

Abstract
The transformation suppressor gene RECK was isolated by cDNA expression cloning (1998), and GPR124/TEM5 was detected as a tumor endothelial marker by differential screening (2000). The importance of Wnt7a/b and Gpr124 in brain angiogenesis was demonstrated by reverse genetics in mice (2008–2010). A series of recent studies using genetically engineered mice and zebrafish as well as luciferase reporter assays in cultured cells led to the discovery of functional interactions among Reck, Gpr124, and Wnt7a/b in triggering canonical Wnt signaling with relevance to embryonic brain angiogenesis and blood–brain barrier formation.
Keywords: Angiogenesis, Gpr124, Reck, Wnt7a/b, zebrafish
Prologue
In Shakespeare's comedy “The Winter's Tale”, the infant princess Perdita (lost one) was abandoned and her mother dies due to the king's outrageous jealousy of his rival king. After 16 years, the grown‐up orphan travels to her homeland with her boyfriend Florizel (flower; the rival king's son trying to establish himself) and unexpectedly learns about her origin in a magical happy ending.
A series of recent studies has set the stage for a dramatic encounter of an orphan G protein‐coupled receptor, Gpr124, accompanied by the Reck tumor suppressor, with a particular pair of the Wnt family, Wnt7a/b, which together trigger canonical Wnt signaling to induce brain angiogenesis and blood–brain barrier (BBB) formation in vertebrates.1
Gpr124: An orphan with multiple names
The story begins with the provocative paper published in 2000 by St. Croix et al. describing the isolation of vascular endothelial cells through an elaborate series of negative and positive selections from colon tumor and normal mucosa and the comparison of gene expression profiles between these cells using the then‐current technique called serial analysis of gene expression (SAGE). This screen allowed the researchers to identify nine previously unknown genes that showed more than 16‐fold higher expression in tumor endothelial cells than the normal counterpart and were therefore termed tumor endothelial markers (TEMs).2 One of these genes, TEM5, was found to encode an orphan G protein‐coupled receptor (GPCR)3 and later termed GPR1244 which was eventually classified into the adhesion G protein‐coupled receptor (ADGR) family, the second largest subfamily of GPCRs consisting of 33 members, and hence conferred yet another name, ADGRA2.5 Most ADGRs are characterized by a long NH2‐terminal ectodomain followed by the seven transmembrane domain and the COOH‐terminal cytoplasmic tail (Fig. 1, left side).5 While membrane distal extracellular regions of different ADGRs can vary greatly, homology and family affiliation are confined and determined, respectively, by a highly conserved membrane proximal domain termed the GPCR autoproteolysis‐inducing domain and the highly conserved seven‐pass transmembrane domain.
Figure 1.
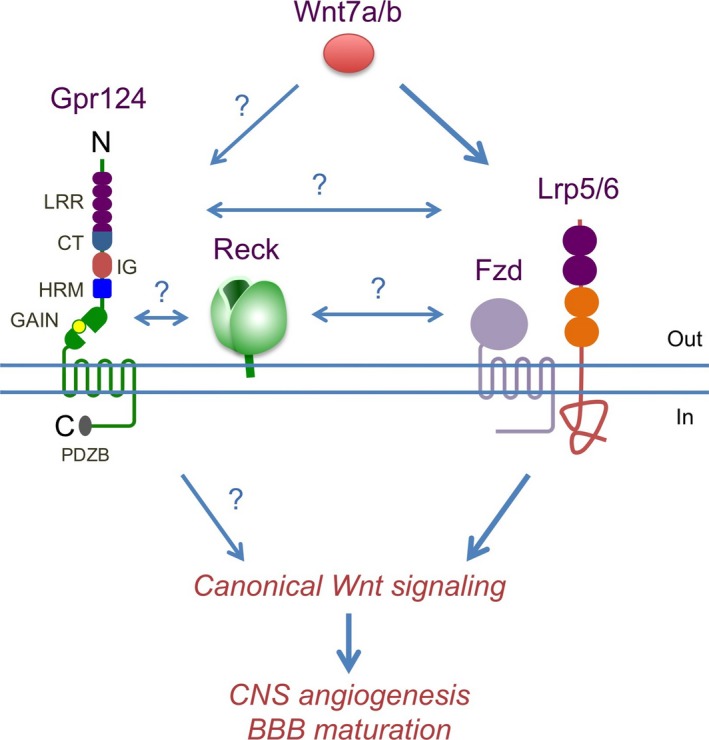
Functional interactions among Gpr124, Reck, and Wnt7a/b‐triggered canonical Wnt signaling which promotes central nervous system (CNS) angiogenesis and blood–brain barrier (BBB) formation. New genetic studies in mouse and zebrafish indicate functions of Gpr124 and Reck to enhance canonical Wnt signaling induced by Wnt7a/b. Question marks denote possible molecular interactions. C, COOH‐terminus; CT, C‐terminal domain; GAIN, G protein‐coupled receptor autoproteolysis‐inducing; HRM, hormone receptor motif; IG, immunoglobulin domain; LRR, leucine‐rich repeat; N, NH2‐terminus; PDZB, PDZ domain‐binding motif.3, 5, 14
An important function of Gpr124 (Tem5) was revealed when this gene was inactivated in mice.6 Gpr124‐null mice show lethality from embryonic day 15.5 (E15.5) onward with hemorrhage in the forebrain and neural tube. In these embryos, angiogenic sprouting from the perineural vascular plexus into the forebrain and ventral neural tube is severely impaired and formation of hemorrhagic glomeruloid vascular malformations at the periphery of the neuroepithelium is observed. Furthermore, filopodia are misoriented in tip‐like cells belonging to invading angiogenic sprouts.6 In wild‐type mice, although Gpr124 (Tem5) is highly expressed in the central nervous system (CNS) vasculature, it is also detectable in other organs and in multiple cell types. It is therefore remarkable that the mutant phenotype (vascular malformation) appears to be confined to the CNS. These mice also failed to establish an effective BBB. These findings were underscored by two papers reporting similar phenotypes of Gpr124‐null mice established independently.7, 8
The BBB is a special property of CNS endothelium, induced and maintained by a multicellular neurovascular unit. It separates the CNS from the peripheral blood circulation and maintains an environment that allows neurons to function properly. Endothelial cells of the neurovascular unit have special characteristics, such as continuous tight junctions, lack of fenestrations, and low rates of transcytosis, which greatly limits the movement of molecules through the endothelial layer.9 Instead, passage of molecules through the BBB is regulated by a series of specific transporters that allow delivery of nutrients to the brain and extrusion of unwanted molecules. One such transporter, Glut1/Slc2a1, has been used as a marker for the BBB.1 Indeed, Glut1 expression was absent in Gpr124‐deficient endothelium in the forebrain,6, 7, 8 indicating that the mutation not only causes anatomical vascular defects, as evidenced by massive brain hemorrhage, but also affects endothelial differentiation leading to the formation of a functional BBB.
Wnt7a/b: The royal couple
In E12.5 mouse embryos, canonical Wnt signaling (as detected by the TOP‐GAL reporter transgene) co‐localized with endothelium in CNS but not in other areas. Wnt7a and Wnt7b were the canonical Wnt ligands expressed in the forebrain and on the ventral neural tube where CNS angiogenesis initiates.10 The phenotype of Wnt7a/Wnt7b double‐knockout mice10, 11 was reminiscent of that of Gpr124‐null mice.6, 7, 8
An actual link between Wnt7a/b and Gpr124 was reported by Zhou and Nathans12 and by Posokhova et al.13 in mice and by Vanhollebeke et al.14 in zebrafish using reporter assays in vitro and genetic studies in vivo. These studies established the role of Gpr124 as a ligand (Wnt7a/b)‐specific coactivator of canonical Wnt/β‐catenin signaling.
Reck: The prince likened to a flower
Reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) was first isolated as a cDNA inducing morphological reversion when expressed in a mouse embryo fibroblast cell line transformed by a RAS oncogene.15 Despite the in vitro screen used for isolation, the gene soon attracted the attention of many clinicians, as its expression tends to be downregulated in various cancer tissues, including those of the lung, colon, breast, pancreas, and testis.16, 17, 18 It was found that RECK expression was suppressed by various stimuli and signals, such as growth factors, low cell density, hypoxia, and oncogenes.15, 19, 20, 21 Restored RECK expression in cancer cells has been reported to suppress cell proliferation, tumor angiogenesis, invasion, and metastasis, depending on the cell lines and assay systems used.21, 22, 23 Frequency of spontaneous tumors increases in mice with reduced Reck expression (Sachiyo Yamaguchi, Tomoko Matsuzaki, Makoto Noda, et al., unpublished data), demonstrating the function of Reck as a bona fide tumor suppressor.
RECK encodes a glycosylphosphatidylinositol‐anchored glycoprotein of approximately 125 kDa that forms a tulip‐shaped dimer (Fig. 2)24 and can negatively regulate multiple extracellular proteases, such as several members of the MMP family, Adam10, and CD13.15, 22, 25, 26 At the cellular level, RECK can regulate directional cell migration27, 28 and cell cycle progression.23
Figure 2.
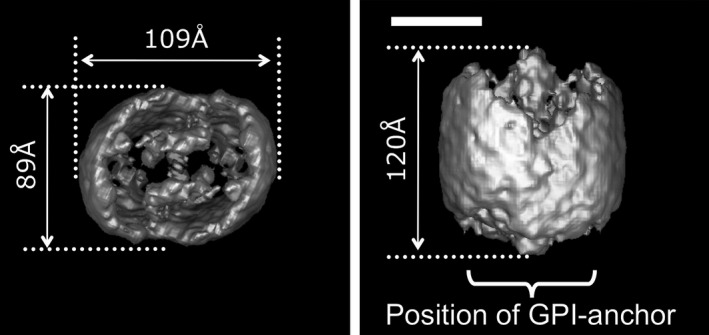
Surface representation of the RECK protein. Three‐dimensional reconstruction of the RECK‐His dimer viewed from the top (a) and the side (b). GPI, glycosylphosphatidylinositol. Scale bar = 50 Å.24
RECK is not found in Caenorhabditis elegans but is conserved from fruit fly to human as a single gene. Reck‐deficient mice die at approximately E10.5 with reduced tissue integrity, abdominal hemorrhage,22 and precocious neuronal differentiation.26 In wild‐type embryos at this stage, Reck is abundantly expressed in blood vessels (both endothelial and mural components) and the neural precursor cells.22, 26, 29, 30, 31 Experiments using tamoxifen‐inducible Reck knockout mice indicated that global inactivation of Reck starting from E11 resulted in hemorrhage and vascular defects in the brain by E15.5 and embryonic death before birth.29 Hence, Reck is essential for mouse embryogenesis.
Mice expressing Reck at a level <25% of the normal level are viable but show a defect in anterior–posterior limb patterning most similar to that found in Wnt7a‐null mice,32 albeit with peculiar asymmetry (right‐dominant, forelimb‐specific).33
News from the aquarium
In 2012, Prendergast et al.34 announced an important breakthrough in better understanding the physiological functions of RECK. They carried out a forward genetic screen in zebrafish for mutations causing deficiencies in dorsal root ganglia (DRG). They isolated four mutants lacking all DRG and found that these mutants carry independent mutations in the reck gene. Morpholino‐mediated knockdown of reck phenocopied the DRG deficiency, supporting its involvement and the recessive (loss‐of‐function type) nature of the mutations. Transplantation experiments indicated that reck was required in a cell‐autonomous fashion in DRG neurons. The reck deficiency led to slower migration and mislocalization of the neural crest‐derived DRG precursor cells.34 Interestingly, this migration requires Mmp17, a glycosylphosphatidylinositol‐anchored member of the MMP family, co‐expressed with Reck in DRG precursor cells.35
More surprise from the aquarium
Vanhollebeke and coworkers introduced mutations in the zebrafish gpr124 gene using transcription activator‐like effector nucleases (TALEN).14 These mutant fish recapitulate the brain vascular defects found in mice, although the defects seem to be less life‐threatening, for half of the mutants reached adulthood and some were even fertile. In addition, they found a lack of DRG in these mutant fish. To test the possibility that common molecules underlie these two events, they knocked down genes known to be involved in DRG formation, including erbb3, sorbs3, and reck, and found that reck morpholino could induce brain vascular defects reminiscent of that induced by gpr124 mutations.
Furthermore, the TOP‐flash Wnt reporter assay showed that Wnt7a/b‐triggered canonical Wnt signaling in vitro was markedly enhanced when Gpr124 and Reck co‐exist with the Wnt receptor, for example, Fzd4/Lrp6. They also used double immunofluorescence staining and a proximity ligation assay to show co‐localization of overexpressed Gpr124 and Reck on the surface of HEK293T cells. However, although the proximity ligation assay technique demonstrates proximity, it does not unequivocally reveal direct physical interaction between Gpr124 and Reck, which yet remains to be determined.
Through elegant experiments in vivo with zebrafish, Vanhollebeke and colleagues further demonstrated the following points: (i) Wnt signaling is required for brain angiogenesis and DRG neurogenesis (through pharmacological and genetic approaches); (ii) both gpr124 and reck are required for endothelial‐specific Wnt signaling; and (iii) Gpr124/Reck‐mediated Wnt signaling is required in a tip cell‐specific manner during angiogenic sprouting in the brain (through cell transplantations, mosaic vessel quantification, and live imaging).
Based on these findings, Vanhollebeke et al. proposed three alternative models of how Gpr124 and Reck contribute to Wnt signaling: (i) Reck protects an Fzd/Lrp/Gpr124 complex from proteolysis; (ii) Fzd/Lrp/Gpr124/Reck form a high affinity Wnt7a/b receptor; or (iii) Gpr124/Reck constitute an independent Wnt7a/b receptor (Fig. 1).14
The first model seems to be relevant to the previous findings that the GPR124 (TEM5) ectodomain can be shed by MMP9 and mediates endothelial cell survival by linking integrin αvβ3 to glycosaminoglycans36 and that RECK regulates MMP9.15, 37 Vanhollebeke et al., however, argued against this model, based on the difficulty to rescue reck morphant phenotypes by treating embryos with various MMP/ADAM inhibitors. They instead suggested the involvement of other activities of Reck, for instance, regulation of endocytic trafficking,25 focal adhesions, and cell polarity27 that could influence canonical Wnt signaling.38, 39
No food for thought
Another forward genetics study using zebrafish has recently provided evidence in vivo for the involvement of Reck in Wnt signaling and BBB formation. Shaw et al.40 previously mutagenized zebrafish expressing GFP in endothelial cells and screened mutants with defects in vascular patterning. In one such line, no food for thought (nft), a missense (C254Y) mutation in reck was found to be responsible for its phenotypes including loss of intracerebral central arteries (CtAs) and downregulation of Glut1 as well as Wnt signaling in cerebral vasculature.41 The mutant phenotype was phenocopied by inactivating Wnt signaling (by overexpression of a dominant negative form of TCF3).
The loss of CtAs in the nft mutant is accompanied by hyperplasia of the primordial hindbrain channels, which is reminiscent of the disorganized perineural vascular plexus found in Reck KO mice,29 suggesting inefficient vascular sprouting from primordial hindbrain channels. Transplantation of wild‐type endothelial cells into nft mutant zebrafish embryos resulted in phenotypic suppression and chimeric CtAs formation, suggesting a non‐cell autonomous contribution of Reck to intracerebral vascularization.
The heart bleeds, but here is more matter for a hot brain
Another recent study using tissue‐selective Reck knockout mice indicated that the lack of Reck expression in mural cells mediated by Sm22‐Cre largely recapitulates the phenotypes of global Reck knockout mice, that is, death around E10.5 accompanied by abdominal hemorrhage and loss of the pericardial membrane.31 In contrast, the lack of Reck expression in endothelial cells mediated by Tie2‐Cre leads to embryonic death at much later stages (between E15.5 and P0). Of note, the most prominent phenotype in these mice is found in the brain: dilated blood vessels accompanied by massive hemorrhage and impaired cortical structure.31
Aortic tissue explants from young mice with conditionally inactivated Reck gene (tamoxifen‐induced, Reck + cell‐specific knockout) showed (in addition to the increased collagenolytic activity) excessive sprouting of microvessels, which tended to aggregate and form thicker vessels than the control;31 this vessel instability may explain why Reck‐deficient mice show dilated blood vessels. It was unclear, however, why the phenotype of endothelial Reck knockout mice was confined to the brain. A feasible answer to this question was provided by the new findings in zebrafish14 (see above).
Also in this system, vascular cells lacking Reck expression failed to associate properly with microvessels: namely, the endothelial tip cells were mislocalized while the mural cells failed to tightly associate with microvessels31 (Fig. 3a). According to the model proposed by Senger and Davis,42 the association of mural cells to endothelial tubules triggers fibronectin (FN) deposition followed by vascular basement membrane deposition required for vascular stability (Fig. 3b). Interestingly, exogenous FN could ameliorate the instability of Reck conditional knockout (cKO) microvessels but not the poor association of mural cells with microvessels,31 an event upstream of FN deposition in this model (Fig. 3b).
Figure 3.
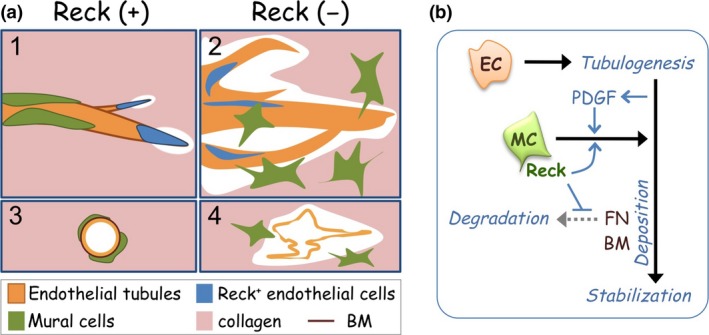
Effects of Reck in sprouting angiogenesis.31 (a) Summary of findings by aortic ring assay. When Reck is present (+; panels 1 and 3), endothelial sprouting led by tip cells (blue) is appropriately regulated; tight association of mural cells (green) with endothelial tubules (orange) is promoted, and individual vessels are stabilized. When Reck is absent or reduced (−; panels 2 and 4), endothelial sprouting and collagenolysis are activated; tip cells and mural cells localize inappropriately, and microvessels are destabilized, permitting lateral fusion and ectopic anastomoses. (b) A model to explain how Reck promotes vascular stabilization. Reck promotes (blue arrow) tight association of mural cells (MC) to the tubule‐forming endothelial cells (EC), an event known to trigger perivascular fibronectin (FN) deposition.2, 3 Reck also protects FN from degradation, thereby promoting downstream events, including perivascular basement membrane (BM) deposition, which helps establish vascular stability. PDGF, platelet‐derived growth factor.
What animals lack from head to heel
A common phenotype found in Gpr124‐deficient and Reck‐deficient animals (both mice and zebrafish) is the lack of blood vessels, at least in a certain part of the brain, often accompanied by brain hemorrhage. A major difference between Gpr124‐deficient and Reck‐deficient animals is the timing of death: global Reck mutants die earlier (embryonic or post‐natal) than Gpr124 mutants (some zebrafish mutants even survive to adulthood), although endothelial‐specific Reck mutants and Gpr124 mutants look similar in their survival curves. More subtle differences are found in the morphology of mouse blood vessels: forebrain and neural tube vessels in Gpr124‐deficient mice show glomeruloid malformations, whereas the vessels in Reck‐deficient mice show larger, irregularly shaped, and often broken luminal space.
It is remarkable that independent observations by imaging (i.e., intravital imaging in zebrafish, and murine cells or tissue explants in culture) let us focus on the same phenomenon: misbehaviors of endothelial (tip) cells that lack Gpr124 or Reck (Table 1). The activity of these molecules to make the tip cells function and/or localize properly seem to be cell‐autonomous,14 whereas their activity to promote proper vascular sprouting seems to be non‐cell‐autonomous.14, 41 These findings are consistent with the model that Gpr124 and Reck on the tip cell surface enhance Wnt7a/b signaling in tip cells, which instruct the behavior of stalk cells that do not require Gpr124 and/or Reck. It should be noted, however, that the misbehaviors of tip cells could be caused by several different events: failures in proper cell–cell or cell–matrix association,31 process extension,14 and directional migration.6 It was previously found that forebrain cortical cells secrete guidance cues that stimulate Gpr124‐dependent migration of brain endothelial cells.6 Wnt7b, known as a forebrain marker,43 is an obvious candidate for such a guidance cue. In contrast, a wide range of functions have been assigned to Wnt7a, based mainly on the neuronal phenotypes of Wnt7a knockout mice.44, 45 In light of the recent findings discussed here, however, some of the previous interpretations may need to be re‐evaluated whether or not the neuronal phenotypes represent a secondary manifestation of vascular defects.
Table 1.
Comparison of findings on Gpr124, Reck, and their functional relationship in different experimental systems
| In vivo | In vitro | ||||
|---|---|---|---|---|---|
| Mouse | Zebrafish | HEK‐293 cells | |||
| Phenotype | Phenotype | TOP‐flash assay | |||
| Gpr124 KO | Reck cKO (Tie2) *cKO (Reck‐CreERT2) | gpr124 KO | reck MO/†KO | Gpr124, Reck, Fzd, Lrp, Wnt7a/b | |
| Brain vasculature |
‐ CNS hemorrhage ‐ Glomeruolid malformation ‐ PNVP thickening ‐ Avascular telencephalon |
‐ Cranial hemorrage ‐ Dilated vessels |
Avascular brain | ‐ Avascular brain | |
| BBB |
‐ Low Glut‐1 expression ‐ Breakdown (tracer; Cullen) |
? | ? | ? | |
| DRG | ? | ? | Absent | Absent, cell‐autonomous† (Prendargast) | |
| Endothelial cells | Failure in chemotactic migration and sprouting in vitro (Cdc42‐dependent, VEGF‐independent, cell‐autonomous) | Tip cells mislocalized in aortic ring assay (Fig. 3)* |
‐ Lack of vascular sprouting into the hindbrain tissue ‐ Loss of tip‐cell sprouts from PHBC (cell autonomous) |
‐ Lack of vascular sprouting into the hindbrain tissue (non cell‐autonomous) ‐ PHBP hyperplasia (Ulrich)† |
|
| Mural cells | ? | Mislocalized in vivo and in aortic ring assay (Fig. 3)* | No change | No change | |
| Wnt signaling | Reduced | ? | Reduced | Reduced | Ligand (Wnt7a/b)‐specific enhancement of canonical Wnt signaling by Gpr124 plus Reck |
| Timing of death | E15‐before adulthood | E15‐P0 | ~50% healthy and fertile | Before day 12† | |
| Other findings | Tg (Tie2) mice: brain hypervascularity |
‐ Defects in cortical development ‐ Reck KO: lethal around E10.5 with abdominal hemorrhage |
‐ Smaller body size ‐ Disrupted skin pigmentation pattern |
‐ Co‐localization of Gpr124 and Reck (PLA; Vanhollebeke) ‐ Gpr124 interacts with DLG1 (Posokhova) |
|
| References | Kuhner, Anderson, Cullen, Zhou, Posokhova | Almeida*, Oh, Chandana | Vanhollebeke | Vanhollebeke, Ulrich†, Prendargast† | Vanhollebeke, Zhou, Posokhova |
KO, global knockout; cKO, conditional or cell type‐specific knockout; MO, morpholino‐mediated knockdown; Tie2, endothelial cell‐specific promoter; PNVP, perineural vascular plexus; Tg, transgenic; PLA, proximity ligation assay. *Results with Reck cKO mice; †Results with reck KO fish.
It would also be worthwhile to clarify which findings in one system have not been confirmed in the other (Table 1, indicated by question marks). First, BBB breakdown accompanied by Glut1 downregulation has not been confirmed in Reck‐deficient mice. Second, DRG deficiency has been found in zebrafish mutants but not in mouse mutants. A possible explanation for this difference might be the lack of Gpr124 expression in mouse DRG.46 Of note in this context, however, Wnt signaling has been implicated in DRG development.47, 48 Third, the importance of mural Reck for vascular development and survival has been documented in mouse.31 What about mural Gpr124? In this respect, the glomeruloid malformations in global Gpr124‐deficient mice are phenocopied by endothelial‐specific deletion of Gpr124. Furthermore, the glomeruloid malformations are covered by pericytes,6 suggesting that Gpr124 is dispensable for pericyte function in vivo. Vanhollebeke et al.14 reported that vascular wall coverage by pericytes was unaffected in gpr124‐deficient zebrafish. Fourth, is Wnt signaling also affected in Reck‐deficient mice? Finally, adult zebrafish gpr124 mutants and a mouse Reck hypomorphic mutant show smaller body sizes. What is the reason for this phenotype? Addressing these questions is not just filling the space in the table but may yield more clues to new discoveries.
Toward a happy ending
Several major questions are in sight at this stage. First, how do Reck and Gpr124 cooperate in Wnt signaling? To address this question, we need to better understand the nature of molecular interactions: which protein directly interacts with which through which domains? The role(s) of DLG1, identified as a Gpr124‐interactor,13 in CNS angiogenesis and other events also needs to be investigated.
A second important question is how far we can extend our knowledge of the functions of Reck and Gpr124 along this line. In other words, is enhancement of Wnt7a/b signaling the only function of Reck and Gpr124? This question has to do with a more general question of what we could learn from phenotypes of knockout animals. Phenotypes of recessive mutants usually appear in the places where functional redundancy is poor or absent. In this regard, the earlier death of Reck mutants with broader spectra of deficiencies than the Gpr124 mutants (Table 1) may indicate either a broader spectrum of Reck's functions or the absence of redundancy for Reck in broader areas of the body. Likewise, the similarity in phenotype between endothelial Reck cKO and Gpr124 KO mice and the earlier death of global Reck KO and mural Reck cKO mice may indicate either the importance of Reck in both cell types or the absence of redundancy for Gpr124 in endothelial cells. Nevertheless, it seems unlikely that all characteristics of Reck‐null microvessels in culture (Fig. 3a) are due to the abortive canonical Wnt signaling.
Gpr124 might exhibit redundancy via its closest paralogue Gpr125,5 which has been characterized as a spermatogonial progenitor marker,49 and is expressed in choroid plexus.50 Gpr125, however, regulates non‐canonical Wnt/PCP signaling in zebrafish51 and shows little effects on canonical Wnt signaling in vitro.9, 10 Importance of the Gpr124 ectodomain in mediating canonical Wnt7 signaling has been reported.10 Some of the other ADGR family members share the same motifs with Gpr124 (e.g., hormone receptor motif) in their ectodomain.5 Comparison between precise expression patterns and knockout phenotypes of these genes during embryogenesis may provide some clues to this issue. Efforts to address the first question (e.g., elucidating the partners and domains involved in molecular interactions) may also provide information useful for designing further genetic experiments. A previous study with Reck hypomorphic mice suggested a functional link between Reck and Wnt7a in limb patterning.32, 33 Whether this reflects an extra‐CNS and Gpr124‐independent role of Reck in Wnt7a signaling or a totally different mechanism is another interesting question to be addressed in future studies. Additionally, it will be interesting to explore the role of Gpr124 and Reck in adult pathologic states.
But now, let us celebrate the happy reunion of the orphan (Gpr124) and her tulip‐shaped partner (Reck) with the royal couple (Wnt7a/b)!
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The Noda group has been supported by Japan Society for the Promotion of Science KAKENHI Grants‐in‐Aid for Creative Scientific Research, Scientific Research on Priority Areas, and Scientific Research on Innovative Areas. The Kuo group has been supported by the National Institutes of Health (NS064517 and CA158528).
Cancer Sci 107 (2016) 576–582
Funding Information
Japan Society for the Promotion of Science; National Institutes of Health (CA158528, NS064517).
References
- 1. Vallon M, Chang J, Zhang H, Kuo CJ. Developmental and pathological angiogenesis in the central nervous system. Cell Mol Life Sci 2014; 71: 3489–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. St Croix B, Rago C, Velculescu V et al Genes expressed in human tumor endothelium. Science 2000; 289: 1197–202. [DOI] [PubMed] [Google Scholar]
- 3. Carson‐Walter EB, Watkins DN, Nanda A, Vogelstein B, Kinzler KW, St Croix B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res 2001; 61: 6649–55. [PubMed] [Google Scholar]
- 4. Fredriksson R, Gloriam DE, Hoglund PJ, Lagerstrom MC, Schioth HB. There exist at least 30 human G‐protein‐coupled receptors with long Ser/Thr‐rich N‐termini. Biochem Biophys Res Commun 2003; 301: 725–34. [DOI] [PubMed] [Google Scholar]
- 5. Hamann J, Aust G, Arac D et al International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein‐coupled receptors. Pharmacol Rev 2015; 67: 338–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuhnert F, Mancuso MR, Shamloo A et al Essential regulation of CNS angiogenesis by the orphan G protein‐coupled receptor GPR124. Science 2010; 330: 985–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cullen M, Elzarrad MK, Seaman S et al GPR124, an orphan G protein‐coupled receptor, is required for CNS‐specific vascularization and establishment of the blood‐brain barrier. Proc Natl Acad Sci USA 2011; 108: 5759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anderson KD, Pan L, Yang XM et al Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein‐coupled receptor. Proc Natl Acad Sci USA 2011; 108: 2807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood–brain barrier. Nat Med 2013; 19: 1584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta‐catenin signaling is required for CNS, but not non‐CNS, angiogenesis. Proc Natl Acad Sci USA 2009; 106: 641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ‐specific assembly and differentiation of CNS vasculature. Science 2008; 322: 1247–50. [DOI] [PubMed] [Google Scholar]
- 12. Zhou Y, Nathans J. Gpr124 controls CNS angiogenesis and blood‐brain barrier integrity by promoting ligand‐specific canonical wnt signaling. Dev Cell 2014; 31: 248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Posokhova E, Shukla A, Seaman S et al GPR124 functions as a WNT7‐specific coactivator of canonical beta‐catenin signaling. Cell Rep 2015; 10: 123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vanhollebeke B, Stone OA, Bostaille N et al Tip cell‐specific requirement for an atypical Gpr124‐ and Reck‐dependent Wnt/beta‐catenin pathway during brain angiogenesis. Elife 2015; 4. doi: 10.7554/eLife.06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takahashi C, Sheng Z, Horan TP et al Regulation of matrix metalloproteinase‐9 and inhibition of tumor invasion by the membrane‐anchored glycoprotein RECK. Proc Natl Acad Sci USA 1998; 95: 13221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark JC, Thomas DM, Choong PF, Dass CR. RECK–a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev 2007; 26: 675–83. [DOI] [PubMed] [Google Scholar]
- 17. Noda M, Takahashi C. Recklessness as a hallmark of aggressive cancer. Cancer Sci 2007; 98: 1659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Noda M, Takahashi C, Matsuzaki T, Kitayama H. What we learn from transformation suppressor genes: lessons from RECK. Future Oncol 2010; 6: 1105–16. [DOI] [PubMed] [Google Scholar]
- 19. Sasahara RM, Takahashi C, Noda M. Involvement of the Sp1 site in ras‐mediated downregulation of the RECK metastasis suppressor gene. Biochem Biophys Res Commun 1999; 264: 668–75. [DOI] [PubMed] [Google Scholar]
- 20. Hatta M, Matsuzaki T, Morioka Y, Yoshida Y, Noda M. Density‐ and serum‐dependent regulation of the Reck tumor suppressor in mouse embryo fibroblasts. Cell Signal 2009; 21: 1885–93. [DOI] [PubMed] [Google Scholar]
- 21. Loayza‐Puch F, Yoshida Y, Matsuzaki T, Takahashi C, Kitayama H, Noda M. Hypoxia and RAS‐signaling pathways converge on, and cooperatively downregulate, the RECK tumor‐suppressor protein through microRNAs. Oncogene 2010; 29: 2638–48. [DOI] [PubMed] [Google Scholar]
- 22. Oh J, Takahashi R, Kondo S et al The membrane‐anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001; 107: 789–800. [DOI] [PubMed] [Google Scholar]
- 23. Yoshida Y, Ninomiya K, Hamada H, Noda M. Involvement of the SKP2‐p27(KIP1) pathway in suppression of cancer cell proliferation by RECK. Oncogene 2012; 31: 4128–38. [DOI] [PubMed] [Google Scholar]
- 24. Omura A, Matsuzaki T, Mio K et al RECK forms cowbell‐shaped dimers and inhibits matrix metalloproteinase‐catalyzed cleavage of fibronectin. J Biol Chem 2009; 284: 3461–9. [DOI] [PubMed] [Google Scholar]
- 25. Miki T, Takegami Y, Okawa K, Muraguchi T, Noda M, Takahashi C. The reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) interacts with membrane type 1 matrix metalloproteinase and CD13/aminopeptidase N and modulates their endocytic pathways. J Biol Chem 2007; 282: 12341–52. [DOI] [PubMed] [Google Scholar]
- 26. Muraguchi T, Takegami Y, Ohtsuka T et al RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat Neurosci 2007; 10: 838–45. [DOI] [PubMed] [Google Scholar]
- 27. Morioka Y, Monypenny J, Matsuzaki T et al The membrane‐anchored metalloproteinase regulator RECK stabilizes focal adhesions and anterior‐posterior polarity in fibroblasts. Oncogene 2009; 28: 1454–64. [DOI] [PubMed] [Google Scholar]
- 28. Yuki K, Yoshida Y, Inagaki R, Hiai H, Noda M. E‐cadherin‐downregulation and RECK‐upregulation are coupled in the non‐malignant epithelial cell line MCF10A but not in multiple carcinoma‐derived cell lines. Sci Rep 2014; 4: 4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chandana EP, Maeda Y, Ueda A et al Involvement of the Reck tumor suppressor protein in maternal and embryonic vascular remodeling in mice. BMC Dev Biol 2010; 10: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang H, Imamura Y, Ishibashi R, Chandana EP, Yamamoto M, Noda M. The Reck tumor suppressor protein alleviates tissue damage and promotes functional recovery after transient cerebral ischemia in mice. J Neurochem 2010; 115: 385–98. [DOI] [PubMed] [Google Scholar]
- 31. Almeida GM, Yamamoto M, Morioka Y, Ogawa S, Matsuzaki T, Noda M. Critical roles for murine Reck in the regulation of vascular patterning and stabilization. Sci Rep 2015; 5: 17860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parr BA, McMahon AP. Dorsalizing signal Wnt‐7a required for normal polarity of D‐V and A‐P axes of mouse limb. Nature 1995; 374: 350–3. [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto M, Matsuzaki T, Takahashi R et al The transformation suppressor gene Reck is required for postaxial patterning in mouse forelimbs. Biol Open 2012; 1: 458–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prendergast A, Linbo TH, Swarts T et al The metalloproteinase inhibitor Reck is essential for zebrafish DRG development. Development 2012; 139: 1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leigh NR, Schupp MO, Li K et al Mmp17b is essential for proper neural crest cell migration in vivo. PLoS ONE 2013; 8: e76484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vallon M, Essler M. Proteolytically processed soluble tumor endothelial marker (TEM) 5 mediates endothelial cell survival during angiogenesis by linking integrin alpha(v)beta3 to glycosaminoglycans. J Biol Chem 2006; 281: 34179–88. [DOI] [PubMed] [Google Scholar]
- 37. Takagi S, Simizu S, Osada H. RECK negatively regulates matrix metalloproteinase‐9 transcription. Cancer Res 2009; 69: 1502–8. [DOI] [PubMed] [Google Scholar]
- 38. Fonar Y, Frank D. FAK and WNT signaling: the meeting of two pathways in cancer and development. Anticancer Agents Med Chem 2011; 11: 600–6. [DOI] [PubMed] [Google Scholar]
- 39. Feng Q, Gao N. Keeping Wnt signalosome in check by vesicular traffic. J Cell Physiol 2014; 230: 1170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shaw KM, Castranova DA, Pham VN et al fused‐somites‐like mutants exhibit defects in trunk vessel patterning. Dev Dyn 2006; 235: 1753–60. [DOI] [PubMed] [Google Scholar]
- 41. Ulrich F, Carretero‐Ortega J, Menendez J et al Reck enables cerebrovascular development by promoting canonical Wnt signaling. Development 2015; 143: 147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Senger DR, Davis GE. Angiogenesis. Cold Spring Harb Perspect Biol 2011; 3: a005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parr BA, Shea MJ, Vassileva G, McMahon AP. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 1993; 119: 247–61. [DOI] [PubMed] [Google Scholar]
- 44. Qu Q, Sun G, Murai K et al Wnt7a regulates multiple steps of neurogenesis. Mol Cell Biol 2013; 33: 2551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fernando CV, Kele J, Bye CR et al Diverse roles for Wnt7a in ventral midbrain neurogenesis and dopaminergic axon morphogenesis. Stem Cells Dev 2014; 23: 1991–2003. [DOI] [PubMed] [Google Scholar]
- 46. Homma S, Shimada T, Hikake T, Yaginuma H. Expression pattern of LRR and Ig domain‐containing protein (LRRIG protein) in the early mouse embryo. Gene Expr Patterns 2009; 9: 1–26. [DOI] [PubMed] [Google Scholar]
- 47. Hari L, Brault V, Kleber M et al Lineage‐specific requirements of beta‐catenin in neural crest development. J Cell Biol 2002; 159: 867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee HY, Kleber M, Hari L et al Instructive role of Wnt/beta‐catenin in sensory fate specification in neural crest stem cells. Science 2004; 303: 1020–3. [DOI] [PubMed] [Google Scholar]
- 49. Seandel M, Falciatori I, Shmelkov SV, Kim J, James D, Rafii S. Niche players: spermatogonial progenitors marked by GPR125. Cell Cycle 2008; 7: 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pickering C, Hagglund M, Szmydynger‐Chodobska J et al The adhesion GPCR GPR125 is specifically expressed in the choroid plexus and is upregulated following brain injury. BMC Neurosci 2008; 9: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li X, Roszko I, Sepich DS et al Gpr125 modulates Dishevelled distribution and planar cell polarity signaling. Development 2013; 140: 3028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]