

Abstract
Medulloblastoma (MB) is the most common malignant pediatric brain tumor. Despite great improvements in the therapeutic regimen, relapse and leptomeningeal dissemination still pose great challenges to the long‐term survival of MB patients. Developing more effective strategies has become extremely urgent. In recent years, a number of malignancies, including MB, have been found to contain a subpopulation of cancer cells known as cancer stem cells (CSCs), or tumor initiating/propagating cells. The CSCs are thought to be largely responsible for tumor initiation, maintenance, dissemination, and relapse; therefore, their pivotal roles have revealed them to be promising targets in MB therapy. Our growing understanding of the major medulloblastoma molecular subgroups and the derivation of some of these groups from specific stem or progenitor cells adds additional layers to the CSC knowledge base. Herein we review the current knowledge of MB stem cells, highlight the molecular mechanisms relating to MB relapse and leptomeningeal dissemination, and incorporate these with the need to develop more effective and accurate therapies for MB patients.
Keywords: Cancer stem cells, medulloblastoma, medulloblastoma stem cells, stemness, therapeutic resistance
Medulloblastoma is a primitive neuroepithelial tumor originating from the hindbrain and represents one of the leading causes of pediatric tumor‐related death.1, 2 The 2007 WHO classification defines four histological variants of MB: classic, LCA, desmoplastic/nodular, and MB with extensive nodularity.3 In 2010, several large‐scale transcriptional profiling studies led to a consensus that MB contains four distinct major subgroups at the molecular level: Wnt, Shh, Group 3, and Group 4.4 Each differs with respect to demographics, histological phenotypes, somatic mutations, and clinical outcomes.4 Current clinical practice stratifies a high risk group as follows: infants <3 years old, patients with a post‐surgical residual tumor >1.5 cm2, LMD at diagnosis, or LCA histology.5, 6 Indeed, relapsed disease and LMD are still major causes of poor outcomes.6 With gradually increasing understanding of MB at the pathological and molecular levels, along with the continuous refinements in clinical risk stratification, therapeutic regimens have immensely improved, and the overall survival rate of MB patients has increased from 20% to approximately 80% in the last 35 years.6
The molecular classification has provided key guidance in the development of new targeted therapies. For example, vismodegib (GDC‐0449), an Shh pathway inhibitor targeting Smo, triggers the rapid regression of an MB patient's metastatic tumors, but it is soon followed by relapse due to mutations in Smo causing therapeutic resistance.7, 8 Molecular classification can also guide the reduced treatment of medulloblastoma patients with less aggressive disease. While aggressive treatment strategies usually result in improved survival, it is unfortunately at the cost of severely affecting the quality of life of MB survivors. The devastating side‐effects include cognitive deficits, endocrine disorders, and increased incidence of secondary cancers later in life.2
Recently, the intratumor heterogeneity revealed in malignancies such as breast cancer9 and glioblastoma10 has offered new insights into MB relapse. The spatial and temporal patterns of genetic, phenotypic, and functional diversity enable cancer cells to respond differently to targeted agents, with less sensitive or even insensitive populations surviving and leading to tumor relapse.11 Among multiple theories proposed to interpret the intratumor heterogeneity,12 the CSC model established by studies of malignancies such as leukemia,13 breast cancer,14 and brain tumors15 is of intense interest. Cancer stem cells show unlimited self‐renewal and differentiation capacity, leading to phenotypically and functionally heterogeneous progenies and fueling tumor initiation, maintenance, and progression.12 Additional layers of heterogeneity may come from clonal evolution and environmental differences within tumors, giving rise to distinct subpopulations (Fig. 1).16
Figure 1.

Role of medulloblastoma stem cells (MBSCs) in medulloblastoma (MB) heterogeneity and relapse. MBSCs possess the ability to self‐renew while differentiating into phenotypical and functional heterogeneous progenies. The possible existence of MBSC subpopulations further contributes to MB heterogeneity. When treated with chemoradiotherapy, insensitive MB cells will survive and lead to MB relapse. Meanwhile, the transition between epithelial and mesenchymal properties of MB cells through the epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition (MET) programs also cause MB relapse by endowing MB cells with stemness and multidrug resistance features.
Cancer stem cells survive chemoradiotherapy and further contribute to treatment failure through multiple drug resistance mechanisms (i.e., the activation of pro‐survival/anti‐apoptosis pathways).17 Cancer stem cells have also been found to be tightly associated with tumor dissemination through molecular networks such as EMT, which increases the invasion and motility of cancer cells, and confers them a stem‐like property essential for distant colonization.18 Elucidating the molecular mechanisms of MBSCs will definitely contribute to the discovery of more efficient agents that will notably improve the survival rate and quality of life of MB patients.
Medulloblastoma Cell‐of‐Origin versus MBSCs
The CSCs required for long‐term neoplastic growth and xenograft initiation are sometimes confused with the “cell‐of‐origin” from within specific tumors. The term “CSC” refers to stem‐like tumor cells containing multiple mutations, whereas the “cell‐of‐origin” refers to a normal cell that undergoes an initial transforming event.19
The cells of origin for human MBs are difficult to identify, as the earliest steps of the neoplastic process occur in a “black box.” However, the establishment of a series of GEM models of MB provided important insights into the cellular origins of these tumors (Fig. 2).20 Granule neuron progenitors in the external germinal layer on the surface of the developing cerebellum have long been proposed as cells of origin for MB, and it is now clear that Shh‐driven MB arises from this group.21, 22 In contrast, the Wnt group is now thought to originate from progenitors in the dorsal brainstem.23 Group 3, which is the most aggressive subgroup of MB and is characterized by high levels of MYC amplification or overexpression, appears to derive from cerebellar stem cells.24, 25 The origins of Group 4, the most prevalent MB subgroup, remain unknown.26, 27
Figure 2.

Cells of origin of medulloblastomas (MBs) and a summary of the related genetically engineered mouse (GEM) models. The top panels are adapted from the review by Rusert et al.2 EGL, external granular layer; GNP, granular neuroprogenitor; RL, rhombic lip; SHH, Sonic Hedgehog; VZ, ventricular zone; WNT, Wingless.
Sorting and Identifying MBSCs
The last decade has produced great advances in sorting and identifying MBSCs using markers, such as CD133 and CD15, and varying methods, such as flow cytometry, xenograft models, and lineage tracing.
In 2003, a CD133+/Nestin+ cell from human MB tissue was first reported, which shows a marked capacity for proliferation, self‐renewal, and differentiation in vitro.15 Further exploration showed that, in an immunodeficient mouse, 100 CD133+ cells were sufficient to produce a tumor resembling the original human tumor, whereas 100 000 CD133− cells could not,28 indicating that CD133 serves as a marker of MBSCs. However, these findings were called into question by later reports showing that tumor‐initiating properties were also possessed by CD133− MB cells.29, 30 These discrepancies suggest more efforts need to be made to effectively single out MBSCs.
The GEM models established based on the molecular classification of MB facilitated the identification of MBSCs. In 2009, a tumor‐propagating CD15+ cell was isolated from a Patched haploinsufficient (Ptch+/−) MB model.29 These cells were suggested to represent progenitor‐like cells as they did not display multilineage differentiation or form neurospheres in stem‐cell growth conditions.29 However, this was challenged by a study showing that CD15+ MB cells from Ptch+/− mice can be propagated long term in similar conditions while showing stem‐like and tumor‐initiating capacity.31 Recently, quiescent Sox2+ MB cells from post‐irradiated Ptch+/− mice were found to be tumorigenic.32 Interestingly, more than 80% of Sox2+ cells were also CD15+, while Sox2+ cells formed a minority (<10%) of the CD15+ population; these results indicated that MB relapse may actually be propagated by a subset of CD15+ cells that are also Sox2+.32
Medulloblastoma stem cells identified in the more aggressive Group 3 MB have rarely been reported. Recently, tumor‐derived neurosphere cell lines from a MYCN‐driven Group 3 MB were established.33 These highly tumorigenic cells displayed features of partially committed neural stem and progenitor cells (i.e., upregulation of CD133, Nestin, and Musashi.33 As MB patients harboring stem‐like tumor cells that display stemness signatures are often characterized by poor prognoses,15, 28, 34 more research needs to be focused on identifying MBSCs in the more aggressive MB subtypes, Group 3 and Group 4.
Additionally, MBSCs can also be sorted out by markers such as CD27135 and ABCG2 (marker of the SPs),36 each representing a subpopulation efficient in tumor initiation or propagation.
Regulation of MBSC Key Properties on MB Relapse and LMD
Accumulating research on MBSCs indicates that the key properties related to MB relapse and LMD are stemness, therapeutic resistance, invasion, and motility. The stemness property is extremely vital, as it allows MBSCs to reproduce tumors in both post‐surgical residual disease and LMD.
Critical regulator candidates of MBSC stemness
Shh signaling pathway
The Shh pathway is the major mitogenic regulator that promotes granule neuron progenitor proliferation.37 Excessive activation of Shh pathways had been reported to cause MB tumorigenesis.37
Unlike the CSCs in human leukemia, which are more quiescent than the “blasts” that make up the majority of the tumor, brain CSCs seem to show higher proliferation than do non‐CSCs from the same tumor.15, 28 Consistent with this, when the aforementioned CD15+ MBSCs were compared with those of CD15− cells in Ptch+/− MB, CD15+ cells expressed increased levels of Shh target genes Gli1 and CyclinD1, and showed higher incorporation levels of thymidine in vitro and BrdU in vivo.29, 32 This evidence suggests that the increased proliferative capacity of CD15+ MBSCs is closely related with increased Shh pathway activation.
The transcription factors Gli1/2 the main effectors of the Shh pathway, have been reported to interact with stem‐related factors, such as Nanog,38 MYCN,39 and Bmi‐1, in the self‐renewal regulation of MBSCs. The reprogramming factor Nanog was found highly expressed in stem cells from postnatal cerebellum and MB, and its specific cis‐regulatory sequences were direct targets of Gli1/2.38 When the Shh pathway was blocked by Smo‐antagonist KAAD‐cyclopamine, the expression of Nanog was reduced, and the self‐renewal of MB neurospheres was also significantly inhibited38 (Fig. 3).
Figure 3.
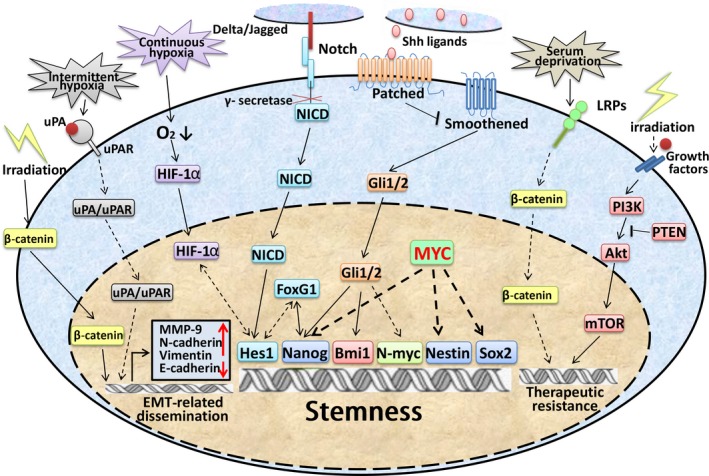
Regulation mechanisms of medulloblastoma stem cell key properties in relation to medulloblastoma relapse and leptomeningeal dissemination. Akt, protein kinase B; EMT, epithelial–mesenchymal transition; FoxG1, Forkhead box 1; HIF‐1α, hypoxia‐inducible factor 1α; LRP, low‐density lipoprotein receptor‐related protein; mTOR, mammalian target of rapamycin; MYC, v‐Myc avian myelocytomatosis viral oncogene homolog; NCID, Notch1 intracellular domain; PI3K, phosphatidylinositol 3‐kinase; PTEN, phosphatase and tensin homolog; Shh, Sonic Hedgehog; uPA, urokinase‐type plasminogen activator; uPAR, uPA receptor.
A member of the polycomb protein family, Bmi‐1, is a key regulator of stemness.40 A direct feedback mechanism between downregulators of bmi‐1 and Shh signaling was found in CD133+ MBSCs, indicating that bmi‐1 and Shh are mutually indispensable in MBSC maintenance.41 In addition, bmi‐1 was also found to promote the self‐renewal and tumorigenicity of CD15+ MBSCs in vivo through reciprocal promoter occupancy with FoxG1.42 Together, these findings suggest that the Shh pathway plays a key role in regulating the self‐renewal and maintenance of MBSCs (Fig. 3).
MYC family
Group 3 MB is characterized by elevated expression of MYC and LCA histopathology. Medulloblastoma patients with MYC amplification have a particularly high risk of relapse and the poorest prognosis of all MB patients.2
Early in 2006, when co‐overexpressed with a neuronal differentiation repressor gene REST/NRSF, MYC was shown to block the differentiation of NSCs derived from the external germinal layer and cause MB formation.43 Recently, JQ1, a small molecule inhibitor uniquely targets BRD4 (a key mediator of MYC‐driven transcription programs), could strongly repress the expression of critical stem‐related factors Sox2, Nestin, and Nanog in MB cells.44 Moreover, JQ1 strongly suppressed the ability of these cells to form neurospheres in vitro and tumors in vivo.44 These results indicate that MYC is tightly associated with MBSC stemness.
Another MYC family member, MYCN, was shown to be aberrantly amplified across MB subtypes.45 Notably, Ahmad et al. recently established a MYCN‐driven MB displaying the large‐cell histopathology and expression profiles resembling Group 3 MB. Furthermore, CD133+ neurosphere cell lines were established, the self‐renewal and growth of which were highly dependent on MYCN.33 Specifically, the neurospheres showed restricted growth and reduced Ki‐67, Nestin, and CD133 after 48 h of MYCN withdrawal,33 indicating that MYCN is critical in the development and maintenance of aggressive MBSCs.
Notch signaling pathway
The Notch pathway is known for its regulation of stem cells in both normal and cancerous brain tissue. Hes1, the main effector of the Notch pathway, was found upregulated exponentially in CD133+ MB cells. When it was downregulated by Notch pathway γ‐secretase inhibitors, the CD133+ MB cells and SP were largely reduced,46 indicating that the Notch pathway plays an important role in MBSC self‐renewal. Furthermore, Notch signaling was shown to interact with hypoxia‐inducible factor‐1α to maintain NSCs and MBSCs in an undifferentiated status.47 Particularly, stimulating Notch1 with Dll4 ligand under hypoxic conditions drives the expansion of MB‐derived CD133+/Nestin+ precursors, while treating with γ‐secretase inhibitors promoted the neuronal differentiation of these cells (Fig. 4).47
Figure 4.

Relationship between medulloblastoma (MB) leptomeningeal dissemination, MB stem cells (MBSCs), and epithelial–mesenchymal transition. The epithelial–mesenchymal transition program allows the epithelial MB cells to acquire a mesenchymal phenotype with increased motility and invasiveness, facilitating the initiation of MB dissemination. Inversely, the mesenchymal–epithelial transition (MET) program enables mesenchymal MB cells to settle and self‐renew, leading to medulloblastoma leptomeningeal dissemination and relapse. CSF, cerebrospinal fluid.
MicroRNA
MicroRNA, an endogenous, 19–25‐nt long, non‐coding RNA, has been broadly shown to be involved in MB tumorigenesis and progression,48 and its roles in MBSCs are becoming gradually illuminated. For example, miR‐199b‐5p was found to regulate the self‐renewal of CD15+ and CD133+ MBSCs by interacting with Hes1 through reciprocal binding on the promoter regions.49, 50 The famous stem‐related miR‐34a also regulates MBSC maintenance by inhibiting the PI3K/Akt/PTEN pathway.51 When miR‐34a was overexpressed, the proportion of active Akt decreased, and the subpopulations of CD15+ and CD133+ cells were significantly reduced.51 Moreover, the tumorigenic properties of MBSCs can also be strongly inhibited by miR‐135a.52 Together, these data suggest that microRNAs may serve as powerful tools in eradicating MBSCs.
Therapeutic resistance regulation mechanisms of MBSCs
The resistance characteristics possessed by CSCs, such as quiescence, activation of pro‐survival/anti‐apoptosis pathways, and interaction with microenvironmental factors (i.e., hypoxia),17 constitute an additional level of mechanisms that lead to treatment failure.
Targeting the Shh pathway seemed to be an effective way to eradicate MBSCs. However, the results were disappointing. Although vismodegib was found to be effective in eliminating most proliferating MB cells, it also significantly increased the frequency of quiescent Sox2+ MB cells in Ptch+/− MB mouse models, strongly indicating that the quiescence feature can serve as an efficacious defense mechanism for MBSCs in resisting chemoradiotherapy.32
Another example is the activation of the PI3K/Akt signaling pathway, which promotes tumor cell survival by regulating multiple apoptosis‐related proteins (i.e., the Bcl‐2 family).53 Hambardzumyan et al. discovered a Nestin+ MBSC in the perivascular niche in different post‐irradiated Shh‐MB mouse models that could re‐enter the cell cycle (72 h after radiation) through radiation‐induced PTEN loss and the subsequent activation of the PI3K/Akt pathway.54 When pretreated with the PI3K/Akt inhibitor perifosine, Nestin+ MBSCs displayed increased sensitivity to radiation‐induced apoptosis,54 indicating that activation of the PI3K/Akt pathway helps Nestin+ MBSCs to survive radiotherapy.
The Wnt pathway may also play a role in the therapeutic resistance of MBSCs. The LRPs are membrane co‐receptors of the Wnt/β‐catenin pathways55 and have been shown to mediate the resistance phenotypes of many malignancies.56 Annabi et al.56 found that DAOY CD133+ MB cells mainly express LRP5/8, whereas under nutrient derivation, these cells acquire an increase of CD133+ cells expressing LRP1/1b/5, indicating that the LRP expression pattern mediates MBSC adaptation to nutrient derivation.
Leptomeningeal dissemination of MB, EMT, and MBSCs
Leptomeningeal dissemination, the metastatic spread of tumor cells through the cerebrospinal fluid to the brain and spinal cord, is a defining characteristic of MB and is associated with short survival time for MB patients.57 Recently, the characteristics shared by CSCs and cancer cells in the EMT in many malignancies provided new insights into the elusive mechanisms of LMD.58 The EMT program is a process characterized by the repression of epithelial markers (i.e., E‐cadherin), the acquisition of mesenchymal makers (i.e., vimentin, N‐cadherin) and the loss of cell adhesion. Together with its reverse process, mesenchymal–epithelial transition, the EMT has been found to play vital roles in tumor metastasis (Fig. 4).59
The nuclear translocation of β‐catenin (Wnt pathway key effector) was found to be induced by irradiation therapy in MB cells.60 Interestingly, in the nucleus, β‐catenin was then found to activate the EMT effector MMPs, such as MMP‐9, and to promote MB invasion and migration.60 The uPA/uPAR complex, which is a major regulator of ECM degradation,61 has been found to be activated by intermittent hypoxia and can induce the activation of the EMT programs of MB cells.61 It has been reported that uPAR+ cells in small‐cell lung cancer are multidrug resistant and highly clonogenic and co‐express CSC markers CD44 and multidrug resistance protein 1 (MDR1).62 Thus, uPA/uPAR may also be responsible for MB cell acquisition of therapeutic resistance and stem‐like properties.
In spite of these findings, it is critical to define the role of MYC or MYCN in the more malignant MB subtypes, Group 3 and Group 4, the metastasis rates of which are up to 45% and 40%, respectively.2
Approaches for Eradicating MBSCs
Cancer stem cells can be eliminated through a plethora of methods, such as blocking pro‐survival/anti‐apoptosis pathways, targeting the mechanisms maintaining quiescence, or disrupting CSC interaction with the microenvironment.63, 64 The molecular classification of MB has undoubtedly helped develop therapeutic strategies for targeting unique signaling pathways, specific oncogenes, or tumor suppressor genes aberrantly regulated in specific MB subtypes. However, many of these targeted therapies poorly eliminate MBSCs.32, 33
To date, most studies aiming to eradicate MBSCs have focused on targeting specific signaling pathways. The preceding sections have discussed that when the Notch pathway was blocked by γ‐secretase inhibitors, miR‐199b‐5p, or exposure to 20% oxygen concentration, the fractions of stem‐like CD133+, CD15+ MB cells and SPs were significantly reduced. Inhibiting the PI3K/Akt pathway also seems to be effective in removing MBSCs in some contexts. Apart from the aforementioned perifosine, GDC‐0941, which is another highly specific PI3K inhibitor, could significantly reduce the stem‐like CD133+ MB subpopulations as well as their clonogenicity.65 More importantly, it strongly delayed tumor growth of a mouse that orthotopically received the most aggressive Group 3 MB xenograft.65
In addition, the STAT3 pathway was found to be enhanced in CD133+ MB cells, and the STAT3 inhibitors, cucurbitacin I or celecoxib, can strongly inhibit the stem‐like properties of CD133+ MBSCs, as well as improve the chemoradiosensitivity of, and survival time, with CD133+ MBSCs xenografts.66, 67 Taken together, these data showed that developing agents targeting these pathways is promising for the elimination of MBSCs.
Conclusion and Perspectives
Given the pivotal role of stem‐like tumor cells in MB relapse and LMD, improved treatment strategies combining traditional and MBSC‐targeted therapies may be required to significantly improve the survival time and quality of life of MB patients. However, despite great efforts and multiple encouraging discoveries, the current knowledge of MBSCs is still not sufficient and the optimal clinical application of MBSC‐targeted agents awaits future groundbreaking work. Goals to achieve include: (i) finding more reliable MBSC markers and gaining a comprehensive knowledge of the discrepancies between MBSCs and NSCs/neural progenitor cells to develop agents that uniquely target MBSCs; (ii) investigating MBSC subclones and the essential molecular mechanisms they share, or with which they interact; and (iii) understanding further CSC plasticity and the possibility that stemness can reflect a transient state rather than a metastable state, although the flux of non‐CSCs into the CSC compartment appears relatively low.68
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- Akt
protein kinase B
- CSC
cancer stem cell
- EMT
epithelial–mesenchymal transition
- GEM
genetically engineered mouse
- LCA
large cell/anaplastic
- LMD
leptomeningeal dissemination
- LRP
low‐density lipoprotein receptor‐related protein
- MB
medulloblastoma
- MBSC
medulloblastoma stem cell
- miR
microRNA
- MYC
v‐Myc avian myelocytomatosis viral oncogene homolog
- MYCN
v‐Myc avian myelocytomatosis viral oncogene neuroblastoma‐derived homolog
- NSC
neural stem cell
- PI3K
phosphatidylinositol 3‐kinase
- PTEN
phosphatase and tensin homolog
- Shh
Sonic Hedgehog
- Smo
Smoothen
- SP
side population
- STAT3
signal transducer and activator of transcription 3
- uPA
urokinase‐type plasminogen activator
- uPAR
urokinase‐type plasminogen activator receptor
- Wnt
Wingless
Acknowledgments
We thank Professor Charles G. Eberhart from The Johns Hopkins University School of Medicine (Baltimore, MD, USA) for useful suggestions and carefully commenting on this review. This work was supported by the National Natural Science Foundation of China (Grant Nos. NSFC81272783 and NSFC30973075) and a grant from Xinqiao Hospital of the Third Military Medical University (Chongqing, China) (Grant No. 2014YCL25).
Cancer Sci 107 (2016) 583–589
Funding Information
National Natural Science Foundation of China; Xinqiao Hospital, Third Military Medical University, China.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 2. Rusert JM, Wu X, Eberhart CG, Taylor MD, Wechsler‐Reya RJ. SnapShot: Medulloblastoma. Cancer Cell 2014; 26: 940.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Ohgaki H, Wiestler OD et al The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor MD, Northcott PA, Korshunov A et al Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012; 123: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Northcott PA, Jones DT, Kool M et al Medulloblastomics: the end of the beginning. Nat Rev Cancer 2012; 12: 818–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gottardo NG, Hansford JR, McGlade JP et al Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol 2014; 127: 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yauch RL, Dijkgraaf GJ, Alicke B et al Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009; 326: 572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rudin CM, Hann CL, Laterra J et al Treatment of medulloblastoma with hedgehog pathway inhibitor GDC‐0449. N Engl J Med 2009; 361: 1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Russnes HG, Navin N, Hicks J, Borresen‐Dale AL. Insight into the heterogeneity of breast cancer through next‐generation sequencing. J Clin Invest 2011; 121: 3810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sottoriva A, Spiteri I, Piccirillo SG et al Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 2013; 110: 4009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Renovanz M, Kim EL. Intratumoral heterogeneity, its contribution to therapy resistance and methodological caveats to assessment. Front Oncol 2014; 4: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 2012; 21: 283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–7. [DOI] [PubMed] [Google Scholar]
- 14. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singh SK, Clarke ID, Terasaki M et al Identification of a cancer stem cell in human brain tumors. Cancer Res 2003; 63: 5821–8. [PubMed] [Google Scholar]
- 16. Wang A, Chen L, Li C, Zhu Y. Heterogeneity in cancer stem cells. Cancer Lett 2015; 357: 63–8. [DOI] [PubMed] [Google Scholar]
- 17. Cojoc M, Mabert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol 2015; 31: 16–27. [DOI] [PubMed] [Google Scholar]
- 18. Mani SA, Guo W, Liao MJ et al The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol 2005; 15: 494–501. [DOI] [PubMed] [Google Scholar]
- 20. Swartling FJ, Bolin S, Phillips JJ, Persson AI. Signals that regulate the oncogenic fate of neural stem cells and progenitors. Exp Neurol 2014; 260: 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schuller U, Heine VM, Mao J et al Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh‐induced medulloblastoma. Cancer Cell 2008; 14: 123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang ZJ, Ellis T, Markant SL et al Medulloblastoma can be initiated by deletion of Patched in lineage‐restricted progenitors or stem cells. Cancer Cell 2008; 14: 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gibson P, Tong Y, Robinson G et al Subtypes of medulloblastoma have distinct developmental origins. Nature 2010; 468: 1095–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawauchi D, Robinson G, Uziel T et al A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 2012; 21: 168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pei Y, Moore CE, Wang J et al An animal model of MYC‐driven medulloblastoma. Cancer Cell 2012; 21: 155–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swartling FJ, Grimmer MR, Hackett CS et al Pleiotropic role for MYCN in medulloblastoma. Genes Dev 2010; 24: 1059–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swartling FJ, Savov V, Persson AI et al Distinct neural stem cell populations give rise to disparate brain tumors in response to N‐MYC. Cancer Cell 2012; 21: 601–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh SK, Hawkins C, Clarke ID et al Identification of human brain tumour initiating cells. Nature 2004; 432: 396–401. [DOI] [PubMed] [Google Scholar]
- 29. Read TA, Fogarty MP, Markant SL et al Identification of CD15 as a marker for tumor‐propagating cells in a mouse model of medulloblastoma. Cancer Cell 2009; 15: 135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Srivastava VK, Nalbantoglu J. Flow cytometric characterization of the DAOY medulloblastoma cell line for the cancer stem‐like phenotype. Cytometry A 2008; 73: 940–8. [DOI] [PubMed] [Google Scholar]
- 31. Ward RJ, Lee L, Graham K et al Multipotent CD15 + cancer stem cells in patched‐1‐deficient mouse medulloblastoma. Cancer Res 2009; 69: 4682–90. [DOI] [PubMed] [Google Scholar]
- 32. Vanner RJ, Remke M, Gallo M et al Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014; 26: 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahmad Z, Jasnos L, Gil V et al Molecular and in vivo characterization of cancer‐propagating cells derived from MYCN‐dependent medulloblastoma. PLoS ONE 2015; 10: e0119834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hemmati HD, Nakano I, Lazareff JA et al Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA 2003; 100: 15178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morrison LC, McClelland R, Aiken C et al Deconstruction of medulloblastoma cellular heterogeneity reveals differences between the most highly invasive and self‐renewing phenotypes. Neoplasia 2013; 15: 384–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu J, Chi N, Zhang JY, Zhu W, Bian YS, Chen HG. Isolation and characterization of cancer stem cells from medulloblastoma. Genet Mol Res 2015; 14: 3355–61. [DOI] [PubMed] [Google Scholar]
- 37. Hatten ME, Roussel MF. Development and cancer of the cerebellum. Trends Neurosci 2011; 34: 134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Po A, Ferretti E, Miele E et al Hedgehog controls neural stem cells through p53‐independent regulation of Nanog. EMBO J 2010; 29: 2646–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marino S. Medulloblastoma: developmental mechanisms out of control. Trends Mol Med 2005; 11: 17–22. [DOI] [PubMed] [Google Scholar]
- 40. Leung C, Lingbeek M, Shakhova O et al Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004; 428: 337–41. [DOI] [PubMed] [Google Scholar]
- 41. Wang X, Venugopal C, Manoranjan B et al Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor‐initiating cells. Oncogene 2012; 31: 187–99. [DOI] [PubMed] [Google Scholar]
- 42. Manoranjan B, Wang X, Hallett RM et al FoxG1 interacts with Bmi1 to regulate self‐renewal and tumorigenicity of medulloblastoma stem cells. Stem Cells 2013; 31: 1266–77. [DOI] [PubMed] [Google Scholar]
- 43. Su X, Gopalakrishnan V, Stearns D et al Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol 2006; 26: 1666–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Venkataraman S, Alimova I, Balakrishnan I et al Inhibition of BRD4 attenuates tumor cell self‐renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 2014; 5: 2355–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roussel MF, Robinson GW. Role of MYC in Medulloblastoma. Cold Spring Harb Perspect Med 2013; 3: a014308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fan X, Matsui W, Khaki L et al Notch pathway inhibition depletes stem‐like cells and blocks engraftment in embryonal brain tumors. Cancer Res 2006; 66: 7445–52. [DOI] [PubMed] [Google Scholar]
- 47. Pistollato F, Rampazzo E, Persano L et al Interaction of hypoxia‐inducible factor‐1alpha and Notch signaling regulates medulloblastoma precursor proliferation and fate. Stem Cells 2010; 28: 1918–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhi F, Wang SN, Wang R, Xia XW, Yang YL. From small to big: microRNAs as new players in medulloblastomas. Tumor Biol 2013; 34: 9–15. [DOI] [PubMed] [Google Scholar]
- 49. Garzia L, Andolfo I, Cusanelli E et al MicroRNA‐199b‐5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLoS ONE 2009; 4: e4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Andolfo I, Liguori L, De Antonellis P et al The micro‐RNA 199b‐5p regulatory circuit involves Hes1, CD15, and epigenetic modifications in medulloblastoma. Neuro Oncol 2012; 14: 596–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Antonellis P, Medaglia C, Cusanelli E et al MiR‐34a targeting of Notch ligand delta‐like 1 impairs CD15+/CD133+ tumor‐propagating cells and supports neural differentiation in medulloblastoma. PLoS ONE 2011; 6: e24584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hemmesi K, Squadrito ML, Mestdagh P et al miR‐135a inhibits cancer stem cell‐driven medulloblastoma development by directly repressing Arhgef6 expression. Stem Cells 2015; 33: 1377–89. [DOI] [PubMed] [Google Scholar]
- 53. Hambardzumyan D, Becher OJ, Holland EC. Cancer stem cells and survival pathways. Cell Cycle 2008; 7: 1371–8. [DOI] [PubMed] [Google Scholar]
- 54. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova‐Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008; 22: 436–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. MacDonald BT, Tamai K, He X. Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Annabi B, Doumit J, Plouffe K, Laflamme C, Lord‐Dufour S, Beliveau R. Members of the low‐density lipoprotein receptor‐related proteins provide a differential molecular signature between parental and CD133 + DAOY medulloblastoma cells. Mol Carcinog 2010; 49: 710–7. [DOI] [PubMed] [Google Scholar]
- 57. Faria CC, Golbourn BJ, Dubuc AM et al Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res 2015; 75: 134–46. [DOI] [PubMed] [Google Scholar]
- 58. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug‐resistance. Oncotarget 2015; 6: 10697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun Y, Ma L. The emerging molecular machinery and therapeutic targets of metastasis. Trends Pharmacol Sci 2015; 36: 349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Asuthkar S, Nalla AK, Gondi CS et al Gadd45a sensitizes medulloblastoma cells to irradiation and suppresses MMP‐9‐mediated EMT. Neuro Oncol 2011; 13: 1059–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gupta R, Chetty C, Bhoopathi P et al Downregulation of uPA/uPAR inhibits intermittent hypoxia‐induced epithelial‐mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int J Oncol 2011; 38: 733–44. [DOI] [PubMed] [Google Scholar]
- 62. Gutova M, Najbauer J, Gevorgyan A et al Identification of uPAR‐positive chemoresistant cells in small cell lung cancer. PLoS ONE 2007; 2: e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016; 107: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Islam F, Gopalan V, Smith RA, Lam AK. Translational potential of cancer stem cells: a review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp Cell Res 2015; 335: 135–47. [DOI] [PubMed] [Google Scholar]
- 65. Ehrhardt M, Craveiro RB, Holst MI, Pietsch T, Dilloo D. The PI3K inhibitor GDC‐0941 displays promising in vitro and in vivo efficacy for targeted medulloblastoma therapy. Oncotarget 2015; 6: 802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chang CJ, Chiang CH, Song WS et al Inhibition of phosphorylated STAT3 by cucurbitacin I enhances chemoradiosensitivity in medulloblastoma‐derived cancer stem cells. Childs Nerv Syst 2012; 28: 363–73. [DOI] [PubMed] [Google Scholar]
- 67. Yang MY, Lee HT, Chen CM, Shen CC, Ma HI. Celecoxib suppresses the phosphorylation of STAT3 protein and can enhance the radiosensitivity of medulloblastoma‐derived cancer stem‐like cells. Int J Mol Sci 2014; 15: 11013–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009; 15: 1010–2. [DOI] [PubMed] [Google Scholar]