

Abstract
Objective:
To examine associations between aggregate genetic risk and Alzheimer disease (AD) markers in stages preceding the clinical symptoms of dementia using data from 2 large observational cohort studies.
Methods:
We computed polygenic risk scores (PGRS) using summary statistics from the International Genomics of Alzheimer's Project genome-wide association study of AD. Associations between PGRS and AD markers (cognitive decline, clinical progression, hippocampus volume, and β-amyloid) were assessed within older participants with dementia. Associations between PGRS and hippocampus volume were additionally examined within healthy younger participants (age 18–35 years).
Results:
Within participants without dementia, elevated PGRS was associated with worse memory (p = 0.002) and smaller hippocampus (p = 0.002) at baseline, as well as greater longitudinal cognitive decline (memory: p = 0.0005, executive function: p = 0.01) and clinical progression (p < 0.00001). High PGRS was associated with AD-like levels of β-amyloid burden as measured with florbetapir PET (p = 0.03) but did not reach statistical significance for CSF β-amyloid (p = 0.11). Within the younger group, higher PGRS was associated with smaller hippocampus volume (p = 0.05). This pattern was evident when examining a PGRS that included many loci below the genome-wide association study (GWAS)–level significance threshold (16,123 single nucleotide polymorphisms), but not when PGRS was restricted to GWAS-level significant loci (18 single nucleotide polymorphisms).
Conclusions:
Effects related to common genetic risk loci distributed throughout the genome are detectable among individuals without dementia. The influence of this genetic risk may begin in early life and make an individual more susceptible to cognitive impairment in late life. Future refinement of polygenic risk scores may help identify individuals at risk for AD dementia.
The asymptomatic stage of Alzheimer disease (AD) is thought to last over a decade, during which the pathophysiologic processes are under way in the absence of clinical symptoms.1 Given that current clinical trials are testing whether antiamyloid therapies slow cognitive decline among clinically normal individuals at risk for AD dementia,2,3 it is critical to understand the influence of risk factors before overt symptoms of dementia are present.
Genetic variants have a large influence in sporadic AD dementia, with heritability estimates exceeding 60%.4 In addition to the APOE gene,5 to date 21 common genetic variants have been associated with AD in large genome-wide association study (GWAS) meta-analyses.6 However, the effect sizes of these variants are small (odds ratios <1.22), and recent work suggests that numerous additional loci distributed throughout the genome explain a much larger portion of the variance than the select few that surpass GWAS-level significance thresholds.7–9 For instance, GWAS-confirmed loci account for 2% of the variance in discriminating patients with AD dementia and controls, beyond the 6% accounted for by APOE, whereas examination across the remaining 2 million common genetic variants explains an additional 25% of the variance.9 Thus, aggregation across a large number of loci is likely a more sensitive method to establish underlying genetic risk to AD dementia than solely examining loci surpassing stringent GWAS-level significance thresholds.
Although the accumulation of the β-amyloid (Aβ) protein is likely a central event in AD development,10 mechanistic pathways not directly linked to Aβ have been increasingly implicated in common genetic risk loci of AD, such as processes related to the immune system and cytoskeletal function.11–14 Given the involvement of nonamyloid pathways in common AD genetic risk loci, it is possible that genetic variants influence AD markers early in the lifespan, before amyloid accumulation has begun.
We sought to derive measures of polygenic risk based on the results from the recent large meta-analysis of AD dementia from the International Genomics of Alzheimer's Project (IGAP) and determine whether these polygenic risk scores (PGRS) were associated with AD-relevant markers during the stages preceding dementia among older participants, but also early in the lifespan among younger participants. Understanding the early influence of genetic risk will provide important insights into mechanisms of AD development throughout the lifespan as well as improve detection of at-risk individuals before clinical symptoms and widespread neuronal damage has ensued.
METHODS
Participants.
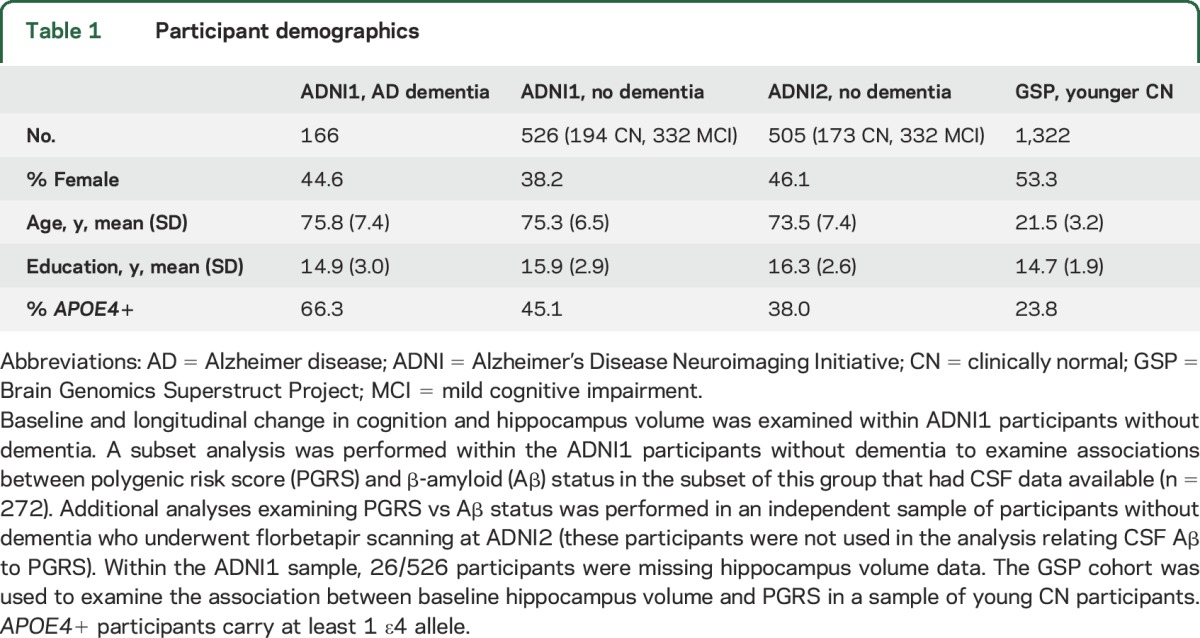
Our analyses included older clinically normal (CN) participants, patients with mild cognitive impairment (MCI), and patients with AD dementia from the Alzheimer's Disease Neuroimaging Initiative study (ADNI),15 as well as younger CN participants from the Brain Genomics Superstruct Project (GSP)16,17 (table 1). Enrollment began in 2004 for ADNI phase 1 and in 2011 for ADNI phase 2. Enrollment for the GSP occurred between 2008 and 2012. In brief, older CN participants from ADNI had Mini-Mental State Examination (MMSE) score ≥24, Clinical Dementia Rating (CDR) 0, and were within the normal range on education-adjusted Logical Memory delayed recall cutoffs. ADNI MCI had MMSE ≥24, CDR 0.5, and fell below education-adjusted logical memory delayed recall cutoffs. ADNI patients with AD dementia meet the National Institute of Neurologic and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria for probable AD, had memory complaints, MMSE 20–26, and CDR ≥0.5. Diagnoses were determined using each participant's baseline visit (clinical progression from baseline was not considered). ADNI1 participants were included in the current analysis if they had genotyping data available and were of European ancestry (n = 166 AD, 332 MCI, and 194 CN). ADNI2 participants were included if they had genotyping data available, were of European ancestry, underwent florbetapir PET, and were not included in ADNI1 analyses (n = 332 MCI and 173 CN). GSP participants were healthy volunteers in a Boston area imaging study and between 18 and 35 years old. GSP participants were included if they had genotyping data available, were of European ancestry, and had structural MRI data available (n = 1,322).
Table 1.
Participant demographics
Standard protocol approvals, registrations, and patient consents.
Institutional review boards approved study procedures across participating institutions. Written informed consent was obtained from all participants.
GWAS processing.
Genotyping procedures are described elsewhere for the ADNI18 and GSP.19 Genotyping data from the Illumina Human610-Quad BeadChip was used for ADNI1, Illumina Omni 2.5 M platform for ADNI2, and Illumina Infinium Human OmniExpress for GSP. All analyses were restricted to participants with non-Hispanic European ancestry, as identified with multidimensional scaling analysis performed in combination with the 1000 Genomes sample20 and performed within each genotyping platform separately.
Standard quality control procedures were applied to GWAS data using PLINK v1.9 (https://www.cog-genomics.org/plink2). Individuals were excluded for missing genotype rates >5% and sex inconsistency. Single nucleotide polymorphisms (SNPs) were excluded if minor allele frequency was <0.01, genotype call rate was <95%, Hardy-Weinberg Equilibrium deviation (p < 1 × 10−6), and ambiguous strand information. Nongenotyped SNPs were imputed using MiniMac35, with the 1000 Genomes European participants as the reference sample and following the Minimac cookbook instructions.21
Computation of PGRS.
We computed PGRS using PLINK's profile function, which computes the sum of reference allele counts at each SNP weighted by the log odds ratio from the stage 1 analysis of the IGAP GWAS, which contrasted 17,008 patients with AD dementia with 37,154 CN controls.6 Critically, the summation was constrained to independent loci with an IGAP stage 1 p value below a threshold. The independent loci were identified using PLINK's linkage disequilibrium (LD) clumping procedure (with 0.5 as the LD threshold), which reveals correlated sets of SNPs. LD clumping ensures that large blocks of correlated SNP sets do not overwhelm the PGRS computation. To determine the appropriate p value threshold, we iterated over a range of values (the GWAS-level significance threshold of p = 5 × 10−8, p = 0.0001, p = 0.001, p = 0.01, p = 0.02, p = 0.03, p = 0.04, p = 0.05, p = 0.10, p = 0.20, p = 0.30, p = 0.40, and p = 0.50). Conservative p value thresholds (e.g., p = 5 × 10−8) result in fewer SNPs contributing to the PGRS, while liberal p value thresholds (e.g., p = 0.5) result in a large number of contributing SNPs (table e-1 on the Neurology® Web site at Neurology.org). For all primary analyses, chromosome 19 SNPs were excluded to preclude any influence of the APOE gene. However, we also repeated analyses using PGRS calculations that included chromosome 19. Importantly, the ADNI participants used in the current analysis were not included in the IGAP stage 1 analysis that generated the summary statistics file. To select a PGRS for subsequent analyses within CN and MCI, we examined the p value threshold that best differentiated patients with AD dementia from older CN participants within the ADNI1 sample. PGRS were z-transformed based on the distribution of values within CN.
Aβ markers.
Associations with Aβ were examined in 2 independent groups without dementia from ADNI. First, we examined this association within a subset of ADNI1 participants with CSF data22 (n = 272). Given the small sample size of this subset, we additionally explored associations with Aβ within 505 participants without dementia who underwent florbetapir PET imaging in ADNI2.23 Participants were only included in this analysis if they were not used in the ADNI1 CSF analysis to ensure an independent sample. Since ADNI1 and ADNI2 samples underwent genotyping on different platforms, analyses were performed separately within each group.
Structural MRI acquisition and processing.
Acquisition and processing of structural MRI scans have been described previously.19,24 A longitudinal Freesurfer version 4.3 pipeline was used to extract hippocampus volume (HV) for ADNI1, while a cross-sectional Freesurfer version 4.5 pipeline was used for GSP. HV was adjusted by total intracranial volume and additionally adjusted for coil type within GSP (12 vs 32 channel).
Statistical models.
Statistical analyses were performed using R v3.0 (http://www.r-project.org/). Linear mixed models were used to assess baseline and longitudinal effects in cognitive factor scores (memory and executive function)25,26 and HV (mean neuropsychological follow-up 4.58 ± 2.74 years; mean follow-up for HV 2.97 ± 1.22 years). Linear mixed models included a random intercept and slope for each participant. Risk of clinical progression (CN to MCI/AD or MCI to AD) within 3 years of follow-up was examined with logistic regression. Multiple regression was used to assess the association between PGRS and HV within GSP where only baseline data were available.
All analyses controlled for APOE4, age, and sex, as well as 5 principal components from a multidimensional scaling analysis to account for population heterogeneity. Analyses within ADNI participants without dementia additionally controlled for diagnosis (CN vs MCI). The interaction between each covariate and time was included in linear mixed models. Education was controlled in models examining cognition. The p values were 2-sided and no multiple comparisons correction was performed.
RESULTS
Participant demographics are listed in table 1. Given that the number of SNPs used to calculate a PGRS is arbitrary (table e-1), we first examined the ability of different PGRS iterations to discriminate between 166 patients with AD dementia and 194 older CN from ADNI1. Interestingly, discrimination between patients with AD dementia and older CN participants from ADNI1 dramatically increased between very stringent p values and p = 0.01, and reached a plateau after p = 0.01 (figure 1). Based on this pattern, we examined a liberally defined PGRS using the p = 0.01 threshold in follow-up analyses.
Figure 1. Polygenic risk score (PGRS) discrimination between patients with Alzheimer disease (AD) dementia and older clinically normal (CN) participants.
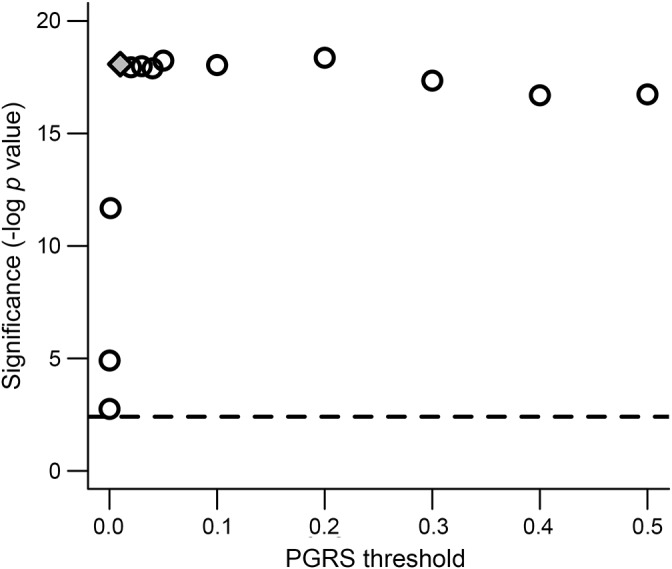
Multiple PGRS iterations were examined that incorporated different quantities of loci based on p value significance thresholds from the large International Genomics of Alzheimer's Project (IGAP) meta-analysis. Significance is shown on the y-axis (negative log p value), and corresponds to the independent contribution of PGRS in predicting diagnosis in our analysis of Alzheimer's Disease Neuroimaging Initiative patients with AD dementia compared to CN participants (controlling for APOE4, age, sex, and 5 multidimensional scaling principal components). The x-axis shows the p value threshold applied to the IGAP summary statistics file to determine which single nucleotide polymorphisms (SNP) to include in each PGRS iteration. The effect using a threshold of p = 0.01 is shown by the gray filled diamond. The horizontal dashed line reflects the significance value corresponding to a conservative PGRS that only incorporated 18 SNP that were significant in the IGAP meta-analysis.
Within the ADNI1 group without dementia (older CN and MCI combined, n = 526), linear mixed models were conducted to assess associations with baseline and longitudinal change in memory, executive function, and HV (table 2). There was a main effect of PGRS on memory (p = 0.002), such that elevated PGRS was associated with worse baseline memory performance (figure 2A). The main effect of PGRS on baseline executive function was not significant (p = 0.32; figure 2B). Finally, there was a main effect on HV, such that higher PGRS was associated with smaller baseline HV (p = 0.002, figure 2C). PGRS accounted for 2.3% of the variance in baseline memory and 2.0% of the variance in baseline HV.
Table 2.
Main findings as well as repeated analyses using a conservative polygenic risk score (PGRS)
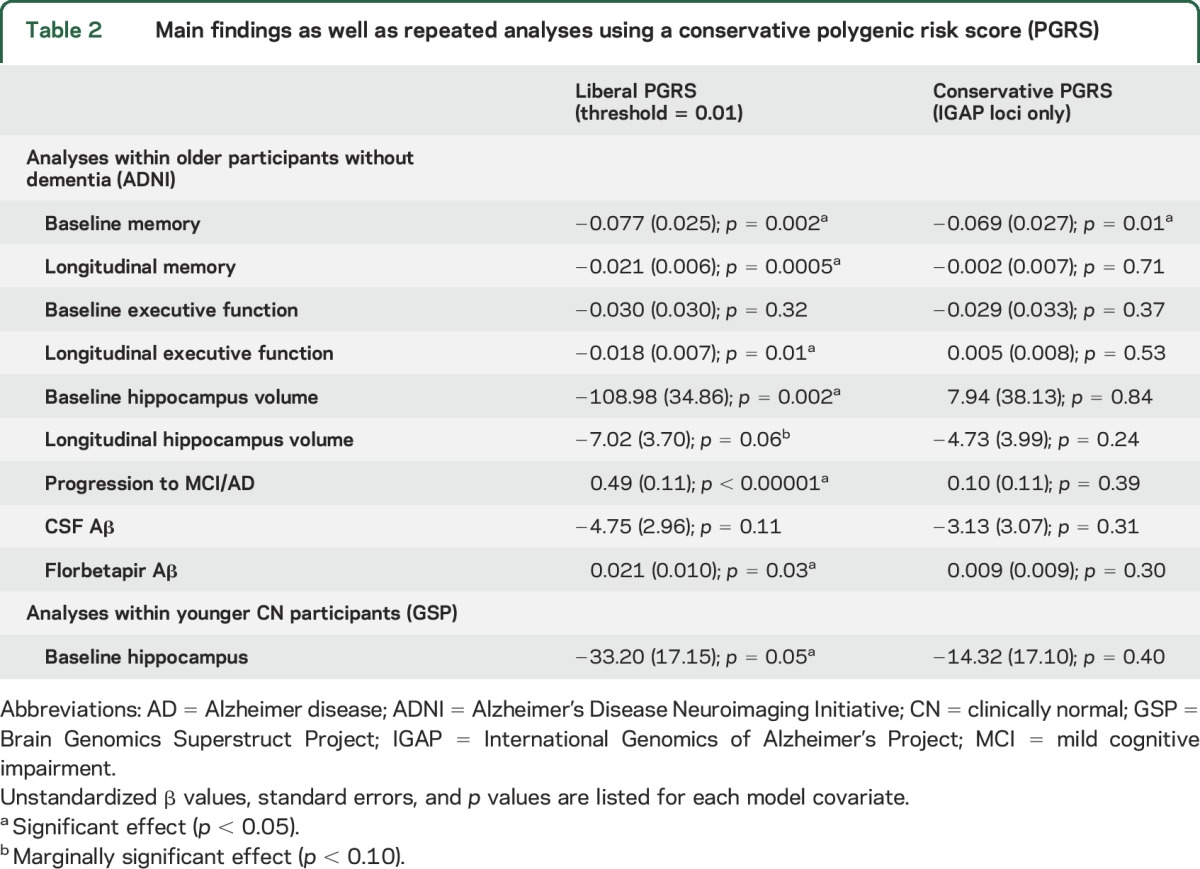
Figure 2. Polygenic risk score (PGRS) vs cognition and hippocampus volume in older participants without dementia.

PGRS vs baseline and longitudinal change in memory (A, D), executive function (EF) (B, E), and hippocampus volume (C, F). Baseline associations are shown in A–C, while longitudinal effects are shown in D–F. Plotted values are residualized by model covariates. aHV = adjusted hippocampus volume.
Within these linear mixed models, the interaction between PGRS and time was significant for both memory (p = 0.0005) and executive function (p = 0.01), such that higher PGRS was associated with greater rate of decline across both domains (figure 2, D and E, table 2). The association between PGRS and change in HV did not reach statistical significance (p = 0.06, figure 2F). PGRS accounted for 3.2% of the variance in longitudinal memory, 1.4% of the variance in longitudinal executive function, and 1.1% of the variance in longitudinal HV.
Examination of clinical progression within 3 years of follow-up (CN to MCI/AD or MCI to AD) revealed progression in 15 of the 194 ADNI1 CN and 143 of the 332 ADNI1 MCI. In a logistic regression model, PGRS was associated with increased risk of progression within 3 years (p < 0.00001, table 2, figure e-1).
Given that PGRS was significantly associated with baseline HV, cognitive decline over time, and clinical progression, models were repeated with PGRS and baseline HV as simultaneous predictors of cognitive decline and clinical progression. This analysis revealed independent contributions of PGRS and baseline HV to change in memory (PGRS: p = 0.0041, HV: p < 0.0001), change in executive function (PGRS: p = 0.07, HV: p < 0.0001), as well as risk of clinical progression (PGRS: p = 0.00004, HV: p < 0.0001).
We next examined associations between PGRS and Aβ in the subset of 272 ADNI1 participants without dementia with CSF data available (table 2). Although higher PGRS were associated with lower (e.g., more AD-like) CSF Aβ levels, this analysis did not reach statistical significance (p = 0.11, figure 3A). Given the smaller sample size of this CSF analysis, we additionally examined an independent sample of 505 participants without dementia who underwent florbetapir PET imaging in ADNI2. This analysis revealed a similar association such that elevated PGRS was associated with greater (e.g., more AD-like) levels of florbetapir Aβ (p = 0.03, figure 3B). Across both analyses, PGRS accounted for 1.0% of the variance in Aβ levels.
Figure 3. Polygenic risk score (PGRS) (residuals) vs β-amyloid (Aβ) in older participants without dementia.

The association between PGRS and Aβ was examined across 2 independent samples of older individuals without dementia: (A) ADNI1 with CSF (n = 272) and (B) ADNI2 with florbetapir PET imaging (n = 505). Plotted values are adjusted for model covariates.
Finally, we examined the association between PGRS and cross-sectional HV within a large younger cohort (age 18–30 years, n = 1,322). This analysis revealed a marginal association, such that higher PGRS was associated with smaller HV (p = 0.05; PGRS explained 0.2% of the variance in HV).
All analyses were repeated using PGRS that included chromosome 19 and revealed similar results (table e-2). Analyses were also repeated using a more conservative PGRS that only incorporated the smaller set of loci meeting statistical significance in the large IGAP meta-analysis (table 2, table e-2). Interestingly, there were no significant associations with cognition or biomarkers using this conservatively defined PGRS, with the exception of baseline memory (p = 0.01).
DISCUSSION
Among older participants without dementia, PGRS were associated with multiple AD markers. Specifically, higher PGRS was associated with worse memory, smaller HV, and AD-like levels of Aβ at baseline. We also found that higher PGRS was associated with greater longitudinal change in both memory and executive function, as well as elevated risk of clinical progression. Within a large sample of young clinically normal adults under age 35 years, we found that higher PGRS was associated with smaller HV, suggesting that an effect of aggregate genetic risk is not specific to processes occurring in late life. Importantly, this pattern of results was only present when PGRS calculations incorporated many loci below GWAS-level significance. Overall, these analyses provide evidence that aggregate genetic risk of AD dementia exerts effects that are detectable before the clinical symptoms of dementia are present, even among young adults.
The added sensitivity gained in our analyses by summing across a large number of genetic loci is consistent with work examining heritability within AD7–9 as well as studies investigating highly heritable psychiatric disorders that are associated with risk loci that individually exert small effects.27,28 This pattern implies that restricting analyses to a small number of risk loci provides a weak signal of underlying genetic risk that is improved by aggregating effects across many loci. Our results might further explain discrepancies reported across exploratory GWAS studies of AD markers. For instance, the top genome-wide significant loci that emerged from an exploratory GWAS analysis of AD pathologies (Aβ plaques and neurofibrillary tangles) were not significant at a liberal statistical significance threshold of p < 0.05 in the IGAP GWAS contrasting AD dementia to CN,6,29 highlighting that the top loci surpassing GWAS level significance thresholds in analyses examining different AD phenotypes are likely to differ. Thus, although it is necessary to apply stringent criteria in discovery style GWAS analyses to reduce the incidence of false-positives, it is highly likely that relevant signal exists below these stringent criteria.9
Within older participants without dementia, PGRS was associated with cognitive decline, highlighting that elevated genetic risk influences longitudinal trajectories even among individuals without dementia. This finding is consistent with work across multiple laboratories linking early cognitive decline to established AD risk factors, such as elevated amyloid30 and the APOE4 genotype,31 as well as work showing an association between cortical thickness and polygenic risk within CN participants (even among Aβ− CN participants).32 However, our findings are at odds with a previous study that did not find an association between liberally defined PGRS and cognitive decline among a large sample of CN participants from the Cognitive Aging Genetics in England and Scotland consortium.33 It is noteworthy that PGRS from that study was computed using an older GWAS of AD dementia with a much smaller discovery sample than the recent IGAP GWAS used in our analysis (3,941 patients with AD dementia vs 17,008 patients with AD dementia).6,34 The power of PGRS analyses is known to increase as discovery sample size increases. Nevertheless, it will be important to replicate the association between PGRS and cognitive decline in cohorts beyond ADNI.
We also found that liberal PGRS was associated with baseline HV within older adults without dementia. Although the association between PGRS and Aβ in the subset of ADNI1 participants who had CSF data available (n = 272) did not reach statistical significance (p = 0.11), an independent analysis using ADNI2 florbetapir data in a much larger sample of participants without dementia (n = 505) did reveal a significant association between PGRS and Aβ. Importantly, the magnitude and direction of the effect was identical across the 2 independent samples, providing strong support for an association between PGRS and Aβ among individuals without dementia and highlights the importance of large samples when performing analyses linking genetics with biomarkers. Although an association between Aβ and PGRS was identified in older participants, we also found that elevated PGRS was modestly associated with HV in healthy individuals under age 35 years, an age at which Aβ accumulation is very unlikely.35,36 Thus, PGRS may also have an effect via non-Aβ pathways. It has become increasingly clear that additional factors beyond Aβ influence the emergence of clinical symptoms of AD dementia, which may include developmental factors, the spread of tau into neocortex, or inflammatory responses.37–39 The involvement of non-Aβ processes in AD risk may explain why a portion of older individuals are able to function normally despite high quantities of Aβ, which has important implications for mechanisms of disease resilience.
Our study has several limitations. Given the relatively small sample size, it will be important to replicate these findings in independent samples. The associations identified between PGRS and AD markers were small, accounting for 1.0%–3.2% of the variance among the older group without dementia, and only 0.2% of the variance in HV within the younger group. However, these effect sizes are consistent with other biomarker studies assessing common genetic variants among older40 and younger participants.41 The smaller sample size within ADNI may have provided insufficient power to detect a statistically significant effect on Aβ in the ADNI1 CSF sample. However, examination of the larger florbetapir ADNI dataset provided support for an association between PGRS and Aβ among individuals without dementia.
Supplementary Material
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CDR
Clinical Dementia Rating
- CN
clinically normal
- GSP
Brain Genomics Superstruct Project
- GWAS
genome-wide association study
- HV
hippocampus volume
- IGAP
International Genomics of Alzheimer's Project
- LD
linkage disequilibrium
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- PGRS
polygenic risk score
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Alzheimer's Disease Neuroimaging Initiative, Michael Weiner, Paul Aisen, Michael Weiner, Paul Aisen, Ronald Petersen, Clifford R. Jack, Jr, William Jagust, John Q. Trojanowki, Arthur W. Toga, Laurel Beckett, Robert C. Green, Andrew J. Saykin, John Morris, Enchi Liu, Robert C. Green, Tom Montine, Ronald Petersen, Paul Aisen, Anthony Gamst, Ronald G. Thomas, Michael Donohue, Sarah Walter, Devon Gessert, Tamie Sather, Laurel Beckett, Danielle Harvey, Anthony Gamst, Michael Donohue, John Kornak, Clifford R. Jack, Jr, Anders Dale, Matthew Bernstein, Joel Felmlee, Nick Fox, Paul Thompson, Norbert Schuff, Gene Alexander, Charles DeCarli, William Jagust, Dan Bandy, Robert A. Koeppe, Norm Foster, Eric M. Reiman, Kewei Chen, Chet Mathis, John Morris, Nigel J. Cairns, Lisa Taylor-Reinwald, J.Q. Trojanowki, Les Shaw, Virginia M.Y. Lee, Magdalena Korecka, Arthur W. Toga, Karen Crawford, Scott Neu, Andrew J. Saykin, Tatiana M. Foroud, Steven Potkin, Li Shen, Zaven Kachaturian, Richard Frank, Peter J. Snyder, Susan Molchan, Jeffrey Kaye, Joseph Quinn, Betty Lind, Sara Dolen, Lon S. Schneider, Sonia Pawluczyk, Bryan M. Spann, James Brewer, Helen Vanderswag, Judith L. Heidebrink, Joanne L. Lord, Ronald Petersen, Kris Johnson, Rachelle S. Doody, Javier Villanueva-Meyer, Munir Chowdhury, Yaakov Stern, Lawrence S. Honig, Karen L. Bell, John C. Morris, Beau Ances, Maria Carroll, Sue Leon, Mark A. Mintun, Stacy Schneider, Daniel Marson, Randall Griffith, David Clark, Hillel Grossman, Effie Mitsis, Aliza Romirowsky, Leyla deToledo-Morrell, Raj C. Shah, Ranjan Duara, Daniel Varon, Peggy Roberts, Marilyn Albert, Chiadi Onyike, Stephanie Kielb, Henry Rusinek, Mony J de Leon, Lidia Glodzik, Susan De Santi, P. Murali Doraiswamy, Jeffrey R. Petrella, R. Edward Coleman, Steven E. Arnold, Jason H. Karlawish, David Wolk, Charles D. Smith, Greg Jicha, Peter Hardy, Oscar L. Lopez, MaryAnn Oakley, Donna M. Simpson, Anton P. Porsteinsson, Bonnie S. Goldstein, Kim Martin, Kelly M. Makino, M. Saleem Ismail, Connie Brand, Ruth A. Mulnard, Gaby Thai, Catherine Mc-Adams-Ortiz, Kyle Womack, Dana Mathews, Mary Quiceno, Ramon Diaz-Arrastia, Richard King, Myron Weiner, Kristen Martin-Cook, Michael DeVous, Allan I. Levey, James J. Lah, Janet S. Cellar, Jeffrey M. Burns, Heather S. Anderson, Russell H. Swerdlow, Liana Apostolova, Po H. Lu, George Bartzokis, Daniel H.S. Silverman, Neill R Graff-Radford, Francine Parfitt, Heather Johnson, Martin R. Farlow, Ann Marie Hake, Brandy R. Matthews, Scott Herring, Christopher H. van Dyck, Richard E. Carson, Martha G. MacAvoy, Howard Chertkow, Howard Bergman, Chris Hosein, Sandra Black, Bojana Stefanovic, Curtis Caldwell, Ging-Yuek Robin Hsiung, Howard Feldman, Benita Mudge, Michele Assaly, Andrew Kertesz, John Rogers, Dick Trost, Charles Bernick, Donna Munic, Diana Kerwin, Marek-Marsel Mesulam, Kristina Lipowski, Chuang-Kuo Wu, Nancy Johnson, Carl Sadowsky, Walter Martinez, Teresa Villena, Raymond Scott Turner, Kathleen Johnson, Brigid Reynolds, Reisa A. Sperling, Keith A. Johnson, Gad Marshall, Meghan Frey, Jerome Yesavage, Joy L. Taylor, Barton Lane, Allyson Rosen, Jared Tinklenberg, Marwan Sabbagh, Christine Belden, Sandra Jacobson, Neil Kowall, Ronald Killiany, Andrew E. Budson, Alexander Norbash, Patricia Lynn Johnson, Thomas O. Obisesan, Saba Wolday, Salome K. Bwayo, Alan Lerner, Leon Hudson, Paula Ogrocki, Evan Fletcher, Owen Carmichael, John Olichney, Charles DeCarli, Smita Kittur, Michael Borrie, T-Y Lee, Dr Rob Bartha, Sterling Johnson, Sanjay Asthana, Cynthia M. Carlsson, Steven G. Potkin, Adrian Preda, Dana Nguyen, Pierre Tariot, Adam Fleisher, Stephanie Reeder, Vernice Bates, Horacio Capote, Michelle Rainka, Douglas W. Scharre, Maria Kataki, Earl A. Zimmerman, Dzintra Celmins, Alice D. Brown, Godfrey D. Pearlson, Karen Blank, Karen Anderson, Andrew J. Saykin, Robert B. Santulli, Eben S. Schwartz, Kaycee M. Sink, Jeff D. Williamson, Pradeep Garg, Franklin Watkins, Brian R. Ott, Henry Querfurth, Geoffrey Tremont, Stephen Salloway, Paul Malloy, Stephen Correia, Howard J. Rosen, Bruce L. Miller, Jacobo Mintzer, Crystal Flynn Longmire, Kenneth Spicer, Elizabether Finger, Irina Rachinsky, John Rogers, Andrew Kertesz, Dick Drost, Nunzio Pomara, Raymundo Hernando, Antero Sarrael, Susan K. Schultz, Laura L. Boles Ponto, Hyungsub Shim, Karen Elizabeth Smith, Norman Relkin, Gloria Chaing, Lisa Raudin, Amanda Smith, Kristin Fargher, and Balebail Ashok Raj
AUTHOR CONTRIBUTIONS
E.C.M.: conception and design of the study, analysis of data, statistical analysis, drafting the manuscript. R.A.S.: conception and design of the study. A.J.H.: conception and design of the study, analysis of data. R.L.B.: conception and design of the study. P.L.D.: conception and design of the study. J.W.S.: conception and design of the study. M.R.S.: conception and design of the study, analysis of data, statistical analysis, drafting the manuscript.
STUDY FUNDING
This work was funded by NIH grants F32AG044054 (E.C.M.), P01AG036694 (R.A.S.), K01MH099232 (A.J.H.), R01AG036836 (P.L.D.), K24MH094614 (J.W.S.), and K25EB013649 (M.R.S.). Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of Southern California. The data from younger participants were collected as part of the Brain Genomics Superstruct Project (http://neuroinformatics.harvard.edu/gsp/).
DISCLOSURE
E. Mormino received funding from NIH grant F32AG044054 and P01 AG036694. R. Sperling has served as a paid consultant for Abbvie, Biogen, Bracket, Genentech, Lundbeck, Roche, and Sanofi. She has served as a coinvestigator for Avid, Eli Lilly, and Janssen Alzheimer Immunotherapy clinical trials. She has spoken at symposia sponsored by Eli Lilly, Biogen, and Janssen. R. Sperling receives research support from Janssen Pharmaceuticals and Eli Lilly and Co. These relationships are not related to the content in the manuscript. She also receives research support from the following grants: P01 AG036694, U01 AG032438, U01 AG024904, R01 AG037497, R01 AG034556, K24 AG035007, P50 AG005134, U19 AG010483, R01 AG027435, Fidelity Biosciences, Harvard NeuroDiscovery Center, and the Alzheimer's Association. A. Holmes received funding from K01MH099232. R. Buckner reports personal fees from Pfizer that are not related to the content in the manuscript. P. De Jager received funding from R01AG036836. J. Smoller received funding from K24MH094614. M. Sabuncu received funding from K25EB013649. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Villemagne VL, Burnham S, Bourgeat P, et al. . Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 2013;12:357–367. [DOI] [PubMed] [Google Scholar]
- 2.Reiman EM, Langbaum JB, Fleisher AS, et al. . Alzheimer's prevention initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis 2011;26(suppl 3):321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling RA, Rentz DM, Johnson KA, et al. . The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014;6:228fs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gatz M, Reynolds CA, Fratiglioni L, et al. . Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174. [DOI] [PubMed] [Google Scholar]
- 5.Corder EH, Saunders AM, Strittmatter WJ, et al. . Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 6.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. . Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escott-Price V, Sims R, Bannister C, et al. . Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain 2015;138:3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SH, Harold D, Nyholt DR, et al. . Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer's disease, multiple sclerosis and endometriosis. Hum Mol Genet 2013;22:832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridge PG, Mukherjee S, Crane PK, Kauwe JS; Alzheimer's Disease Genetics Consortium. Alzheimer's disease: analyzing the missing heritability. PLoS One 2013;8:e79771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 11.Desikan RS, Schork AJ, Wang Y, et al. . Polygenic overlap between C-reactive protein, plasma lipids, and Alzheimer disease. Circulation 2015;131:2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Genomics of Alzheimer's Disease Consortium. Convergent genetic and expression data implicate immunity in Alzheimer's disease. Alzheimers Dement 2015;11:658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 2015;77:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan K. The three new pathways leading to Alzheimer's disease. Neuropathol Appl Neurobiol 2011;37:353–357. [DOI] [PubMed] [Google Scholar]
- 15.Aisen PS, Petersen RC, Donohue M, Weiner MW; Alzheimer's Disease Neuroimaging Initiative. Alzheimer's Disease Neuroimaging Initiative 2 clinical core: progress and plans. Alzheimers Dement 2015;11:734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ge T, Nichols TE, Lee PH, et al. . Massively expedited genome-wide heritability analysis (MEGHA). Proc Natl Acad Sci USA 2015;112:2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes AJ, Hollinshead MO, O'Keefe TM, et al. . Brain genomics Superstruct project initial data release with structural, functional, and behavioral measures. Sci Data 2015;2:150031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saykin AJ, Shen L, Yao X, et al. . Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimers Dement 2015;11:792–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmes AJ, Lee PH, Hollinshead MO, et al. . Individual differences in amygdala-medial prefrontal anatomy link negative affect, impaired social functioning, and polygenic depression risk. J Neurosci 2012;32:18087–18100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genomes Project C, Abecasis GR, Auton A, et al. . An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 2012;44:955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. . Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landau SM, Mintun MA, Joshi AD, et al. . Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CR Jr, Bernstein MA, Fox NC, et al. . The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crane PK, Carle A, Gibbons LE, et al. . Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 2012;6:502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbons LE, Carle AC, Mackin RS, et al. . A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav 2012;6:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dima D, Breen G. Polygenic risk scores in imaging genetics: usefulness and applications. J Psychopharmacol 2015;29:867–871. [DOI] [PubMed] [Google Scholar]
- 28.International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. . Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beecham GW, Hamilton K, Naj AC, et al. . Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet 2014;10:e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim YY, Maruff P, Pietrzak RH, et al. . Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer's disease. Brain 2013;137:221–231. [DOI] [PubMed] [Google Scholar]
- 31.Caselli RJ, Dueck AC, Osborne D, et al. . Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med 2009;361:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabuncu MR, Buckner RL, Smoller JW, et al. . The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb Cortex 2012;22:2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris SE, Davies G, Luciano M, et al. . Polygenic risk for Alzheimer's disease is not associated with cognitive ability or cognitive aging in non-demented older people. J Alzheimers Dis 2014;39:565–574. [DOI] [PubMed] [Google Scholar]
- 34.Harold D, Abraham R, Hollingworth P, et al. . Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:351–357. [DOI] [PubMed] [Google Scholar]
- 36.Kok E, Haikonen S, Luoto T, et al. . Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 2009;65:650–657. [DOI] [PubMed] [Google Scholar]
- 37.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 1999;45:358–368. [DOI] [PubMed] [Google Scholar]
- 38.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2013;2:a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolf AB, Valla J, Bu G, et al. . Apolipoprotein E as a beta-amyloid-independent factor in Alzheimer’s disease. Alzheimers Res Ther 2013;5:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shulman JM, Chen K, Keenan BT, et al. . Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol 2013;70:1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hibar DP, Stein JL, Renteria ME, et al. . Common genetic variants influence human subcortical brain structures. Nature 2015;520:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.