

Abstract
Sweet basil (Ocimum basilicum), one of the most popular cultivated herbs worldwide, displays a number of varieties differing in several characteristics, such as the color of the leaves. The development of a reference transcriptome for sweet basil, and the analysis of differentially expressed genes in acyanic and cyanic cultivars exposed to natural sunlight irradiance, has interest from horticultural and biological point of views. There is still great uncertainty about the significance of anthocyanins in photoprotection, and how green and red morphs may perform when exposed to photo-inhibitory light, a condition plants face on daily and seasonal basis. We sequenced the leaf transcriptome of the green-leaved Tigullio (TIG) and the purple-leaved Red Rubin (RR) exposed to full sunlight over a four-week experimental period. We assembled and annotated 111,007 transcripts. A total of 5,468 and 5,969 potential SSRs were identified in TIG and RR, respectively, out of which 66 were polymorphic in silico. Comparative analysis of the two transcriptomes showed 2,372 differentially expressed genes (DEGs) clustered in 222 enriched Gene ontology terms. Green and red basil mostly differed for transcripts abundance of genes involved in secondary metabolism. While the biosynthesis of waxes was up-regulated in red basil, the biosynthesis of flavonols and carotenoids was up-regulated in green basil. Data from our study provides a comprehensive transcriptome survey, gene sequence resources and microsatellites that can be used for further investigations in sweet basil. The analysis of DEGs and their functional classification also offers new insights on the functional role of anthocyanins in photoprotection.
Introduction
The green color is most frequently associated to land plants, as early land plants evolved from green Chlorophytes [1]. However, the radiation of Angiosperms, dated approximately 200 MYA, was accompanied by a variety of colors that characterize actual plant landscapes. In contrast to the chromatic variations that characterize plant reproductive organs, leaves are usually green and this is not surprising given the optical properties of chlorophyll. Nonetheless, red leaves or red leaf portions are observed frequently in a range of species, particularly at specific developmental stages (e.g., in juvenile and senescence stages, [2,3]) or in response to different abiotic stresses [4–6]. Red coloration may be also a constitutive trait in some species, likely as the result of selection for aesthetic purposes [7].
Anthocyanins are the pigments responsible for leaf red coloration in the majority of plants, although in some members of Caryophyllales, betalains [8] confer red coloration to leaves. Anthocyanins have the potential to serve multiple functional roles in plants challenged against a wide range of stress agents of abiotic and biotic origin [3,9–12]. Epidermal anthocyanins confer greater capacity to red than to green individuals to withstand a severe, even transient excess of solar irradiance, because of their ability to absorb over the blue and the green portions of the solar spectrum [13–15]. Red varieties have received increasing interest over the last decade because of substantially greater content of health promoting substances as compared to green varieties, especially when plants grow under limited light irradiance. Genes encoding enzymes as well as regulatory genes involved in flavonoid and anthocyanin biosynthesis, have been characterized [16,17]. This has not resulted, however, into a corresponding in-depth knowledge about how cyanic and acyanic individuals of the same species perform when suffering from supernumerary photons, a condition plant face on daily, not only on seasonal basis [18].
Genomic and transcriptomic tools may help unveil the transcriptional regulation of genes involved in light-induced metabolic and signaling pathways, in leaves that differ in their ability to absorb over the visible portion of the solar spectrum [19]. Rapid advances in next-generation sequencing (NGS) technologies and associated bioinformatics tools provide novel opportunities for gene expression analysis [20]. The high-throughput RNA-sequencing (RNA-Seq) has emerged as a powerful method for transcriptome analysis of a complete set of expressed mRNA sequences in specific tissues as well as in the whole organism, and may have important applications in plant biology [21–23]. Since RNA-Seq is independent on prior knowledge of gene sequences, the reconstruction of transcriptome has key significance for molecular and genetic studies in non-model species [24,25]. De novo transcriptome analysis may help dissect mechanisms of photoprotection in acyanic and cyanic individuals, in the absence of mutants defective in early steps of the anthocyanin branch-pathway.
Our study explores the transcriptome of sweet basil (O. basilicum) one of the most popular cultivated herbs worldwide that displays a huge number of varieties differing in the leaf shape and aroma, as well as in the color of the leaf [26]. Recently, NGS technologies have been applied in sweet basil [27], but RNA-Seq analysis of cultivars that differs in the leaf color is still lacking. In particular, we applied RNA-Seq technology for in-depth sequencing of purple-leaved “Red Rubin” and green-leaved “Tigullio” transcriptomes, to provide adequate genomic resources for further investigation of the functional role of anthocyanins in photoprotection. We obtained about 128 million clean pair-end reads, which were further used for the de novo assembly of transcriptome of O. basilicum. The sequencing coverage was comprehensive enough to identify most unigenes and major metabolic pathways.
Results and Discussion
Sample preparation and Illumina sequencing
In our study, we carried out the RNA-Seq of four cDNA libraries, constructed from leaves of Red Rubin (RR) and Tigullio (TIG) basil cultivars, which were further sequenced with the Illumina platform. We obtained 128,551,512 paired-end (PE) clean reads by filtering and removing adapter sequences from raw data (Table 1). The output was greater than a previous study on sweet basil transcriptome which generated a total of about 50 millions high quality reads [27]. All clean reads derived from our analysis have been deposited at the Sequence Read Archive (SRA) of the National Centre of Biotechnology Information (NCBI) under the accession number SRA313233.
Table 1. Assembly statistics for Trinity RR-TIG.
| Trinity | |
|---|---|
| Reads | 128,551,512 |
| N50 | 1,613 |
| N75 | 2,488 |
| GC content | 41.3% |
| Shortest transcript length | 201 |
| Longest transcript length | 15,660 |
| Mean Size | 988 |
| Total assembled bases | 166,663,118 |
| Count | 168,627 |
| Count (after CD-HIT 95%) | 134,194 |
| Count (after IsoPct selection) | 111,007 |
De novo assembly of the transcriptome and assessment of assembly
Clean reads of RR and TIG were assembled de novo to generate a non-redundant dataset of gene sequences for sweet basil. We used individual reads of RR and TIG to produce de novo transcriptome assembly of each cultivar (S1 Table).
The assembly of de novo transcriptomes was performed with the de Bruijn's graph approach, which is particularly suitable for transcript reconstruction using Illumina reads [28]. Full-length transcripts reconstruction was carried out with Trinity, CLC Genomics Workbench (CLC), and SOAPdenovo-trans assemblers (all these assemblers adopt de Bruijn's graph approach). Trinity has been widely used for full-length transcripts reconstruction, but the use of a single k-mer (size = 25) may both introduce chimeric assemblies and be unable to cover the whole breadth of transcriptome expression [29]. CLC and SOAPdenovo-trans assemblers use k-mer of different sizes, thus providing a wider representation of different transcript isoforms and reducing perturbation graph topology that is originated from noises in the library sequencing [30]. In our study, k-mer sizes of 45-mer, 41-mer and 37-mer were best fitted for de Bruijn graph construction of for RR, TIG and RR-TIG, respectively.
In our study, CLC assembled 128,684 contigs, with a mean length of 658 bp and N50 of 962 bp (S1 File); SOAPdenovo-trans assembled 294,479 total transcripts, with a mean length of 352bp and N50 of 607 bp (S2 File), and Trinity yielded 168,627 transcripts, with a mean size of 988 bp and N50 of 1,613 bp. The quality of individual de novo assemblers was assessed based upon the length-weighted medians (N50 and N75), the number of both returned contigs and conserved core eukaryotic proteins (CEGMA, complete and partial proteins, [31]). We identified more than 87% highly conserved CEGMA core eukaryotic genes in each transcriptome, which supports assembly completeness. The number of conserved CEGMA proteins was higher in Trinity and, as expected, similar results were observed for the de novo assembly of the transcriptomes in each basil cultivar (S1 Table). Trinity turned out the most suitable de novo assembly program for our datasets, with higher N50 and N75 statistics and transcript numbers, and recovering more full-length transcripts of higher quality. Therefore, Trinity transcripts were retained for further analyses, confirming previous reports by Villarino et al. [32] and Grabherr et. al. [33]. We observed that a large percentage of transcripts were small-sized, since 36.4% of contigs were in the 200–400 bp size range (S1 Fig). Transcripts were therefore clustered using a sequence similarity threshold of 95%, to join further sequences and remove any redundant and/or highly similar contigs. The resulting 134,194 transcripts were further processed based on reads abundance, by removing contigs with < 1% of reads per component (Table 1, S3 File). The final unigene dataset consisted of 111,007 transcripts.
Annotation and characterization of de novo transcripts
Transcripts were subjected to BLASTX homology search against the RefSeq plant database at the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) and the Arabidopsis protein database, allowing annotation of 91,231 (82.2%) and 57,663 (51.9%) transcripts, respectively (S2 Table). The percentage of annotated unigenes was similar to previous study in O. basilicum (81.1%) [27], suggesting that the assembled unigenes have captured the majority of sweet basil transcriptome. The E-value distribution of the top hits from RefSeq plant database indicates that 57.5% has strong homology (E-value smaller than 1.0e⁻⁵⁰), while 41.6% of homologous hits has E-value ranging from 1.0e⁻⁵ to 1.0e⁻⁵⁰ (S2 Fig). Similarity distribution (S2 Fig) shows that 44.6% of the query sequences had similarity > 80%, whereas similarity ranged from 40% to 80% in 55.3% of hits. About 52% of annotated transcripts of purple and green basil were assigned (with the best score), to the sequences of top four species, namely Sesamum indicum (40.7%), Nicotiana sylvestris (4.2%), Nicotiana tomentosiformis (4.0%) and Vitis vinifera (2.9%) (S2 Fig). Approximately, 28% of annotation descriptions against RefSeq were uninformative, containing “hypothetical” (7234) or “uncharacterized” (18,601) terms, likely due to insufficient knowledge of O. basilicum genome. The high rate (49%) of sequences without a significant homologous hit and informative description (as compared with other non-model plant species, [34–36]) can be partly due to both potentially novel sequences not yet reported in other crop species and the occurrence of highly divergent genes.
BLASTX hits were used for transcript mapping and subsequent assignment of gene ontology (GO) annotations, using Blast2GO PRO. Among the 29,941 transcripts with at least one mapped GO term, only 11,692 were annotated and categorized into 46 functional groups, belonging to three main GO ontologies: molecular function (25.1%), cellular component (31.3%) and biological process (43.6%) (Fig 1, S4 File). This low rate of BLAST hits annotation to GO terms has been previously reported in sweet basil study of de novo transcriptome, where highest percentage of genes were classified under “unknown groups” [27], possibly because O. basilicum does not belong to model organisms family. “Catalytic activity” (6,151 transcripts; GO:0003824), “binding” (5,806; GO:0005488), and “transporter activity” (633; GO:0005215) are GO terms mostly represented in the molecular function category. Biological process are mostly enriched in “metabolic process” (7,028; GO:0008152), “cellular process” (6,231; GO:0009987), “localization” (1,504; GO:0051179) GO terms. Finally, GO terms. “cell” and “cell part” (5,295; GO:0005623, GO:0044464), “organelle” (2,938; GO:0043226), mostly represent the cellular component function category.
Fig 1. Gene Ontology assignments for RR-TIG transcriptome.

The results are summarized in terms of three functional categories: cellular component, molecular function and biological process. The GO terms were visualized using WEGO (http://wego.genomics.org.cn).
All transcripts were compared against InterPro database to find possible matches of domains/families. This annotation step allows retrieving protein domains that might be not accounted by simple sequence-based alignments. In Table 2 we show the ranking of 30 most abundant InterPro domains/families, based on the number of RR-TIG transcripts in each InterPro domain. About 42% of 6,411 domains/families had an occurrence of 1–3 sequences, whereas a small proportion gathered a high number of sequences. Among these protein domains/families, the most represented domain is IPR027417 (P-loop containing nucleoside triphosphate hydrolase) with 2,805 annotated transcripts, and "Protein kinase" and its subcategories containing “Serine/threonine-protein kinase", which are well-known regulators of cellular pathways. Moreover, highly represented are “Cytochrome P450” (Cyt_P450) and “WD40-repeat” domains, both associated with signal transduction and transcription, and “Zinc-finger related RING protein” (Znf-RING) domain.
Table 2. The 30 most representative GO terms revealed by the InterProScan annotation.
| GO_ID | GO_Name | Aspect | Accession_Count |
|---|---|---|---|
| GO:0005524 | ATP binding | Molecular_Function | 4901 |
| GO:0016772 | transferase activity, transferring phosphorus-containing groups | Molecular_Function | 2719 |
| GO:0006468 | protein phosphorylation | Biological_Process | 2374 |
| GO:0004672 | protein kinase activity | Molecular_Function | 2370 |
| GO:0055114 | oxidation-reduction process | Biological_Process | 2328 |
| GO:0016020 | membrane | Cellular_Component | 2259 |
| GO:0003824 | catalytic activity | Molecular_Function | 2250 |
| GO:0003676 | nucleic acid binding | Molecular_Function | 2204 |
| GO:0016021 | integral component of membrane | Cellular_Component | 2046 |
| GO:0008270 | zinc ion binding | Molecular_Function | 2006 |
| GO:0003677 | DNA binding | Molecular_Function | 2003 |
| GO:0008152 | metabolic process | Biological_Process | 1781 |
| GO:0004674 | protein serine/threonine kinase activity | Molecular_Function | 1359 |
| GO:0006355 | regulation of transcription, DNA-templated | Biological_Process | 1340 |
| GO:0055085 | transmembrane transport | Biological_Process | 1270 |
| GO:0005634 | nucleus | Cellular_Component | 1252 |
| GO:0016491 | oxidoreductase activity | Molecular_Function | 1213 |
| GO:0000166 | nucleotide binding | Molecular_Function | 1039 |
| GO:0005622 | intracellular | Cellular_Component | 935 |
| GO:0006508 | proteolysis | Biological_Process | 891 |
| GO:0005975 | carbohydrate metabolic process | Biological_Process | 848 |
| GO:0006810 | transport | Biological_Process | 788 |
| GO:0046872 | metal ion binding | Molecular_Function | 758 |
| GO:0003700 | sequence-specific DNA binding transcription factor activity | Molecular_Function | 718 |
| GO:0005488 | binding | Molecular_Function | 670 |
| GO:0005506 | iron ion binding | Molecular_Function | 666 |
| GO:0020037 | heme binding | Molecular_Function | 643 |
| GO:0003723 | RNA binding | Molecular_Function | 642 |
| GO:0005737 | cytoplasm | Cellular_Component | 616 |
| GO:0003735 | structural constituent of ribosome | Molecular_Function | 613 |
We conducted KEGG pathway/based analysis (on 111,007 RR-TIG transcripts) to investigate the biological functions of novel transcripts of sweet basil. Overall, 11,199 sequences were assigned to 170 KEGG pathways. The most represented pathways are “metabolic pathways” (8,1%), “biosynthesis of secondary metabolites” (4,2%), “microbial metabolism in diverse environment” (3.7%), “biosynthesis of antibiotics” (3.2%) and “ribosome” (2.3%) (Fig 2). The 633 transcripts in the “biosynthesis of secondary metabolites” category expressed in sweet basil cultivars may have interest in defining pathways of synthesis and turnover of compounds, which have potential beneficial effects to human health [37]. Thus, an accurate knowledge of basil secondary metabolism opens interesting opportunities for plant breeding. A total of 277 transcripts of flavonoid/anthocyanin metabolism were identified in the examined basil cultivars: glutathione metabolism (0.66%), phenylpropanoid biosynthesis (0.47%), phenylalanine metabolism (0.42%), flavonoid biosynthesis (0.15%), ABC transporters (0.11%), anthocyanin biosynthesis (0.03%) and flavone and flavonol biosynthesis (0.01%) (S3 Table). Data of our study provide, therefore, new insights to dissect molecular mechanisms that are at the base of flavonoid and anthocyanin accumulation in green and red basil morphs in response to high solar irradiance.
Fig 2. Top 20 identified KEGG pathways.

The top 20 distribution of categorization of basil transcripts to KEGG biochemical pathways.
Identification of SSR Markers
Simple sequence repeats (SSRs), also known as microsatellites, have been proved to be highly informative and widely used molecular markers for evolutionary and genetics studies. Within these well-established molecular markers, gene-derived SSRs are a valuable resource because they have higher rates of transferability between species as compared to random genomic SSRs. In this study, we predicted potential SSRs in the assembled unigenes of RR and TIG basil cultivars. By estimating the expression level of Trinity transcripts, unigenes of both cultivars with a low coverage (FPKM ≤ 1.5) were excluded to increase the reliability of SSRs identification. Thus, two sets of 45,244 and 51,493 unigenes for TIG and RR, respectively, were searched for repeat motifs to explore their SSRs profiles. A total of 5,468 and 5,969 potential SSRs have been identified in TIG and RR cultivars, respectively. Most SSR motifs are dinucleotides (3,089 and 3,381 in TIG and RR, respectively), accounting for 56.5% and 56.6% of all the predicted SSRs, followed by trinucleotides (2,257, 41.3% in TIG and 2,434, 40.8% in RR), tetranucleotides (95, 1.7% in TIG and 113, 1.9% in RR), and penta/hexanucleotides (5, 0.1% in TIG and 41, 0.7% in RR, S3 Fig). The most abundant repeat type is AG/CT, followed by AC/GT and AAG/CTT in both cultivars (S3 Fig). The overall frequency of SSRs (excluded mononucleotides) is around 1/12kb in both TIG and RR. Checking for potential polymorphisms between cultivar loci, microsatellites of two transcriptomes were compared through Blast search: 406 unigenes have been identified as the same loci, having the same flanking sequences and the same type of repeat motifs. The alignments of the homologous SSRs-containing loci of RR and TIG identified in silico 66 polymorphic microsatellites (S4 Table). The cultivar specific identification of SSRs from RNA-Seq data provides a new way to develop polymorphic SSR markers and new potential tools for genetic diversity assessments, variety protection and molecular mapping.
Comparative DEG profiling in RR and TIG grown in full sunlight
The suitability of statistical analysis to identify differentially expressed genes (DEGs) was checked by a quality control test of overall reads distribution and variability (S4 Fig). Genes differentially expressed in the examined O. basilicum cultivars grown under full solar irradiance, were identified by aligning separately reads generated upon RR and TIG sequencing to the 111,007 transcripts, derived from RR-TIG total assembly. The abundance of gene expression was estimated as RPKM (reads per kilo base of exon model per million mapped reads, S5 File). A total of 2,372 transcripts has been identified as differentially expressed based on absolute fold change greater than 2 and P-value <0.05. There are 1,568 down-regulated and 804 up-regulated transcripts in green as compared to purple basil (S5 Table). To validate the RNA-Seq results, nine DEGs (PAL, CHS, FLS, ANS, DFR, 3GT, PSY, VDE, ZEP) with different expression patterns were selected for quantitative real-time PCR (qRT-PCR) analysis. All nine genes exhibited similar expression pattern to those obtained by sequencing (Fig 3). This offers further experimental validation to the reliability of our RNA-Seq analysis.
Fig 3. Validation of RNA-Seq data by qRT-PCR.

Comparison of expression level of nine DEGs between RNA-Seq data and qRT-PCR assay. PCR primers and unigene IDs are listed in S7 Table.
Among the BLAST hits of the 2,372 DGEs, approximately 28% are uninformative (“not available”, “predicted protein”, “uncharacterized” and “hypothetical”). The potential functions of DEGs were further examined using the Gene Ontology classification system. This annotation reveals 29 functional groups, which are distributed under the following categories: 14 Biological Process, 5 Cellular Component and 10 Molecular Function (GO level 4, S5 Fig). Nine GO categories are only down-regulated in TIG: e.g. “ribonucleprotein complex”, “anthocyanin metabolic process” and “positive regulation of biosynthetic process”. There are four GO-categories that are uniquely up-regulated in green basil, and corresponds to “glucosinolate metabolic process”, “chlorophyll metabolic process”, “terpenoid metabolic process” and “enzyme regulator”. GO categories such as “phenylpropanoid metabolic process”, “pigment biosynthetic process”, “secondary metabolic process” have been identified in both up- and down-regulated expressed genes.
Genes differentially expressed between the red and green morphs of sweet basil were subjected to functional gene enrichment analysis (GEA) to explore whether specific categories are over- or under- represented compared to the reference transcriptome. We identified 222 GO functional categories that were significantly enriched in DEGs (p-value <0.05, see for full list of GO terms S6 Table). After using the blast2GO tool to reduce the GO enriched terms, the most specific terms (the highest level GO terms of a parental line) are reported in Fig 4. Among the significantly enriched GO terms, DEGs involved in “flavonoid biosynthetic process” (GO:0009813) and “chalcone isomerase activity” (GO:0045430) are noteworthy and suggest a possible relevant role of these secondary metabolites in the acclimation strategies adopted by the two basil morphs to high solar irradiance.
Fig 4. Enriched GO terms for RR-TIG DGEs.

The bar chart shows for each significant GO termthe amount (percentage) of annotated transcripts Blue bars correspond to the sequences of the test-set and red bars correspond to the reference transcriptome dataset.
A detailed functional analysis of genes differentially expressed in the examined basil cultivars, which was performed with the MapMan software, allowed drawing an overview of DEGs involved in various metabolic pathways (Fig 5). Some of the DEGs discussed in this section are summarized in Table 3. The MapMan diagram of secondary metabolism in sweet basil shows that all transcripts coding for enzymes of the MEP pathway, referred as to non-mevalonate pathways (Fig 5), are significantly up regulated in TIG. These mostly regard enzymes (or genes) involved in the early committed steps of the MEP pathway, namely 1-deoxy-D-xylulose-5-phosphate (DOXP) synthase (DXS) and DOXP reducto-isomerase (DXR) (Table 3). In contrast, all transcripts of the shikimate pathway are up-regulated in RR.
Fig 5. MapMan-based visualization of the DEGs involved in “secondary metabolism”.
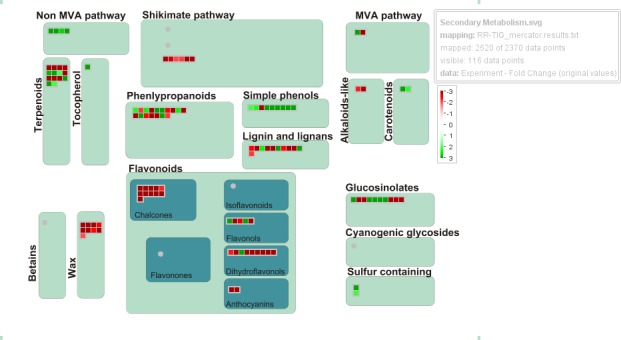
In the display, each BIN or subBIN is represented as a block where each transcript is displayed as a square, which is either colored red if this transcript is up-regulated in RR or green if this transcript is up-regulated in TIG.
Table 3. DEGs identified by comparing RR and TIG basil varieties.
| Transcript ID | Length (bp) | Description (Blast hit) | RR—RPKM | TIG—RPKM | Fold Change |
|---|---|---|---|---|---|
| MEP pathway | |||||
| comp43024_c0_seq1 | 2379 | 1-deoxy-D-xylulose-5-phosphate synthase (DXS) | 6.25 | 31.47 | 5.03 |
| comp49578_c0_seq12 | 3470 | 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) | 5.55 | 19.50 | 3.51 |
| Carotenoids | |||||
| comp48796_c1_seq3 | 1345 | phytoene synthase (PSY) | 6.52 | 44.31 | 6.80 |
| comp50273_c0_seq2 | 1927 | violaxanthin de-epoxidase (VDE) | 20.10 | 44.72 | 2.23 |
| Tocopherol | |||||
| comp48509_c0_seq4 | 1254 | 4-hydroxyphenylpyruvate dioxygenase (HDP) | 4.31 | 13.30 | 3.09 |
| Waxes | |||||
| comp35858_c1_seq1 | 827 | protein ECERIFERUM 1-like (CER1) | 9.49 | 0.52 | -18.23 |
| comp39531_c1_seq1 | 1761 | protein ECERIFERUM 1-like (CER1) | 28.35 | 11.49 | -2.47 |
| comp45710_c0_seq1 | 2116 | protein ECERIFERUM 1-like (CER1) | 206.70 | 68.03 | -3.04 |
| comp47826_c2_seq1 | 2001 | protein ECERIFERUM 1-like (CER1) | 15.28 | 1.18 | -12.92 |
| comp45316_c1_seq9 | 1011 | protein ECERIFERUM 3-like (CER3) | 9.17 | 0.64 | -14.37 |
| comp47720_c1_seq2 | 1357 | 3-ketoacyl-CoA synthase 6-like (KCS6) | 57.90 | 18.63 | -3.11 |
| comp45887_c5_seq2 | 2127 | O-acyltransferase WSD1-like (WSD1) | 13.80 | 3.49 | -3.95 |
| Flavonoids | |||||
| comp39915_c0_seq2 | 1722 | flavonol synthase (FLS) | 4.32 | 18.31 | 4.24 |
| comp30546_c3_seq1 | 1434 | flavonol synthase (FLS) | 66.87 | 150.05 | 2.24 |
| comp40361_c0_seq2 | 1380 | flavonol synthase (FLS) | 49.18 | 108.52 | 2.21 |
Members of Lamiaceae, especially basil, display leaf surfaces rich in peltate glands (trichomes) that synthesize phenyl-propenes, mono- and sesqui-terpenes that impart distinct flavors and aromas [38]. The MEP and MVA pathways produce isopentenyl diphosphate and its isomer dimethyl-allyl diphosphate, which are precursors of terpene biosynthesis [39]. The deviation of upstream metabolites into the MEP pathway and the reduced metabolic flow into the shikimate/phenylpropanoid pathway observed in green basil, possibly as the result of breeding practices designed to select plant rich in aromatic compounds, may have great impact on the plant’s ability to cope with pathogen and predator attacks [40].
As expected, most genes involved in the biosynthesis of anthocyanins are up regulated in RR, the reverse being the case of genes involved in carotenoid (phytoene synthase (PSY), violaxanthin de-epoxidase (VDE) and tocopherol biosynthesis (4-hydroxyphenylpyruvate dioxygenase (HDP)) (Table 3). Carotenoids, particularly xanthophylls play key roles in photoprotection, by dissipating excess radiant energy through non-photochemical quenching as well as behaving as chloroplast antioxidants [41]. This observation taken together with the up-regulation of tocopherol (a well-known chloroplast antioxidant, [42]) biosynthesis, suggests that green basil likely suffered from photo-oxidative stress to greater degree than purple basil did, as previously reported for red and green morphs of several species (for recent review articles, see [2,12]). Data of our study, therefore, add further experimental validation to previous suggestions that anthocyanins may play an effective role in photoprotection [12]. It is also worth noting that the entire cluster of genes related to the synthesis of waxes (Fig 5), CER1 (ECERIFERUM 1, [43]), CER3 [44], KCS6 [45] and WSD1 [46], is greatly over-expressed in RR (Table 3). Waxes are involved in avoiding the entry of photons in the leaf by enhancing reflectance of incident light by leaf surfaces [47]. In some species, epicuticular waxes may greatly contribute in reflecting the shortest solar (UV) wavelengths (and to a lesser extent blue light, [48]), and this may have adaptive significance for plants growing under high solar irradiance [49].
We show that the flavonoid pathway, leading to the synthesis of flavones, flavonols and anthocyanins, greatly differs between purple and green-leaved basil [50], as revealed by DEGs mapping in KEGG pathways. In our annotated O. basilicum RR-TIG transcriptome dataset, 23 transcripts encoding most of the enzymes of flavonoid biosynthesis pathway were identified (S6 Fig). Nineteen of these enzymes (corresponding to nine ko numbers) are down-regulated in green as compared to purple basil (red boxes, Fig 6), consistent with the large flavonoid-anthocyanin production in purple basil. Nonetheless, three genes (corresponding to three ko numbers, green boxes) are up-regulated in green basil. Interestingly, one of these genes corresponds to flavonol synthase (E.C. 1.14.11.23), which is known to catalyze the divergent conversion of dihydro-flavonols to produce flavonols instead of anthocyanins (Table 3). There is recent compelling evidence that the expression of flavonol synthase is inversely related with red coloration and dihydro flavonol reductase expression in crabapples leaves [51]. This is consistent with our MapMan analysis, which indeed shows that genes involved in the biosynthesis of effective UV-screening “simple phenols” and “flavonols” is over-expressed in TIG. Flavonoids, particularly flavonols, the biosynthesis of which is up-regulated because of high sunlight (in the presence or in the absence of UV radiation, [52,53]), play a role in avoiding and countering the photo-oxidative damage driven by an excess of UV radiation. This is of adaptive value in plants challenged against excess light stress on long-term basis. We therefore hypothesize that in RR, an increased biosynthesis of waxes may partially compensate for the reduced biosynthesis of flavonols, which display the greater capacity to absorb over the UV region of the solar spectrum.
Fig 6. KEGG map of the DEGs involved in the flavonoid biosynthesis for the sweet basil leaf.

Red boxes indicate overexpression in purple basil and green boxes overexpression in green basil.
Conclusions
We investigate the transcriptome profile of red and green morphs of sweet basil using Illumina sequencing technology. A high quality transcriptome, consisting of 111,007 unigenes, was functional annotated to number of putative genes related to numerous metabolic and biochemical pathways. We detected a large number of SSR loci, which may constitute a valuable resource for further experimentation on genetic diversity, linkage mapping, and germplasm characterization analysis in Ocimum species (Lamiaceae). In our study, de-novo sequencing of purple-leaved “Red Rubin” and green-leaved “Tigullio” transcriptomes may give new insights on the response mechanisms of cyanic and acyanic leaves suffering from an excess of solar irradiance. A constitutively epidermal cyanic filter may effectively limit photo-oxidative stress by absorbing the excess of supernumerary photons as compared to acyanic leaves. Green morphs counter an excess of unabsorbed visible light by enhancing the thermal dissipation of radiant energy through a more active carotenoid, particularly xanthophyll biosynthesis. This photoprotective strategy coupled with and enhanced biosynthesis of tocopherols, equip chloroplast membranes in TIG with a more active network of antioxidant defenses.
Materials and Methods
Plant material, library preparation and sequencing
Seeds of O. basilicum cv. ‘Red Rubin’ (RR) and cv. ‘Tigullio’ (TIG) were purchased from Franchi Sementi (Milan, Italy) and voucher samples have been deposited at the Institute for Sustainable Plant Protection (CNR). Seedlings were grown in 1.25 L pots with a commercial potting mix over a 30-day-period, and grown for additional three weeks in screenhouses, constructed with roofs and walls, under full sunlight, in the presence or in the absence of UV radiation, following the experimental set-up reported in Agati et al. [54]. Three-week-old leaves of each cultivar were sampled from plants that grew under different light regimes at different hours of the day (09:00–12:00–15:00 hrs) and pooled together prior to analysis. This partly overcame the different potential of green and red morphs to absorb over the UV or visible portion of the solar spectrum. Samples were frozen in liquid nitrogen and stored at -80°C prior to RNA isolation. Total RNA was extracted from 100 mg of leaf tissue using RNeasy Mini Kit, Qiagen, (Valencia, CA, USA) following manufacturer protocol, with little modifications. Briefly, 10% (v/v) of 20% (w/v) N-lauroyl sarcosine was added to RLC buffer. The sample solution was incubated for 10 min at 70°C under vigorous shaking. Contaminating genomic DNA was removed by adding 2 units of DNase I (Ambion, Life Technologies, Gaithersburg, MD) to sample solution kept at 37°C for 30 min. The integrity of total RNA was determined by running samples on 1% agarose gel. The concentration and the quality of total RNA was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA with RNA Integrity Number (RIN) >7 was used for successive analyses. cDNAs were amplified according to the Illumina RNA-Seq protocol and sequenced using the Illumina HiSeq2000 system (Illumina, Inc., CA, USA). Transcriptome library for sequencing was constructed according to the Illumina TruSeq RNA library protocol outlined in “TruSeq RNA Sample Preparation Guide”. The Illumina reads were trimmed at the ends by quality scores on the basis of the presence of ambiguous nucleotides (typically N, maximum value = 2) using ERNE-FILTER (www.erne.sourceforge.net), a modified version of the PHRED/PHRAP Mott’s trimming algorithm (www.phrap.org/phredphrap/phred.html), using default parameters, with the exception of minimum-size errors-rate, which were fixed at 50 and 25, respectively. All sequences reads were then processed for quality assessment using the FastQC quality control tool (v0.10.0, [55]). In all libraries we maintained a phred-like quality score (Q-score) of 20 for downstream analysis. cDNA Illumina sequencing was performed at IGA Technology Services Srl Service Provider (Udine, Italy).
De novo assembly
The transcriptome assembly was performed on RR (65,661,162) and TIG (62,890,350) paired reads. A total de novo assembly was also performed using combined RR and TIG reads (128,551,512), to increase transcript coverage and build-up a reference transcriptome of sweet basil. To select the optimal k-mer length for the assemblies, k-mer analysis was used as implemented in the code KmerGenie [56]. We used three de novo transcriptome assemblers: Trinity r2013-02-25, SOAPdenovo-Trans v1.01 and CLC Genomics Workbench v.8.5.1 (CLC-Bio, Aarhus, Denmark). The softwares have been developed for transcriptome assembly of short reads, using Brujin graph algorithm [57]. First, clean reads were split into 'k-mers' and then assembled to produce contigs, which were joined into scaffolds, further assembled through gap filling to reconstruct unigenes. The most suitable assembly was selected by comparing contig mean size, number of sequences (N50) and Core Eukaryotic Genes Mapping Approach (CEGMA) output [31] of tested assembly programs. We used contigs longer than 200 nt for further analyses. To reduce assembly redundancy the transcripts were clustered at 95% identity using CD-HIT-EST v4.6.1 [58]. Filtration of likely contig artifacts and low expressed contigs was conducted by preliminarily estimating reads abundance for each contig using RSEM (RNASeq by Expectation Maximization) software package [59], with Bowite2 [60] bam file output. Contigs representing more than 1% of the per-component (IsoPct) expression level were kept for further analyses.
Annotation and characterization of the de novo transcripts
Analysis of sequence similarity was performed using BLAST (Basic Local Alignment Search Tool) algorithm with an E-value cut-off of 10−5 [61] and Reference Sequence (RefSeq) plant protein collection database to assess the similarity of Trinity clustering transcripts to those of other model and closely related species. We used Blast2GO suite [62] to generate gene ontology (GO) terms based on the RefSeq BLAST, and the WEGO software to visualize distribution of gene functions [63]. BLAST search against InterPro protein families database [64] retrieved putative functions of newly assembled transcripts. Finally, contigs resulting from Trinity assembly were submitted to the Kyoto Encyclopedia of Genes and Genomes (KEGG) Automatic Annotation Server (KAAS) (v1.6a) [65] to gain an overview of gene pathway networks. The alignment was performed using KAAS default settings with single-directional best hit (SBH) method and databases including all available plant organisms (Arabidopsis thaliana, Arabidopsis lyrata, Theobroma cacao, Glycine max, Fragaria vesca, Cucumis sativus, Vitis vinifera, Solanum lycopersicum, Oryza sativa japonica).
Simple sequence repeats (SSRs) identification
SSRs mining and primers design were performed following the method described previously [66]. In this study, more than 5 times repeats of di-, tri-, tetra-, penta- and hexa-nucleotide were included in search criteria in MISA script (http://pgrc.ipk-gatersleben.de/misa/).
Analysis of differentially expressed genes
CLC Genomics Workbench was used to map the reads to assemblies, and identify and analyze the differentially expressed genes (DEGs). Reads from each sample were aligned to RR-TIG transcriptome reference with a minimum and maximum insert size of 180 and 1000, respectively. The gene expression level was calculated using RPKM method (Reads per kilobase transcriptome per million mapped reads) [20]. A box plot and a density analysis were conducted using R software (version 3.2.3) to estimate whether the RPKM overall distributions were comparable. Differentially expressed genes were identified by comparing expression values between samples and using Kal’s Z-test of proportions [66] with corrected Bonferroni p values (fold change >|2| and p value <0.05). The Z-test calculates the difference in the proportion of reads observed between two conditions and compares whether each sample was drawn from the same distribution relying on an approximation of the binomial distribution by the normal distribution [67].
Gene enrichment analysis (GEA) was performed on DEGs by Blast2GO using Fisher’s exact test (p<0.05) in combination with False Discovery Rate (FDR) correction for multiple testing [68]. Most specific terms were obtained removing parent terms of already existing, statistically significant, child GO terms through filtering enriched GO terms with FDR adjusted p-value < 0.05.
Functional categorization and pathway visualization of DEGs was performed using both KEGG orthologs (using KAAS with default parameters, http://www.genome.jp/kegg/) and the MapMan tool [69]. A MapMan BIN file, with hierarchical ontology system for basil genes, was prepared using Mercator (default options, Blast_cutoff: 50 and IS_DNA; [70], by comparing transcripts against already-classified proteins.
Validation of DEGs by qRT-PCR
Total RNA was extracted from red and green basil morphs as already reported described above and reverse-transcribed by using SuperScript® VILO cDNA Synthesis Kit (Invitrogen, Carlsbad, CA). Nine DEGs, were chosen for validation by quantitative real-time PCR (qRT-PCR) and specific primer pairs (S7 Table) were designed using Primer3 [71]. qRT-PCR was carried out in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA, USA) with SYBR green technologies (Power SYBR green PCR Master Mix; Applied Biosystems) according to the manufacturer’s instruction. Measurements were performed on three replicates and the products were verified by melting curve analysis. The relative expression level of the selected unigenes was calculated using the comparative ΔΔCt method [72], by normalizing the number of target transcripts to the reference Tubulin gene (S7 Table).
Supporting Information
Frequency histogram showing the distribution of transcript length in sweet basil.
(TIF)
(A) The E-value distribution of BLAST hits for the assembled RR-TIG sequences with a cutoff of E-value < 10−5. (B) The similarity distribution of BLAST hit for the assembled RR-TIG sequences with a cutoff of E-value < 10−5. (C) The species distribution of the top BLAST hits for each transcript in the RR-TIG transcriptome assembly from Blast2GO.
(TIF)
A. The profiles of different SSR types in TIG (green) and RR (red). B. The distribution of repeat motifs in TIG (green) and RR (red).
(TIF)
The RPKM overall distribution and variability of cDNA libraries/samples were similar, indicating that they were comparable for identification of differentially expressed genes (DEGs) at the transcriptome level. (A) A box plot analysis with original expression values; (B) A box plot analysis with normalized expression values; (C) Density plot showing the distribution of log2(RPKM) in RR and TIG.
(TIF)
The results are summarized in three main categories: cellular component, molecular function, and biological process.
(TIF)
(TIF)
Fasta file of basil transcripts de novo assembled with CLC Genomics Workbench v.8.5.1.
(FASTA)
Fasta file of basil transcripts de novo assembled with SOAPdenovo-Trans v1.01.
(ZIP)
Fasta file of basil transcripts de novo assembled with Trinity r2013-02-25.
(ZIP)
List of GO terms associated to sweet basil transcripts.
(ZIP)
(ZIP)
Statistics of k-mer processed assemblies with three methods (Clc, So, Tr).
(DOCX)
Blastx annotation of all transcripts against plant RefSeq and Arabidopsis databases with E-values and transcripts sizes.
(XLSX)
(DOCX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
The Authors are grateful to iPlant cyberinfrastructure [73] for the bioinformatic support. We appreciated the technical support for Illumina sequencing from Institute of Applied Genomics at Udine, Italy.
Data Availability
All clean reads files are available from the Sequence Read Archive (SRA) database of the National Centre of Biotechnology Information (NCBI) (accession number SRA313233).
Funding Statement
The authors received no specific funding for this work.
References
- 1.Lewis LA, McCourt RM. Green algae and the origin of land plants. Am J Bot. 2004;91: 1535–56. 10.3732/ajb.91.10.1535 [DOI] [PubMed] [Google Scholar]
- 2.Hughes NM. Winter leaf reddening in “evergreen” species. New Phytol. 2011;190: 573–81. 10.1111/j.1469-8137.2011.03662.x [DOI] [PubMed] [Google Scholar]
- 3.Gould KS. Nature’s Swiss Army Knife: The Diverse Protective Roles of Anthocyanins in Leaves. J Biomed Biotechnol. 2004;2004: 314–320. 10.1155/S1110724304406147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalker-Scott L. Environmental Significance of Anthocyanins in Plant Stress Responses. Photochem Photobiol. 1999;70: 1–9. 10.1111/j.1751-1097.1999.tb01944.x [DOI] [Google Scholar]
- 5.Tignor ME, Davies FS, Sherman WB, Davis JM. Rapid Freeze Acclimation of Poncirus trifoliata Seedlings Exposed to 10 {degrees}C and Long Days. HortScience. 1997;32: 854–857. Available: http://hortsci.ashspublications.org/content/32/5/854.short [Google Scholar]
- 6.Yatsuhashi H, Hashimoto T, Shimizu S. Ultraviolet Action Spectrum for Anthocyanin Formation in Broom Sorghum First Internodes. PLANT Physiol. 1982;70: 735–741. 10.1104/pp.70.3.735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harborne JB. Flavonoids: Distribution and contribution to plant colour In: Goodwin TW, editor. Chemistry and Biochemistry of Plant Pigments. Chemistry. London UK: Academic Press; 1965. pp. 247–278. [Google Scholar]
- 8.Tanaka Y, Sasaki N, Ohmiya A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 2008;54: 733–49. 10.1111/j.1365-313X.2008.03447.x [DOI] [PubMed] [Google Scholar]
- 9.Hatier J-HB, Gould KS. Foliar anthocyanins as modulators of stress signals. J Theor Biol. 2008;253: 625–7. 10.1016/j.jtbi.2008.04.018 [DOI] [PubMed] [Google Scholar]
- 10.Archetti M, Döring TF, Hagen SB, Hughes NM, Leather SR, Lee DW, et al. Unravelling the evolution of autumn colours: an interdisciplinary approach. Trends Ecol Evol. 2009;24: 166–73. 10.1016/j.tree.2008.10.006 [DOI] [PubMed] [Google Scholar]
- 11.Hughes NM, Carpenter KL, Cannon JG. Estimating contribution of anthocyanin pigments to osmotic adjustment during winter leaf reddening. J Plant Physiol. 2013;170: 230–3. 10.1016/j.jplph.2012.09.006 [DOI] [PubMed] [Google Scholar]
- 12.Landi M, Tattini M, Gould KS. Multiple functional roles of anthocyanins in plant-environment interactions. Environ Exp Bot. 2015;119: 4–17. 10.1016/j.envexpbot.2015.05.012 [DOI] [Google Scholar]
- 13.Gould KS, McKelvie J, Markham KR. Do anthocyanins function as antioxidants in leaves? Imaging of H2O2 in red and green leaves after mechanical injury. Plant, Cell Environ. 2002;25: 1261–1269. 10.1046/j.1365-3040.2002.00905.x [DOI] [Google Scholar]
- 14.Tattini M, Velikova V, Vickers C, Brunetti C, Di Ferdinando M, Trivellini A, et al. Isoprene production in transgenic tobacco alters isoprenoid, non-structural carbohydrate and phenylpropanoid metabolism, and protects photosynthesis from drought stress. Plant Cell Environ. 2014;37: 1950–1964. 10.1111/pce.12350 [DOI] [PubMed] [Google Scholar]
- 15.Logan BA, Stafstrom WC, Walsh MJL, Reblin JS, Gould KS. Examining the photoprotection hypothesis for adaxial foliar anthocyanin accumulation by revisiting comparisons of green- and red-leafed varieties of coleus (Solenostemon scutellarioides). Photosynth Res. 2015;124: 267–74. 10.1007/s11120-015-0130-0 [DOI] [PubMed] [Google Scholar]
- 16.Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot. 2011;62: 2465–83. 10.1093/jxb/erq442 [DOI] [PubMed] [Google Scholar]
- 17.Winkel-Shirley B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. PLANT Physiol. 2001;126: 485–493. 10.1104/pp.126.2.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes NM, Smith WK. Attenuation of incident light in Galax urceolata (Diapensiaceae): concerted influence of adaxial and abaxial anthocyanic layers on photoprotection. Am J Bot. 2007;94: 784–90. 10.3732/ajb.94.5.784 [DOI] [PubMed] [Google Scholar]
- 19.Guzman F, Kulcheski FR, Turchetto-Zolet AC, Margis R. De novo assembly of Eugenia uniflora L. transcriptome and identification of genes from the terpenoid biosynthesis pathway. Plant Sci. 2014;229: 238–46. 10.1016/j.plantsci.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 20.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. Nature Publishing Group; 2008;5: 621–8. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 21.Annadurai RS, Jayakumar V, Mugasimangalam RC, Katta MAVSK, Anand S, Gopinathan S, et al. Next generation sequencing and de novo transcriptome analysis of Costus pictus D. Don, a non-model plant with potent anti-diabetic properties. BMC Genomics. BioMed Central Ltd; 2012;13: 663 10.1186/1471-2164-13-663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun X, Zhou S, Meng F, Liu S. De novo assembly and characterization of the garlic (Allium sativum) bud transcriptome by Illumina sequencing. Plant Cell Rep. 2012;31: 1823–8. 10.1007/s00299-012-1295-z [DOI] [PubMed] [Google Scholar]
- 23.Kumar V, Kutschera VE, Nilsson MA, Janke A. Genetic signatures of adaptation revealed from transcriptome sequencing of Arctic and red foxes. BMC Genomics. BioMed Central Ltd; 2015;16: 585 10.1186/s12864-015-1724-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onda Y, Mochida K, Yoshida T, Sakurai T, Seymour RS, Umekawa Y, et al. Transcriptome analysis of thermogenic Arum concinnatum reveals the molecular components of floral scent production. Sci Rep. Nature Publishing Group; 2015;5: 8753 10.1038/srep08753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. Nature Publishing Group; 2011;12: 671–82. 10.1038/nrg3068 [DOI] [PubMed] [Google Scholar]
- 26.Makri O, Kintzios S. Ocimum sp. (Basil): Botany, Cultivation, Pharmaceutical Properties, and Biotechnology. J Herbs Spices Med Plants. Taylor & Francis Group; 2008;13: 123–150. 10.1300/J044v13n03_10 [DOI] [Google Scholar]
- 27.Rastogi S, Meena S, Bhattacharya A, Ghosh S, Shukla RK, Sangwan NS, et al. De novo sequencing and comparative analysis of holy and sweet basil transcriptomes. BMC Genomics. BioMed Central Ltd; 2014;15: 588 10.1186/1471-2164-15-588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci U S A. 2001;98: 9748–53. 10.1073/pnas.171285098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sablok G, Fu Y, Bobbio V, Laura M, Rotino GL, Bagnaresi P, et al. Fuelling genetic and metabolic exploration of C 3 bioenergy crops through the first reference transcriptome of Arundo donax L. Plant Biotechnol J. 2014;12: 554–567. 10.1111/pbi.12159 [DOI] [Google Scholar]
- 30.Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20: 1432–40. 10.1101/gr.103846.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23: 1061–7. 10.1093/bioinformatics/btm071 [DOI] [PubMed] [Google Scholar]
- 32.Villarino GH, Bombarely A, Giovannoni JJ, Scanlon MJ, Mattson NS. Transcriptomic analysis of Petunia hybrida in response to salt stress using high throughput RNA sequencing. PLoS One. Public Library of Science; 2014;9: e94651 10.1371/journal.pone.0094651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson D a, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29: 644–52. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenping H, Yuan Z, Jie S, Lijun Z, Zhezhi W. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics. 2011;98: 272–9. 10.1016/j.ygeno.2011.03.012 [DOI] [PubMed] [Google Scholar]
- 35.Yates SA, Swain MT, Hegarty MJ, Chernukin I, Lowe M, Allison GG, et al. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics. BioMed Central Ltd; 2014;15: 453 10.1186/1471-2164-15-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Z-G, Jiang W, Mantri N, Bao X-Q, Chen S-L, Tao Z-M. Transciptome analysis reveals flavonoid biosynthesis regulation and simple sequence repeats in yam (Dioscorea alata L.) tubers. BMC Genomics. BioMed Central Ltd; 2015;16: 346 10.1186/s12864-015-1547-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clifford M, Brown JE, Andersen ØM, Markham KR. Dietary flavonoids and health—broadening the perspective CRC Press LLC; 2006; 319–370. Available: http://www.cabdirect.org/abstracts/20063011830.html;jsessionid=48B81FBAF6ABF7DBD902B0DB0FD13A41 [Google Scholar]
- 38.Gang DR. An Investigation of the Storage and Biosynthesis of Phenylpropenes in Sweet Basil. PLANT Physiol. 2001;125: 539–555. 10.1104/pp.125.2.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang W, Song H, Liu H, Liu P. Current development in isoprenoid precursor biosynthesis and regulation. Curr Opin Chem Biol. 2013;17: 571–9. 10.1016/j.cbpa.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menzies IJ, Youard LW, Lord JM, Carpenter KL, van Klink JW, Perry NB, et al. Leaf colour polymorphisms: a balance between plant defence and photosynthesis. J Ecol. 2015; n/a–n/a. 10.1111/1365-2745.12494 [DOI] [Google Scholar]
- 41.Esteban R, Moran JF, Becerril JM, García-Plazaola JI. Versatility of carotenoids: An integrated view on diversity, evolution, functional roles and environmental interactions. Environ Exp Bot. 2015;119: 63–75. 10.1016/j.envexpbot.2015.04.009 [DOI] [Google Scholar]
- 42.Havaux M G-PJ. Beyond Non-Photochemical Fluorescence Quenching: The Overlapping Antioxidant Functions of Zeaxanthin and Tocopherols In: Demmig-Adams B, Garab G, Adams W III, editors. Non-Photochemical Quenching and Energy Dissipation in Plants, Algae and Cyanobacteria. Dordrecht: Springer Netherlands; 2014. 10.1007/978-94-017-9032-1 [DOI] [Google Scholar]
- 43.Aarts MG, Keijzer CJ, Stiekema WJ, Pereira a. Molecular characterization of the CER1 gene of arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell. 1995;7: 2115–2127. 10.1105/tpc.7.12.2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jenks MA, Tuttle HA, Eigenbrode SD, Feldmann KA. Leaf Epicuticular Waxes of the Eceriferum Mutants in Arabidopsis. Plant Physiol. 1995;108: 369–377. 10.1104/pp.108.1.369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Millar AA. CUT1, an Arabidopsis Gene Required for Cuticular Wax Biosynthesis and Pollen Fertility, Encodes a Very-Long-Chain Fatty Acid Condensing Enzyme. PLANT CELL ONLINE. 1999;11: 825–838. 10.1105/tpc.11.5.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li F, Wu X, Lam P, Bird D, Zheng H, Samuels L, et al. Identification of the wax ester synthase/acyl-coenzyme A: diacylglycerol acyltransferase WSD1 required for stem wax ester biosynthesis in Arabidopsis. Plant Physiol. 2008;148: 97–107. 10.1104/pp.108.123471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shepherd T, Wynne Griffiths D. The effects of stress on plant cuticular waxes. New Phytol. 2006;171: 469–99. 10.1111/j.1469-8137.2006.01826.x [DOI] [PubMed] [Google Scholar]
- 48.Holmes MG, Keiller DR. Effects of pubescence and waxes on the reflectance of leaves in the ultraviolet and photosynthetic wavebands: a comparison of a range of species. Plant Cell Environ. 2002;25: 85–93. 10.1046/j.1365-3040.2002.00779.x [DOI] [Google Scholar]
- 49.Riederer M MC. Biology of the Plant Cuticle Annual Plant Reviews Volume 23 Wiley Onli. John Wiley & Sons; 2008. Available: http://onlinelibrary.wiley.com/book/10.1002/9780470988718 [Google Scholar]
- 50.Landi M, Pardossi A, Remorini D, Guidi L. Antioxidant and photosynthetic response of a purple-leaved and a green-leaved cultivar of sweet basil (Ocimum basilicum) to boron excess. Environ Exp Bot. 2013;85: 64–75. 10.1016/j.envexpbot.2012.08.008 [DOI] [Google Scholar]
- 51.Tian J, Peng Z, Zhang J, Song T, Wan H, Zhang M, et al. McMYB10 regulates coloration via activating McF3’H and later structural genes in ever-red leaf crabapple. Plant Biotechnol J. 2015;13: 948–61. 10.1111/pbi.12331 [DOI] [PubMed] [Google Scholar]
- 52.Agati G, Stefano G, Biricolti S, Tattini M. Mesophyll distribution of “antioxidant” flavonoid glycosides in Ligustrum vulgare leaves under contrasting sunlight irradiance. Ann Bot. 2009;104: 853–61. 10.1093/aob/mcp177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siipola SM, Kotilainen T, Sipari N, Morales LO, Lindfors A V, Robson TM, et al. Epidermal UV-A absorbance and whole-leaf flavonoid composition in pea respond more to solar blue light than to solar UV radiation. Plant Cell Environ. 2015;38: 941–52. 10.1111/pce.12403 [DOI] [PubMed] [Google Scholar]
- 54.Agati G, Biricolti S, Guidi L, Ferrini F, Fini A, Tattini M. The biosynthesis of flavonoids is enhanced similarly by UV radiation and root zone salinity in L. vulgare leaves. J Plant Physiol. Elsevier GmbH.; 2011;168: 204–12. 10.1016/j.jplph.2010.07.016 [DOI] [PubMed] [Google Scholar]
- 55.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010 [Internet]. 2011. Available: http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/
- 56.Chikhi R, Medvedev P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 2014;30: 31–7. 10.1093/bioinformatics/btt310 [DOI] [PubMed] [Google Scholar]
- 57.Bruijn de NG. A combinatorial problem. Proc K Ned Akad van Wet Ser A. 1946;49: 758. [Google Scholar]
- 58.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28: 3150–2. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. BioMed Central Ltd; 2011;12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2012;9: 357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215: 403–10. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 62.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21: 3674–6. 10.1093/bioinformatics/bti610 [DOI] [PubMed] [Google Scholar]
- 63.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34: W293–7. 10.1093/nar/gkl031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17: 847–848. 10.1093/bioinformatics/17.9.847 [DOI] [PubMed] [Google Scholar]
- 65.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35: W182–5. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Torre S, Tattini M, Brunetti C, Fineschi S, Fini A, Ferrini F, et al. RNA-seq analysis of Quercus pubescens Leaves: de novo transcriptome assembly, annotation and functional markers development. PLoS One. Public Library of Science; 2014;9: e112487 10.1371/journal.pone.0112487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kal AJ, van Zonneveld AJ, Benes V, van den Berg M, Koerkamp MG, Albermann K, et al. Dynamics of Gene Expression Revealed by Comparison of Serial Analysis of Gene Expression Transcript Profiles from Yeast Grown on Two Different Carbon Sources. Mol Biol Cell. 1999;10: 1859–1872. 10.1091/mbc.10.6.1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9: 811–818. 10.1002/sim.4780090710 [DOI] [PubMed] [Google Scholar]
- 69.Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, et al. mapman: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37: 914–939. 10.1111/j.1365-313X.2004.02016.x [DOI] [PubMed] [Google Scholar]
- 70.Lohse M, Nagel A, Herter T, May P, Schroda M, Zrenner R, et al. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014;37: 1250–8. 10.1111/pce.12231 [DOI] [PubMed] [Google Scholar]
- 71.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. 2012;40: 1–12. 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif. Elsevier; 2001;25: 402–408. Available: http://www.ncbi.nlm.nih.gov/pubmed/11846609 [DOI] [PubMed] [Google Scholar]
- 73.Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front Plant Sci. 2011;2: 34 10.3389/fpls.2011.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Frequency histogram showing the distribution of transcript length in sweet basil.
(TIF)
(A) The E-value distribution of BLAST hits for the assembled RR-TIG sequences with a cutoff of E-value < 10−5. (B) The similarity distribution of BLAST hit for the assembled RR-TIG sequences with a cutoff of E-value < 10−5. (C) The species distribution of the top BLAST hits for each transcript in the RR-TIG transcriptome assembly from Blast2GO.
(TIF)
A. The profiles of different SSR types in TIG (green) and RR (red). B. The distribution of repeat motifs in TIG (green) and RR (red).
(TIF)
The RPKM overall distribution and variability of cDNA libraries/samples were similar, indicating that they were comparable for identification of differentially expressed genes (DEGs) at the transcriptome level. (A) A box plot analysis with original expression values; (B) A box plot analysis with normalized expression values; (C) Density plot showing the distribution of log2(RPKM) in RR and TIG.
(TIF)
The results are summarized in three main categories: cellular component, molecular function, and biological process.
(TIF)
(TIF)
Fasta file of basil transcripts de novo assembled with CLC Genomics Workbench v.8.5.1.
(FASTA)
Fasta file of basil transcripts de novo assembled with SOAPdenovo-Trans v1.01.
(ZIP)
Fasta file of basil transcripts de novo assembled with Trinity r2013-02-25.
(ZIP)
List of GO terms associated to sweet basil transcripts.
(ZIP)
(ZIP)
Statistics of k-mer processed assemblies with three methods (Clc, So, Tr).
(DOCX)
Blastx annotation of all transcripts against plant RefSeq and Arabidopsis databases with E-values and transcripts sizes.
(XLSX)
(DOCX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All clean reads files are available from the Sequence Read Archive (SRA) database of the National Centre of Biotechnology Information (NCBI) (accession number SRA313233).