepatitis C and related Flaviviridae viruses. Acta Poloniae Pharmaceutica. 2004;61:26–28. [PubMed] [Google Scholar]
- 52.Paeshuyse J., Vliegen I., Coelmont L., et al. Comparative in vitro anti-hepatitis C virus activities of a selected series of polymerase, protease, and helicase inhibitors. Antimicrobial Agents and Chemotherapy. 2008;52(9):3433–3437. doi: 10.1128/AAC.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porter D. J. T. A kinetic analysis of the oligonucleotide-modulated ATPase activity of the helicase domain of the NS3 protein from hepatitis C virus: the first cycle of interaction of ATP with the enzyme is unique. Journal of Biological Chemistry. 1998;273(23):14247–14253. doi: 10.1074/jbc.273.23.14247. [DOI] [PubMed] [Google Scholar]
- 54.Zhang N., Chen H. M., Koch V., et al. Ring-expanded (“fat”) nucleoside and nucleotide analogues exhibit potent in vitro activity against Flaviviridae NTPases/helicases, including those of the West Nile virus, hepatitis C virus, and Japanese encephalitis virus. Journal of Medicinal Chemistry. 2003;46(19):4149–4164. doi: 10.1021/jm030842j. [DOI] [PubMed] [Google Scholar]
- 55.Kaczor A., Matosiuk D. Structure-based virtual screening for novel inhibitors of Japanese encephalitis virus NS3 helicase/nucleoside triphosphatase. FEMS Immunology and Medical Microbiology. 2010;58(1):91–101. doi: 10.1111/j.1574-695X.2009.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ludwig J. A new route to nucleoside 5'-triphosphates. (Short communication) Acta Biochimica et Biophysica Academiae Scientiarum Hungaricae. 1981;16(3-4):131–133. [PubMed] [Google Scholar]
- 57.Vorbrüggen H., Bennua B. Nucleoside syntheses, XXV1). A new simplified nucleoside synthesis. Chemische Berichte. 1981;114(4):1279–1286. [Google Scholar]
- 58.Vorbrüggen H., Krolikiewicz K., Bennua B. Nucleoside syntheses, XXII1). Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chemische Berichte. 1981;114(4):1234–1255. [Google Scholar]
- 59.Bhat C. C. Synthetic Procedures in Nucleic Acid Chemistry. New York, NY, USA: John Wiley & Sons; 1968. [Google Scholar]
- 60.Hodge R. P., Brush C. K., Harris C. M., Harris T. M. Synthesis of 1- and 1,2,2′-deuteriated deoxyribose and incorporation into deoxyribonucleosides. Journal of Organic Chemistry. 1991;56(4):1553–1564. [Google Scholar]
- 61.Wang L., Bhan A., Hosmane R. S. A short synthesis of a novel ring-expanded purine and its nucleoside analogue containing the imidazo[4,5-e][1,3]diazepine ring skeleton with multiple amino substituents attached to the 7-membered ring. Nucleosides and Nucleotides. 1994;13(10):2307–2320. [Google Scholar]
- 62.Hosmane R. S., Bhadti V. S., Lim B. B. Synthesis of a novel ring-expanded xanthine analogue and several methyl or benzyl derivatives containing the 5:7-fused imidazo[4,5-e][1,2,4]triazepine ring system. Synthesis. 1990;(11):1095–1100. [Google Scholar]
- 63.Zhang N., Chen H. M., Koch V., et al. Potent inhibition of NTPase/helicase of the west nile virus by ring-expanded (“Fat”) nucleoside analogues. Journal of Medicinal Chemistry. 2003;46(22):4776–4789. doi: 10.1021/jm030277k. [DOI] [PubMed] [Google Scholar]
- 64.Baxter R. A., Spring F. S. The application of the hofmann reaction to the synthesis of heterocyclic compounds. Part II. Synthesis of xanthine from glyoxaline-4: 5-dicarboxyamide and of 9-methylxanthine from 1-methylglyoxaline-4: 5-dicarboxyamide. Journal of the Chemical Society. 1945:232–234. [Google Scholar]
- 65.Kolks G., Frihart C. R., Coughlin P. K., Lippard S. J. Synthetic, spectroscopic, and solution studies of imidazolate-bridged dicopper(II) complexes. Inorganic Chemistry. 1981;20(9):2933–2940. [Google Scholar]
- 66.Borowski P., Lang M., Haag A., et al. Characterization of imidazo[4,5-d]pyridazine nucleosides as modulators of unwinding reaction mediated by West Nile virus nucleoside triphosphatase/helicase: evidence for activity on the level of substrate and/or enzyme. Antimicrobial Agents and Chemotherapy. 2002;46(5):1231–1239. doi: 10.1128/AAC.46.5.1231-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen C. S., Chiou C. T., Chen G. S., et al. Structure-based discovery of triphenylmethane derivatives as inhibitors of hepatitis C virus helicase. Journal of Medicinal Chemistry. 2009;52(9):2716–2723. doi: 10.1021/jm8011905. [DOI] [PubMed] [Google Scholar]
- 68.Kuang W. F., Lin YU. C., Jean F., et al. Hepatitis C virus NS3 RNA helicase activity is modulated by the two domains of NS3 and NS4A. Biochemical and Biophysical Research Communications. 2004;317(1):211–217. doi: 10.1016/j.bbrc.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 69.Najda-Bernatowicz A., Krawczyk M., Stankiewicz-Drogoń A., Bretner M., Boguszewska-Chachulska A. M. Studies on the anti-hepatitis C virus activity of newly synthesized tropolone derivatives: identification of NS3 helicase inhibitors that specifically inhibit subgenomic HCV replication. Bioorganic and Medicinal Chemistry. 2010;18(14):5129–5136. doi: 10.1016/j.bmc.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 70.Diana G. D., Bailey R. T. Compounds, compositions and methods for treatment of Hepatitis C. (Viropharma Incorporated) US patent no. 5,633,3888, 1997.
- 71.Phoon C. W., Ng P. Y., Ting A. E., Yeo SU. L., Sim M. M. Biological evaluation of hepatitis C virus helicase inhibitors. Bioorganic and Medicinal Chemistry Letters. 2001;11(13):1647–1650. doi: 10.1016/S0960-894X(01)00263-3. [DOI] [PubMed] [Google Scholar]
- 72.Belon C. A., High Y. D., Lin T. I., Pauwels F., Frick D. N. Mechanism and specificity of a symmetrical benzimidazolephenylcarboxamide helicase inhibitor. Biochemistry. 2010;49(9):1822–1832. doi: 10.1021/bi901974a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gozdek A., Zhukov I., Polkowska A., et al. NS3 peptide, a novel potent hepatitis C virus NS3 helicase inhibitor: its mechanism of action and antiviral activity in the replicon system. Antimicrobial Agents and Chemotherapy. 2008;52(2):393–401. doi: 10.1128/AAC.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borowski P., Heising M. V., Miranda I. B., Liao C. L., Choe J., Baier A. Viral NS3 helicase activity is inhibited by peptides reproducing the Arg-rich conserved motif of the enzyme (motif VI) Biochemical Pharmacology. 2008;76(1):28–38. doi: 10.1016/j.bcp.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 75.Maga G., Gemma S., Fattorusso C., et al. Specific targeting of hepatitis C virus NS3 RNA helicase. Discovery of the potent and selective competitive nucleotide-mimicking inhibitor QU663. Biochemistry. 2005;44(28):9637–9644. doi: 10.1021/bi047437u. [DOI] [PubMed] [Google Scholar]
- 76.Kandil S., Biondaro S., Vlachakis D., et al. Discovery of a novel HCV helicase inhibitor by a de novo drug design approach. Bioorganic and Medicinal Chemistry Letters. 2009;19(11):2935–2937. doi: 10.1016/j.bmcl.2009.04.074. [DOI] [PubMed] [Google Scholar]
- 77.Borowski P., Schalinski S., Schmitz H. Nucleotide triphosphatase/helicase of hepatitis C virus as a target for antiviral therapy. Antiviral Research. 2002;55(3):397–412. doi: 10.1016/S0166-3542(02)00096-7. [DOI] [PubMed] [Google Scholar]
- 78.Zhu K., Henning D., Iwakuma T., Valdez B. C., Busch H. Adriamycin inhibits human RH II/Gu RNA helicase activity by binding to its substrate. Biochemical and Biophysical Research Communications. 1999;266(2):361–365. doi: 10.1006/bbrc.1999.1815. [DOI] [PubMed] [Google Scholar]
- 79.Bachur N. R., Yu F., Johnson R., Hickey R., Wu Y., Malkas L. Helicase inhibition by anthracycline anticancer agents. Molecular Pharmacology. 1992;41(6):993–998. [PubMed] [Google Scholar]
- 80.Krawczyk M., Wasowska-Lukawska M., Oszczapowicz I., Boguszewska-Chachulska A. M. Amidinoanthracyclines—a new group of potential anti-hepatitis C virus compounds. Biological Chemistry. 2009;390(4):351–360. doi: 10.1515/BC.2009.040. [DOI] [PubMed] [Google Scholar]
- 81.Stankiewicz-Drogon A., Palchykovska L. G., Kostina V. G., Alexeeva I. V., Shved A. D., Boguszewska-Chachulska A. M. New acridone-4-carboxylic acid derivatives as potential inhibitors of Hepatitis C virus infection. Bioorganic and Medicinal Chemistry. 2008;16(19):8846–8852. doi: 10.1016/j.bmc.2008.08.074. [DOI] [PubMed] [Google Scholar]
- 82.Manfroni G., Paeshuyse J., Massari S., et al. Inhibition of subgenomic hepatitis C virus RNA replication by acridone derivatives: identification of an NS3 helicase inhibitor. Journal of Medicinal Chemistry. 2009;52(10):3354–3365. doi: 10.1021/jm801608u. [DOI] [PubMed] [Google Scholar]
- 83.Stankiewicz-Drogoń A., Dörner B., Erker T., Boguszewska-Chachulska A. M. Synthesis of new acridone derivatives, inhibitors of NS3 helicase, which efficiently and specifically inhibit subgenomic HCV replication. Journal of Medicinal Chemistry. 2010;53(8):3117–3126. doi: 10.1021/jm901741p. [DOI] [PubMed] [Google Scholar]
- 84.Peiris J. S. M., Lai S. T., Poon L. L. M., et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Drosten C., Günther S., Preiser W., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England Journal of Medicine. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 86.Rota P. A., Oberste M. S., Monroe S. S., et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 87.Marra M. A., Jones S. J. M., Astell C. R., et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300(5624):1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 88.Ksiazek T. G., Erdman D., Goldsmith C. S., et al. A novel coronavirus associated with severe acute respiratory syndrome. New England Journal of Medicine. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 89.Tanner J. A., Zheng BO. J., Zhou J., et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chemistry and Biology. 2005;12(3):303–311. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Balzarini J., Keyaerts E., Vijgen L., et al. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Research. 2006;72(1):20–33. doi: 10.1016/j.antiviral.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Balzarini J., Keyaerts E., Vijgen L., et al. Pyridine N-oxide derivatives are inhibitory to the human SARS and feline infectious peritonitis coronavirus in cell culture. Journal of Antimicrobial Chemotherapy. 2006;57(3):472–481. doi: 10.1093/jac/dki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Keyaerts E., Vijgen L., Pannecouque C., et al. Plant lectins are potent inhibitors of coronaviruses by interfering with two targets in the viral replication cycle. Antiviral Research. 2007;75(3):179–187. doi: 10.1016/j.antiviral.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang C.-W., Lee Y.-Z., Kang I.-J., et al. Identification of phenanthroindolizines and phenanthroquinolizidines as novel potent anti-coronaviral agents for porcine enteropathogenic coronavirus transmissible gastroenteritis virus and human severe acute respiratory syndrome coronavirus. Antiviral Research. 2010;88(2):160–168. doi: 10.1016/j.antiviral.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shah P. P., Wang T., Kaletsky R. L., et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Molecular Pharmacology. 2010;78(2):319–324. doi: 10.1124/mol.110.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carta A., Loriga G., Piras S., et al. Synthesis and in vitro evaluation of the anti-viral activity of N-[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl]alkylcarboxamides. Medicinal Chemistry. 2006;2(6):577–589. doi: 10.2174/1573406410602060577. [DOI] [PubMed] [Google Scholar]
- 96.Carta A., Loriga M., Piras S., et al. Synthesis and anti-picornaviridae in vitro activity of a new class of helicase inhibitors the N,N′-bis[4-(1H(2H)-benzotriazol-1(2)-yl)phenyl] alkyldicarboxamides. Medicinal Chemistry. 2007;3(6):520–532. doi: 10.2174/157340607782360308. [DOI] [PubMed] [Google Scholar]