

Abstract
Objective:
To report a large cohort of patients with antibodies against contactin-associated protein-like 2 (Caspr2) and provide the clinical spectrum of this disorder.
Methods:
Serum and CSF samples were assessed at 2 neuroimmunology centers in Barcelona and Rotterdam. Patients were included if Caspr2 antibodies were confirmed with 2 independent techniques, including brain immunohistochemistry and cell-based assay. Clinical information was obtained by the authors or provided by treating physicians after patients' informed consent.
Results:
Median age at symptom onset was 66 years. Of 38 patients, 34 were male. Median time to nadir of disease was 4 months (in 30% >1 year). The most frequent syndromes included limbic encephalitis (42%) and Morvan syndrome (29%). Seventy-seven percent of the patients had ≥3 of the following symptoms: encephalopathy (cognitive deficits/seizures), cerebellar dysfunction, peripheral nervous system hyperexcitability, dysautonomia, insomnia, neuropathic pain, or weight loss. A tumor, mostly thymoma, occurred in 19% of the patients. Immunoglobulin G4 subclass antibodies were present in all patients; 63% also had immunoglobulin G1 antibodies. Treatment response occurred in 93% of the patients and 25% had clinical relapses.
Conclusions:
Caspr2 antibodies associate with a treatable disorder that predominantly affects elderly men. The resulting syndrome may vary among patients but it usually includes a set of well-established symptoms. Recognition of this spectrum of symptoms and consideration of the protracted clinical course are important for early diagnosis of this disorder. Prompt immunotherapy and tumor therapy (if needed) often result in improvement.
Contactin-associated protein-like 2 (Caspr2) is a membrane protein expressed in the CNS and peripheral nervous system. It is essential for proper localization of voltage-gated potassium channels (VGKC). Antibodies to VGKC were initially reported in patients with neuromyotonia, Morvan syndrome, and limbic encephalitis (LE).1–3 However, while the clinical spectrum emerged, it became clear that the antibodies were not directed against the VGKC subunits but to associated proteins. Two of these proteins were identified in 2010: leucine-rich glioma-inactivated1 (LGI1) and Caspr2.4,5 Antibodies to LGI1 are mainly associated with LE and faciobrachial dystonic seizures, but the clinical spectrum of Caspr2 antibodies is more diverse. Most reports on Caspr2 autoimmunity consist of clinically preselected groups of patients with Morvan syndrome,6 epilepsy,7 or pain syndromes.8 In other reports, patients with Caspr2 antibodies were analyzed along with patients with antibodies to LGI1 or unknown proteins considered within the VGKC complex.9,10 Overall, the clinical spectrum of Caspr2 autoimmunity remains not well-defined. We report the largest series of patients with Caspr2 antibodies and provide a framework for the clinical recognition of this disorder.
METHODS
Patients.
The study population consisted of patients suspected to have autoimmune or paraneoplastic neurologic disorders whose serum or CSF were analyzed at 2 referral centers (Center of Experimental Neuroimmunology, Institut d'Investigacions Biomèdiques August Pi i Sunyer, Hospital Clinic, University of Barcelona, Spain; and Department of Immunology, Erasmus University Medical Center, Rotterdam, the Netherlands) between 1994 and 2015 (not published previously). Patients with confirmed Caspr2 antibodies were included in the study. Serum and CSF (if available) were tested using brain immunohistochemistry (IHC) and cell-based assays (CBA) in parallel in both institutions. Patients were considered to have Caspr2 antibodies if both tests were positive in at least one of the samples. Clinical information was obtained from the treating physicians in a standardized fashion after patient informed consent was received, or patients were seen by one of the authors (n = 15). Peripheral nerve hyperexcitability (PNH) was defined as spontaneous muscle overactivity (i.e., myokymia, fasciculations) identified by the treating neurologist during physical examination or with electrophysiologic studies.11 Morvan syndrome was defined as a combination of (1) cognitive symptoms or seizures and (2) peripheral nerve hyperexcitability and (3) dysautonomia or insomnia. LE was defined as an encephalitis with predominant clinical involvement of the limbic system (short-term memory loss, difficulty forming new memories, behavioral disorder) or MRI fluid-attenuated inversion recovery (FLAIR)/T2 abnormalities in the medial temporal lobes. Pain was considered of neuropathic origin if it was described as burning sensation or painful pins and needles, or had a compatible nerve distribution. Relapse was defined as recurrence of symptoms after full or partial recovery, with sustained improvement for at least 2 months.
Laboratory studies.
The CBAs for determination of Caspr212 and other antibodies, brain tissue IHC,13 radioimmunoassay (RIA) to determine VGKC complex antibodies, and immunoblot studies14 have been reported previously, and are described in supplemental data on the Neurology® Web site at Neurology.org.
Statistical analysis.
Fisher exact test was used for categorical data. Mann-Whitney U was used for the comparison of continuous data.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Institutional Review Boards of the University of Barcelona and the Erasmus University Medical Center, Rotterdam.
RESULTS
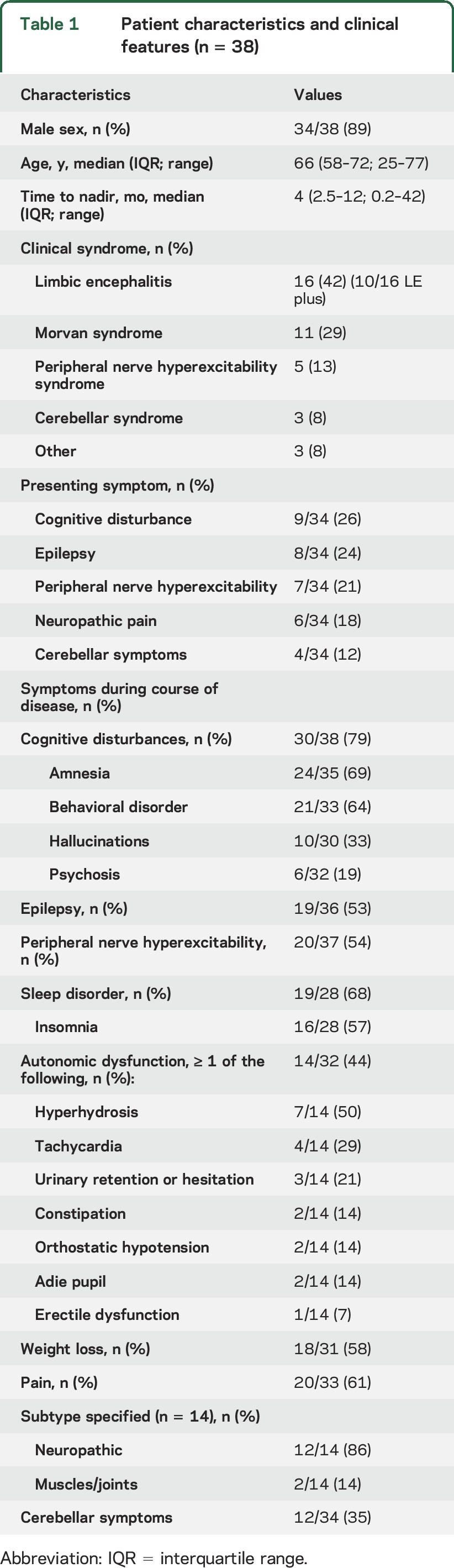
Thirty-eight patients fulfilled the criteria of having Caspr2 antibodies confirmed with more than one test (figure e-1 and table e-1); 10 additional cases had antibodies detected only with CBA but without confirmation with brain IHC (table e-2). The clinical features of patients with confirmed Caspr2 antibodies are shown in table 1. Thirty-four of 38 (89%) were male; the median age at symptom onset was 66 years. Female patients were younger than male patients (median 49 vs 68 years, p = 0.008). The median time to nadir was 4 months; in 10/33 (30%) patients, the nadir of the disease was reached ≥12 months after symptom onset. Morvan syndrome was related to longer time to nadir than other syndromes (p = 0.016).
Table 1.
Patient characteristics and clinical features (n = 38)
Clinical phenotype.
Sixteen patients (42%) developed LE. Ten of them had additional symptoms beyond the limbic system, such as cerebellar dysfunction or pain (LE plus, 26%), and the other 6 had pure LE (16%). Morvan syndrome occurred in 11 (29%) patients, and PNH in 5 (13%). Two of these 5 patients also had insomnia, and another 2 had autonomic dysfunction. Three additional patients (8%) had predominant cerebellar symptoms, and the remaining 3 patients had a single seizure followed by pain syndrome (n = 1), painful polyneuropathy (n = 1), or mild amnestic syndrome with frontal lobe dysfunction (n = 1).
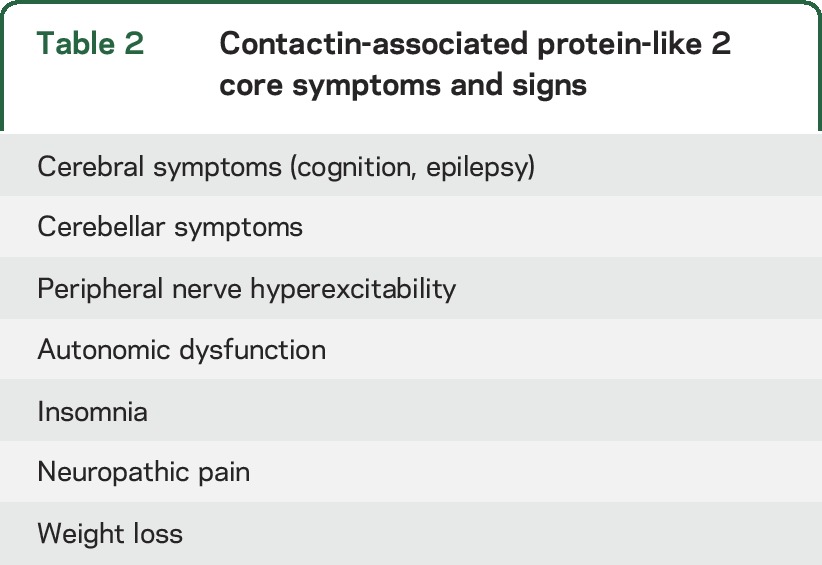
The most common presenting symptoms were cognitive disturbance (26%), seizures (24%), PNH (21%), or neuropathic pain (18%). During the course of disease, cognitive dysfunction was reported by the treating neurologist in 79% of the patients and 53% had seizures. In addition to cerebral symptoms, sleep disorder (68%), pain (61%), weight loss (58%, median 10 kg), PNH (54%), autonomic dysfunction (44%), and cerebellar symptoms (35%) were common. The type of pain most frequently reported was neuropathic (86%), usually described as a burning sensation in the hands or feet; other types of pain included joint and muscle pain, thoracic pain, and lumbocoxalgia. The repertoire of 7 symptoms comprises the spectrum of Caspr2 clinical manifestations (table 2). In 77% of the patients, ≥3 core symptoms were present and 61% had ≥4. Although mainly contributive to patient recognition, these symptoms possibly differentiate Caspr2 patients from patients with other antibodies. Among 35 LGI1 patients and 62 NMDA receptor (NMDAR) patients, only 6 (17%) and 2 patients (3%) had ≥4 core symptoms (both p < 0.001), respectively.
Table 2.
Contactin-associated protein-like 2 core symptoms and signs
Diagnostic tests.
CSF was normal in 65% of the patients (table 3). Seven patients had mild pleocytosis ranging from 6–20 cells/μL. Four of 6 patients with neuropathic pain had unremarkable nerve conduction studies. Needle EMG showed hyperexcitability in all patients with clinical features of PNH. In patients with cognitive decline or seizures, 70% had unremarkable MRI, while 24% showed bilateral T2 hyperintensity of the medial temporal lobes. One patient presenting with ataxia and dysarthria had an area of increased FLAIR/T2 signal in the brainstem. A patient with subacute cerebellar ataxia followed by LE had cerebellar atrophy on the MRI at presentation.
Table 3.
Ancillary testing and laboratory results
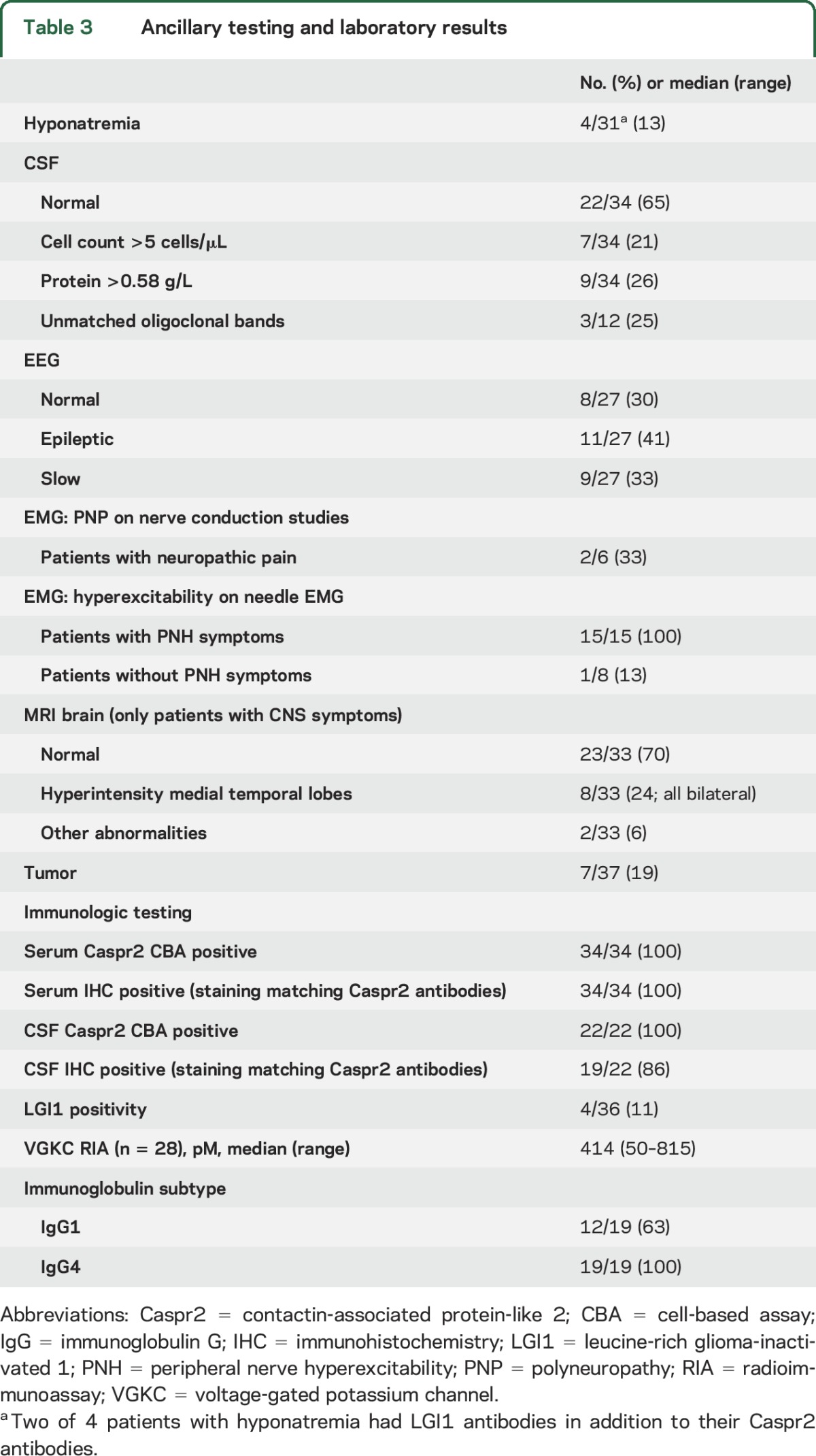
Caspr2 antibodies were detected in serum with both CBA and IHC in all patients. CSF brain IHC was negative in 3 patients, all with a tumor (and presenting with Morvan syndrome). Possibly, CSF antibody titer was lower due to the predominant initial peripheral involvement in this syndrome and the presence of a clear systemic trigger of the immune response. Twenty-eight serum samples were available for VGKC complex RIA. The median titer was 414 pM; 25 patients (89%) had a positive result (titer >100 pM).
Immunoglobulin G (IgG) subtype classification was available in 19 patients. All sera were IgG4 positive (100%) and 12 were also positive for IgG1 subclass antibodies (63%) (figure 1). No clinical correlation with the presence of IgG1 antibodies was detected.
Figure 1. Immunoglobulin G (IgG) subtyping by cell-based immunofluorescence assay.

Serum from patient 1 shows IgG1 and IgG4 reactivity with HEK cells expressing contactin-associated protein-like 2 (Caspr2). Serum from patient 2 shows only IgG4 reactivity. The control serum is from a healthy participant showing absence of IgG1 and IgG4 reactivity with Caspr2.
Comorbidities.
Seven (2 female, 5 male) of 37 (19%) patients had a tumor, including 4 thymoma, 1 adenocarcinoma of the lung, 1 carcinoma in situ of sigmoid, and 1 thoracic mass without pathologic diagnosis (the patient died shortly after presenting neurologic symptoms). Among the 4 patients with thymoma, 1 had tumor resection 2 months before onset of neurologic symptoms, another had an unresectable thymoma that had been stable for several years, and 2 had a tumor relapse by the time of neurologic disease onset. Interestingly, 6/7 tumor patients had PNH syndrome with several additional core symptoms or Morvan syndrome, as compared to 10/30 nontumor patients (uncorrected p = 0.029). Patients with a tumor had a similar progressive disease course as those without tumor (median time to nadir of disease 3 vs 4.5 months, p = 0.72).
In addition to anti-Caspr2-associated symptoms, 3 patients (2 female) had myasthenia gravis (MG). Their anti-Caspr2-related syndromes included Morvan syndrome (n = 2) and PNH syndrome (n = 1). Two of them had recurrent thymoma at the time of presentation and the third had thymic hyperplasia without thymoma.
Four patients (11%) had additional LGI1 antibodies: 1 had LE, 1 PNH syndrome, and 2 Morvan syndrome. Co-occurrence of LGI1 antibodies was present in 2/7 tumor patients (both with thymoma) and in 2/30 nontumor patients (p = 0.15). Serum sodium levels were available in 3 patients with LGI1 antibodies; 2 of them had hyponatremia. Caspr2 antibodies and LGI1 antibodies co-occurred in 2/4 female patients, but this was not different from male patients (2/32, p = 0.053).
Treatment and outcome.
Twenty-eight of 30 patients without tumor were treated with immunotherapy, and treatment effects in the first month were obtained in 23. The median delay between symptom onset and treatment was 6 months, ranging from 10 days to 9 years. Treatments included IV immunoglobulin (IVIg) only (n = 4), IV or oral steroids only (n = 7), plasma exchange only (n = 1), combination of IVIg and steroids (n = 7), combination of IVIg, steroids, and plasma exchange (n = 2), or combination of steroids and plasma exchange (n = 2). Additionally, one patient was treated with azathioprine, and 7 with second-line immunotherapy (cyclophosphamide [n = 2] or rituximab [n = 5]). Full recovery was obtained in 9 patients (39%) and partial response in 12 (52%). Two patients (9%) did not respond to immunotherapy (figure 2A). Six patients required repeated cycles of immunotherapy because symptoms progressed days to weeks after initial response (treatment-related fluctuations). The various therapeutic strategies could not be compared, due to small numbers and selection.
Figure 2. Treatment effect and outcome.

(A) Effect of treatment in 27 patients (23 without and 4 with tumor). (B) Modified Rankin Scale (mRS) at follow-up in 33 patients. 0, no symptoms; 1, no significant disability, able to carry out all usual activities, despite some symptoms; 2, slight disability, able to look after own affairs without assistance, but unable to carry out all previous activities; 3, moderate disability, requires some help, but able to walk unassisted; 4, moderately severe disability, unable to attend to own bodily needs without assistance, and unable to walk unassisted; 5, severe disability, requires constant nursing care and attention, bedridden, incontinent; 6, dead.26
Two patients were not treated. One of them had Morvan syndrome and died before treatment could be started. The other patient was diagnosed recently. He had minor cognitive impairment and refused treatment.
Four of the 7 patients with a tumor were initially treated only with immunotherapy. Two showed transient improvement and the other 2 did not respond. However, all 4 patients had full neurologic recovery after the tumor was identified and were successfully treated with surgery or chemotherapy (figure 2A). The effect of tumor treatment was unknown for the other 3 tumor patients.
The median follow-up was 36 months (range 3–168). Twenty-four of 33 patients (73%) had a favorable outcome at the last follow-up (modified Rankin Scale ≤2) (figure 2B). Four patients died: 2 died at initial stages of the neurologic disease and 1 during a relapse. The fourth patient died after 4 years in a nursing home (with serious cognitive residual symptoms and cardiac disease). Case fatality rate was 3% after 1 year and 10% after 2 years.
Seven of 28 (25%) patients with a ≥1 year follow-up had clinical relapses. Relapses occurred in 3/3 initially untreated patients and 4/26 treated patients (p = 0.010), and presented 8–72 months after the initial episode (median 19 months; interquartile range 9–33). In 3 of these 7 patients, the diagnosis of Caspr2 antibody-associated syndrome was during the relapse. At relapse, 5 patients had symptoms similar to those of the first episode, but the other 2 developed different core symptoms of the disease. A clarifying case is a man who presented at age 61 years with visual hallucinations, behavioral problems, seizures, and ataxia. Six years later, he returned with PNH and dysarthria. The relapse rate did not differ between tumor and nontumor patients (20% vs 25%, p = 1.00).
DISCUSSION
We report 38 patients with Caspr2 antibodies. This is the largest and most detailed description of patients with this disorder and provides several relevant findings: (1) there is a well-defined spectrum of symptoms related to Caspr2 antibodies and most patients have symptoms affecting multiple areas of the nervous system; (2) the symptom development and course of the disease are often less rapid than those of other autoimmune encephalitis; (3) the disorder predominates in males; (4) approximately 25% of the patients had relapses; and (5) all patients had IgG4 subclass Caspr2 antibodies.
The diagnosis of this immune disorder can be complicated by the presentation with a combination of symptoms involving the CNS and peripheral nervous system. Although the clinical picture may vary among patients, it usually includes a set of well-established symptoms. In 77% of the patients, 3 or more core symptoms were present, including cognitive deficits/epilepsy, cerebellar dysfunction, peripheral nerve hyperexcitability, insomnia, autonomic dysfunction, neuropathic pain, or weight loss. This repertoire of symptoms is consistent with the syndromes previously ascribed to this autoantibody in 2 series including 19 and 8 patients.4,12
Two frequent symptoms included neuropathic pain (61%) and cerebellar dysfunction (35%). Pain was previously described in patients with Morvan syndrome and Caspr2 antibodies6,15 and it was attributed to small fiber polyneuropathy.15 Unremarkable nerve conduction studies in 67% of our patients support this hypothesis. Most patients with cerebellar symptoms had additional clinical features, such as LE.12,16,17 Our findings suggest that cerebellar symptoms in patients with LE should raise suspicion of Caspr2 antibodies, although similar clinical features can occur in patients with antibodies to GABAb receptor,18 Hu,19 or in children with NMDAR antibodies.20
In 30% of the patients, the disease evolved in more than 1 year, which is in contrast to the subacute onset of most antibody-associated encephalitis.21 This protracted course of the disease can lead to diagnostic delays or to misdiagnosing the disorder as a primarily neurodegenerative disease, preventing the early use of immunotherapy. The low sensitivity of CSF pleocytosis adds to this difficult distinction. The VGKC RIA does not always test positive either, so Caspr2 should be specifically requested.
Most patients with confirmed Caspr2 antibodies were male (89%), which is in line with earlier reports (84%–88%).4,12 Autoimmune diseases are generally considered to be more frequent in women, but male predominance is also seen in late-onset MG.22 The reason for this male predominance is unclear. Although the expression of Caspr2 mRNA in the prostate was suggested,6 Caspr2 mRNA is also expressed in the ovaries.23 Interestingly, the few women in our study were younger than men, frequently had an underlying tumor (none of them of the ovary), and showed high propensity to autoimmunity (MG and LGI1 antibodies).
Twenty-five percent of the patients had relapses, some of which occurred up to 7 years after the initial episode of the disease. Considering that the overall median follow-up was 3 years, the late relapses of some patients may suggest an even higher relapse rate. In almost half of the cases with relapses, the initial diagnosis of the disease was made during the relapse, suggesting that patients with a monophasic disease may be missed at disease onset, leading to an overestimation of relapse rates. This occurred in other autoimmune encephalitis such as anti-AMPAR24 and anti-NMDAR in which a drop in relapse rate was noted after these disorders were better recognized and promptly diagnosed.20 Similar to these encephalitides, about half of the relapsing cases with Caspr2 antibodies were not appropriately treated in the first episode. The lower relapse rate in treated patients vs untreated patients is similar to that seen in anti-NMDAR encephalitis.20 One should be aware that in patients with Caspr2 antibodies, the symptoms at relapse may involve different parts of the nervous system than those involved in the initial episode (e.g., CNS or peripheral nervous system).
A limited number of autoimmune disorders are associated with IgG4 antibodies. Recently, IgG4 antibodies were demonstrated in several LGI1 patients and in 3 out of 7 Caspr2 patients.6 All our patients had IgG4 antibodies against Caspr2; this finding is important for a better understanding of the pathophysiology of the disease and has treatment implications. IgG4 antibodies have the property of being functionally monovalent through the in vivo exchange of IgG half-molecules (one H- plus one L-chain).25 Therefore, in contrast to IgG1 antibodies (as in anti-NMDAR encephalitis), IgG4 antibodies are unable to crosslink the target leading to its internalization. Moreover, IgG4 antibodies show low affinity for the Fcγ receptor, and are inadequate in activating cellular immune responses and complement. We postulate that IgG4 Caspr2 antibodies may be directly pathogenic by altering Caspr2-related cell-to-cell interactions.
A limitation of this study is the retrospective collection of data obtained from medical records. Therefore, we possibly overestimated the true frequency of some symptoms, as missing information was not taken into account. Nevertheless, the current findings will improve the recognition of the core symptoms associated with Caspr2 antibodies as well as the frequent protracted clinical course and high relapse rate. Early recognition is important because our data confirm that immunotherapy and tumor treatment (if needed) are often effective in this disease.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients for participation; the physicians Luis Bataller, David Genis Batlle, Daniel Costello, Martín Escudero, Koji Fujita, Makito Hirano, Takahiro Iizuka, Henk Kerkhoff, Esther Maan, Antonio Martínez Salio, Hugo Morales, Elvira Munteis, Jerome Posner, Anneke Rampen, Bojan Rojc, Mateus Mistieri Simabukuro, Roland Thijs, and Jan Veldink for providing clinical information of their patients; and Sanae Boukhrissi, Esther Hulsenboom, and Mariska Nagtzaam for technical assistance.
GLOSSARY
- Caspr2
contactin-associated protein-like 2
- CBA
cell-based assay
- FLAIR
fluid-attenuated inversion recovery
- IgG
immunoglobulin G
- IHC
immunohistochemistry
- IVIg
IV immunoglobulin
- LE
limbic encephalitis
- LGI1
leucine-rich glioma-inactivated 1
- MG
myasthenia gravis
- NMDAR
NMDA receptor
- PNH
peripheral nerve hyperexcitability
- RIA
radioimmunoassay
- VGKC
voltage-gated potassium channel
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
A. van Sonderen: study design, acquisition of data, data analysis, medical writing. H. Ariño: study design, acquisition of data, data analysis, medical writing. M. Petit-Pedrol: study design, acquisition of data, data analysis. F. Leypoldt: acquisition of data, revising the manuscript for content. P. Körtvelyessy: acquisition of data, revising the manuscript for content. K.P. Wandinger: acquisition of data, revising the manuscript for content. E. Lancaster: acquisition of data, revising the manuscript for content. P.W. Wirtz: revising the manuscript for content. M.W.J. Schreurs: revising the manuscript for content. P.A.E. Sillevis Smitt: revising the manuscript for content. F. Graus: revising the manuscript for content. J. Dalmau: study design, acquisition of data, data analysis, medical writing. M.J. Titulaer: study design, acquisition of data, data analysis, medical writing.
STUDY FUNDING
M.J.T. was supported by an ErasmusMC fellowship and has received funding from the Netherlands Organization for Scientific Research (NWO, Veni incentive) and from the Dutch Epilepsy Foundation (NEF, project 14–19). J.D. was supported in part by NIH RO1NS077851, Fondo de Investigaciones Sanitarias, FEDER, Spain (FIS PI12/00611, FG; FIS 14/00203), Instituto de Salud Carlos III.
DISCLOSURE
A. Van Sonderen, H. Ariño, M. Petit-Pedrol, F. Leypoldt, P. Körtvelyessy, K. Wandinger, E. Lancaster, P. Wirtz, and M. Schreurs reports no disclosures relevant to the manuscript. P. Sillevis Smitt holds a patent for the detection of anti-DNER and received research support from Euroimmun F. Graus reports no disclosures relevant to the manuscript. J. Dalmau receives royalties from patents from Athena Diagnostics for the use of Ma2 and N-methyl-D-aspartate receptor as autoantibody tests and licensing fees from Euroimmun for a patent for the use of NMDAR as an autoantibody test. M. Titulaer received research funds for serving on a scientific advisory board of MedImmune LLC and a travel grant for lecturing in India from Sun Pharma, India. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Barber PA, Anderson NE, Vincent A. Morvan's syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000;54:771–772. [DOI] [PubMed] [Google Scholar]
- 2.Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001;50:73–78. [DOI] [PubMed] [Google Scholar]
- 3.Shillito P, Molenaar PC, Vincent A, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol 1995;38:714–722. [DOI] [PubMed] [Google Scholar]
- 4.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012;72:241–255. [DOI] [PubMed] [Google Scholar]
- 7.Lilleker JB, Jones MS, Mohanraj R. VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure 2013;22:776–779. [DOI] [PubMed] [Google Scholar]
- 8.Klein CJ, Lennon VA, Aston PA, McKeon A, Pittock SJ. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology 2012;79:1136–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huda S, Wong SH, Pettingill P, O'Connell D, Vincent A, Steiger M. An 11-year retrospective experience of antibodies against the voltage-gated potassium channel (VGKC) complex from a tertiary neurological centre. J Neurol 2015;262:418–424. [DOI] [PubMed] [Google Scholar]
- 10.Klein CJ, Lennon VA, Aston PA, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol 2013;70:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hart IK, Maddison P, Newsom-Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain 2002;125:1887–1895. [DOI] [PubMed] [Google Scholar]
- 12.Lancaster E, Huijbers MG, Bar V, et al. Investigations of Caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005;128:1764–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shams'ili S, Grefkens J, de LB, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 2003;126:1409–1418. [DOI] [PubMed] [Google Scholar]
- 15.Laurencin C, Andre-Obadia N, Camdessanche JP, et al. Peripheral small fiber dysfunction and neuropathic pain in patients with Morvan syndrome. Neurology 2015;85:2076–2078. [DOI] [PubMed] [Google Scholar]
- 16.Balint B, Regula JU, Jarius S, Wildemann B. Caspr2 antibodies in limbic encephalitis with cerebellar ataxia, dyskinesias and myoclonus. J Neurol Sci 2013;327:73–74. [DOI] [PubMed] [Google Scholar]
- 17.Becker EB, Zuliani L, Pettingill R, et al. Contactin-associated protein-2 antibodies in non-paraneoplastic cerebellar ataxia. J Neurol Neurosurg Psychiatry 2012;83:437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: Novel findings in a new case series of 20 patients. Neurology 2013;81:1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason WP, Graus F, Lang B, et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain 1997;120:1279–1300. [DOI] [PubMed] [Google Scholar]
- 20.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graus F, Titulaer MJ, Balu R, et al. Clinical Approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:S1474–S4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent A, Clover L, Buckley C, Grimley EJ, Rothwell PM. Evidence of underdiagnosis of myasthenia gravis in older people. J Neurol Neurosurg Psychiatry 2003;74:1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poliak S, Gollan L, Martinez R, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999;24:1037–1047. [DOI] [PubMed] [Google Scholar]
- 24.Hoftberger R, van SA, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015;84:2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huijbers MG, Querol LA, Niks EH, et al. The expanding field of IgG4-mediated neurological autoimmune disorders. Eur J Neurol 2015;22:1151–1161. [DOI] [PubMed] [Google Scholar]
- 26.van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19:604–607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.