

Abstract
Vasohibin‐1 (VASH1) is a negative feedback regulator of angiogenesis, the first to be discovered, and was identified in vascular endothelial growth factor (VEGF)‐stimulated vascular endothelial cells. Vasohibin‐1 inhibits abnormal vascularization induced by various angiogenic factors including fibroblast growth factor and platelet‐derived growth factor (PDGF), in addition to VEGF. By focusing on this characteristic of VASH1, we investigated the antitumor effects of VASH1 expression on ovarian cancer cells that produce different angiogenic factors. By using a high VEGF‐producing ovarian cancer cell line, SHIN‐3, and a high PDGF‐producing ovarian cancer cell line, KOC‐2S, the cells were transfected with either a VEGF antagonist, soluble VEGF receptor‐1 (sVEGFR‐1, or sFlt‐1), or VASH1 genes to establish their respective cellular expression. The characteristics of these transfectants were compared with controls. We previously reported that the expression of sFlt‐1 inhibited tumor vascularization and growth of high VEGF‐producing ovarian cancer cells, reduced peritoneal dissemination and ascites development, and prolonged the survival time of the host. However, in the current study, the expression of sFlt‐1 had no such effect on the high PDGF‐producing ovarian cancer cells used here, whereas VASH1 expression inhibited tumor vascularization and growth, not only in high VEGF‐producing cells, but also in high PDGF‐producing cells, reduced their peritoneal dissemination and ascites, and prolonged the survival time of the host. These results suggest that VASH1 is an effective treatment for ovarian cancer cells that produce different angiogenic factors.
Keywords: Angiogenesis, ovarian cancer, PDGF, sFlt‐1, vasohibin‐1
Ovarian cancer is the second most prevalent type of malignant tumor diagnosed in the field of gynecology, and is the fifth most frequent cause of cancer death in women.1 Challenges regarding ovarian cancer are mainly associated with early diagnosis of the disease, when it is asymptomatic. Originating from the ovaries, therefore being present in the abdominal cavity, provides quick access for the cancer to spread to major organs. Thus, in over 50% of ovarian cancer patients, peritoneal dissemination and ascites develop by the time a diagnosis is made.2 A combination of debulking surgery and multidrug chemotherapy is standard therapy for treating advanced ovarian cancer. Ovarian cancer is generally considered relatively sensitive to chemotherapy, and many patients achieve cancer remission through a standard therapy described above.3, 4 However, effects of the treatment are only transient, and the cancer often recurs, leading to death in more than half of the instances where remission occurs. Because improving the outcome for ovarian cancer patients with currently available treatment methods is clearly limited, the development of new treatments is imperative.
Vascularization is essential for tumor growth,5 having a strong involvement in ovarian cancer advancement. In particular, vascularization plays a role in the cancer's characteristic proliferation pattern of peritoneal dissemination, which is why it may be targeted by ovarian cancer treatments. We previously reported that the expression of a cytokine with a vascularization‐inhibitory action, interleukin‐10, or a vascular endothelial growth factor (VEGF) antagonist, soluble VEGF receptor‐1 (sVEGFR‐1, or sFlt‐1), suppresses peritoneal dissemination and ascites development, and prolongs survival times in an animal ovarian cancer model.6, 7 The application of an anti‐VEGF mAb, bevacizumab, in ovarian cancer therapy has recently been introduced, and beneficial effects are expected. In parallel, a resistance phenomenon observed in tumors following anti‐VEGF therapy, termed evasive resistance, has been attracting considerable attention.8 The acquisition mechanism of this resistance is thought to involve the induction of angiogenic factors other than VEGF, which indicates that a treatment targeting only a single angiogenic factor would be ineffective as it neglects the range of angiogenic factors involved in tumor vascularization. Therefore, it may be necessary to simultaneously target many angiogenic factors for a substantial therapeutic effect.
Vasohibin‐1 (VASH1), the first negative feedback modulator to be discovered, was identified in VEGF‐stimulated vascular endothelial cells, regulating vascularization by acting on vascular endothelial cells.9, 10 Recently, we showed that VASH1 inhibits ovarian cancer growth and peritoneal dissemination and prolongs host survival by inhibiting angiogenesis in a mouse model of the ovarian cancer cell line SKOV‐3,11 which has previously been reported to produce VEGF.12 Vasohibin‐1 has also been shown to inhibit abnormal vascularization induced by various angiogenic factors, such as fibroblast growth factor (FGF) and platelet‐derived growth factor (PDGF), in addition to VEGF.9, 13 Because this suggests that VASH1 has a diverse target range and that it independently inhibits various angiogenic factors, VASH1 may be superior to VEGF inhibitors in cancer treatment because of its array of treatment targets and the ability to overcome evasive resistance.
In the present study, we investigated the antitumor effects of VASH1 expression in ovarian cancer cells producing different angiogenic factors.
Materials and Methods
Cell culture
Human ovarian serous adenocarcinoma cell lines, SHIN‐314 and KOC‐2S,15 were used in this study as they produce high amounts of VEGF and PDGF, respectively.6, 16 These cell lines were cultured in DMEM/F12 (Life Technologies, Carlsbad, CA, USA), supplemented with 10% FCS (Sigma‐Aldrich, St. Louis, MO, USA) and 1% penicillin/streptomycin (Life Technologies). Human umbilical vein endothelial cells were purchased from Kurabo Industries, Ltd. (Osaka, Japan), and cultured in EBM‐2 medium (Lonza, Walkersville, MD, USA) containing EGM‐2‐MV‐SingleQuots (Lonza) and comprising VEGF, FGF, insulin‐like growth factor‐1, epidermal growth factor, and 2% FCS. All cells were cultured at 37°C in 5% carbon dioxide.
Establishment of sFlt‐1‐ and VASH1‐expressing cell lines
The sFlt‐1 expression vector (pCMV‐sFlt‐1‐IRES‐bsr) was used as described previously.7 A VASH1 expression vector was prepared by inserting human VASH1 cDNA (NCBI database; Gene ID 22846) into the cloning site (Sma‐I, Xba‐I) of pCMV‐IRES‐bsr.17 SHIN‐3 and KOC‐2S were transfected with the sFlt‐1 or VASH1 expression vector, and a luciferase expression plasmid vector (pCMV‐LUC‐IRES‐bsr17) as a control, using Lipofectamine LTX and Plus Reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. The cells were then selectively cultured in a medium containing blasticidin S hydrochloride (Funakoshi Co., Ltd., Tokyo, Japan), and resistant cells were acquired.
Three‐dimensional culture
Tumor cells (1 × 104 cells/well) were seeded on a six‐well plate and cultured in DMEM/F12 with 1% low melting temperature agarose (FMC Bioproducts, Rockland, ME, USA) supplemented with 10% FCS. After 7 days, the diameter of the acini was measured under a light microscope.
Concentrations of VEGF and PDGF in culture supernatants
Tumor cells (1 × 106 cells/well) were seeded on a six‐well plate and cultured in EBM‐2 medium containing no growth factor, and the culture supernatant was collected after 24 h. The VEGF and PDGF concentrations of the culture supernatants were measured with a Quantikine Human VEGF ELISA kit (R&D Systems, Minneapolis, MN, USA) and a Quantikine PDGF‐AA ELISA kit (R&D Systems), following the manufacturer's instructions.
In vitro endothelial cell growth
After seeding the HUVECs in a 96‐well plate (2 × 103 cells/well), the cells were cultured in the above‐described supernatant. An XTT assay (Roche Diagnostics, Mannheim, Germany) was carried out after 48 h of culture, and following an assay‐time period of 24 h, absorbance was then measured at 490 nm.
Western blot analysis
Cells were lysed using lysis buffer (1% NP‐40,150 mM NaCl, 50 mM Tris‐HCl, pH 8.0), and protein was then extracted from the lysate. Tumor cells were cultured at 1 × 106 cells/well on a 6‐well plate in EBM‐2 medium, and the culture supernatant was collected after 24 h. These samples were mixed with 1% SDS sample buffer (10 mM Tris‐HCl [pH 7.5], 150 mM NaCl, 1% SDS, and EDTA‐free Protease Inhibitor Cocktail [Roche]), and were separated by length using 10% PAGE. They were then transferred to a PVDF membrane (Merck Millipore, Billerica, MA, USA). The membrane was placed in Tris buffer (pH 7.6) containing 5% skim milk (Wako Pure Chemical Industries, Tokyo, Japan) at room temperature for 1 h, and then reacted with a rabbit anti‐VEGFR‐1 antibody (Epitomics, Burlingame, CA, USA), mouse anti‐VASH1 antibody,9 rabbit anti‐Akt antibody, rabbit anti‐pAkt (Ser473) antibody, rabbit anti‐ERK antibody, rabbit anti‐pERK (Thr202/Tyr204) antibody (Cell Signaling Technologies, Danvers, MA, USA), or rabbit anti‐actin antibody (Sigma‐Aldrich) at 4°C overnight. After washing three times with PBS–Tween‐20 (PBS‐T), the membrane was incubated with a peroxidase‐labeled anti‐rabbit antibody (GE Healthcare, Little Chalfont, UK) or anti‐mouse antibody (GE Healthcare), at room temperature for 1 h. After washing three times with PBS‐T, chemiluminescence was induced using an ECL kit (Amersham Biosciences, Piscataway, NJ, USA), and luminescence was detected using a cool CCD system (LAS‐4000mini; GE Healthcare).
Animal experiment
BALB/c nude mice, 4–6‐weeks old (Clea Japan, Tokyo, Japan), were used in this study. Mice were maintained under specific pathogen‐free conditions. All animal experiments were approved by the Jichi Medical University (Tochigi, Japan) ethics committee and carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Subcutaneous tumor transplantation model
Tumor cells (5 × 106 cells) were s.c. inoculated into the dorsal region of nude mice to form an s.c. tumor. The tumor size was measured twice a week using calipers to calculate the tumor volume (TV) using the formala: TV = major axis of tumor (mm) × (minor axis of tumor)2 (mm2)/2.
Peritoneal dissemination model and survival time
Tumor cells (5 × 106 cells) were inoculated into the abdominal cavity of nude mice, and the amount of ascites and the peritoneal dissemination were observed. The survival of the animals was confirmed twice a day, and a survival curve was prepared using the Kaplan–Meier method.
Immunohistochemical staining
Tumors were excised from the mice after killing by decapitation. The tumors were then embedded in optimum cutting temperature compound (Sakura Finetek Japan Co., Ltd, Tokyo, Japan) and were frozen, and 7‐μm‐thick sections were subsequently prepared. These sections were fixed in methanol at −20°C for 20 min, followed by blocking with 1% BSA at room temperature. After inactivating endogenous peroxidase using a 3% hydrogen peroxide solution, the sections were reacted with a primary antibody: anti‐CD31 antibody (Pharmingen, San Diego, CA, USA), and then a secondary antibody of peroxidase‐conjugated anti‐rat antibody (Simple Stain Mouse MAX‐PO, Rat; Nichirei, Tokyo, Japan). Color was developed by a reaction with diaminobenzidine. Stained blood vessels were counted under a light microscope at a magnification of 400×.
Statistical analysis
Student's t‐test was used for between‐group comparisons. The log–rank test was used to analyze the Kaplan–Meier plots of the survival curves. A P‐value of <0.05 was regarded as significant. Results values were calculated as mean ± SD.
Results
Establishment of sFlt‐1‐ and VASH1‐expressing cell lines
SHIN‐3/LUC and SHIN‐3/sFlt‐1 cells have been previously reported.7 The results of the Western blot analyses for sFlt‐1 in KOC‐2S, KOC‐2S/LUC, and KOC‐2S/sFlt‐1 cells are shown in Figure 1(a). sFlt‐1 protein was only expressed in KOC‐2S/sFlt‐1 cells, thereby establishing the latter as the sFlt‐1‐expressing ovarian cancer cell line, KOC‐2S/sFlt‐1. The results of the Western blot analyses for VASH1 in SHIN‐3, SHIN‐3/LUC, and SHIN‐3/VASH1 cell groups are shown in Figure 1(b), and those for KOC‐2S, KOC‐2S/LUC, and KOC‐2S/VASH1 cells are shown in Figure 1(c). The VASH1 protein was only expressed in SHIN‐3/VASH1 and KOC‐2S/VASH1, therefore establishing the VASH1‐expressing ovarian cancer cell lines as SHIN‐3/VASH1 and KOC‐2S/VASH1. In addition, the presence of VASH1 in the culture supernatant of each VASH1‐transfected cell line was confirmed by Western blot assay, as shown in Figure 1(d).
Figure 1.
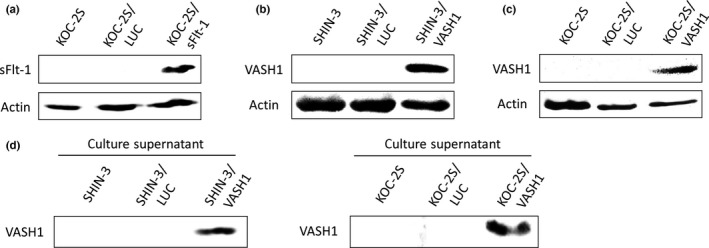
Establishment of soluble vascular endothelial growth factor receptor‐1 (sFlt‐1)‐ and vasohibin‐1 (VASH1)‐expressing cell lines. (a) Western blot with an anti‐vascular endothelial growth factor receptor‐1 antibody. The expression of the sFlt‐1 protein was only observed in KOC‐2S/sFlt‐1 ovarian cancer cells. (b, c) Western blots with an anti‐VASH1 antibody. The expression of the VASH1 protein was only observed in SHIN‐3/VASH1 (b) and KOC‐2S/VASH1 (c) ovarian cancer cells. Actin was used as a positive control. (d) Western blot of the culture supernatant, using an anti‐VASH1 antibody. The expression of the VASH1 protein in the culture supernatant was only observed for SHIN‐3/VASH1 and KOC‐2S/VASH1.
Influence of VASH1 on cell growth and cell signaling pathway
Cell growth in 3D culture was compared between SHIN‐3/LUC and SHIN‐3/VASH1 and between KOC‐2S/LUC and KOC‐2S/VASH1 cell lines (Fig. 2a). No significant difference was observed in the diameter of the acini in either cell line, as seen in Figure 2(b), showing that the expression of VASH1 did not influence the growth of SHIN‐3 or KOC‐2S cells.
Figure 2.
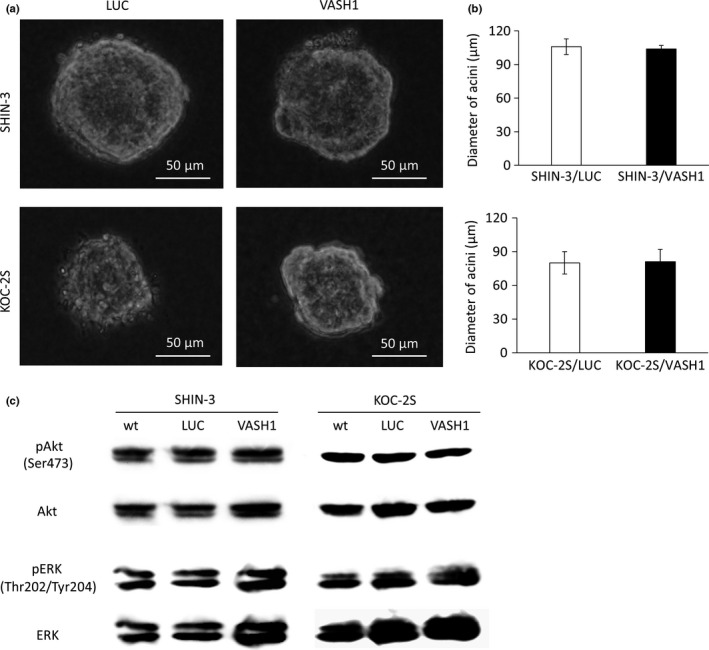
Influence of vasohibin‐1 (VASH1) on cell growth and the cell signaling pathway. (a) In vitro cell growth in 3D cultures of SHIN‐3/LUC and SHIN‐3/VASH1 cells, and of KOC‐2S/LUC and KOC‐2S/VASH1 cells, as observed under a light microscope. (b) No significant differences were noted in the diameter of acini between the two groups for either cell line. (c) Western blots with anti‐protein kinase B (Akt), anti‐pAkt (Ser473), anti‐ERK, and anti‐pERK (Thr202/Tyr204) antibodies. No significant difference was observed in the phosphorylated Akt or ERK levels in either of the cell lines.
The phosphoinositide 3‐kinase/Akt pathways and ERK are involved in physiological functions such as cell proliferation and survival. To investigate whether VASH1 affects these pathways, we assessed the expression levels of pAkt (Ser473) and pERK (Thr202/Tyr204) by Western blot assays. No significant difference was observed in the phosphorylated Akt or ERK levels in either of the cell lines, as seen in Figure 2(c), indicating that the expression of VASH1 did not influence the Akt or ERK pathways of cancer cells.
Effects of VASH1, secreted by SHIN‐3/VASH1 and KOC‐2S/VASH1 cell lines, on vascular endothelial cells
Figure 3(a) shows the VEGF concentrations in the supernatants from SHIN‐3 and KOC‐2S cultures. The wild‐type SHIN‐3 cells were VEGF hypersecretory, whereas the wild‐type KOC‐2S cells were not. No significant differences were observed in the level of VEGF secretion between the VASH1‐transfected SHIN‐3 or KOC‐2S cells and the corresponding controls. Figure 3(b) shows the PDGF concentrations in the culture supernatants. The wild‐type SHIN‐3 cells were PDGF hyposecretory, whereas the wild‐type KOC‐2S cells were PDGF hypersecretory. Similarly, no significant differences were observed in the level of PDGF secretion between the VASH1‐transfected SHIN‐3 or KOC‐2S cells and the corresponding controls. Therefore, VASH1 expression did not influence the VEGF or PDGF secretion by tumor cells.
Figure 3.
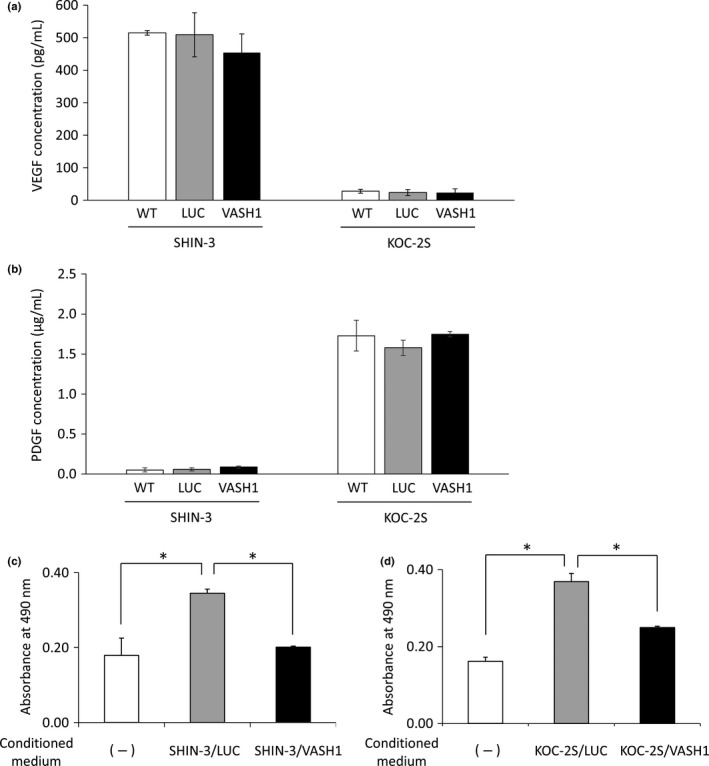
Vascular endothelial growth factor (VEGF) and platelet‐derived growth factor (PDGF) concentrations in the supernatants from SHIN‐3 and KOC‐2S cultures, and HUVEC counts after the addition of the culture supernatant. (a) VEGF concentrations in the supernatants from SHIN‐3 and KOC‐2S cultures. Wild‐type SHIN‐3 cells were VEGF hypersecretory (515 ± 7 pg/mL), whereas WT KOC‐2S cells were not (28 ± 6 pg/mL). No significant difference was observed in the level of VEGF secretion between vasohibin‐1 (VASH1)‐transfected SHIN‐3 or KOC‐2S cell lines and WT or LUC‐transfected cells in each cell line. (b) PDGF concentrations in supernatants from SHIN‐3 and KOC‐2S cultures. Wild‐type SHIN‐3 cells were PDGF hyposecretory (0.049 ± 0.025 μg/mL), whereas WT KOC‐2S cells were PDGF hypersecretory (1.728 ± 0.192 μg/mL). (c) Effects of VASH1, secreted by SHIN‐3/VASH1 and KOC‐2S/VASH1 cells, on HUVECs. Absorbance was measured in order to obtain a relative value for the cell counts. The mean absorbance value was significantly higher for the group cultured with SHIN‐3/LUC culture supernatant (CS) than for the group without, but was significantly lower in the group cultured with SHIN‐3/VASH1 CS than the group with the SHIN‐3/LUC CS (*P < 0.01). (d) The absorbance value for the group cultured with KOC‐2S/LUC CS was significantly higher than for the group without, but was significantly lower for the group cultured with KOC2S/VASH1 CS than that with the KOC‐2S/LUC CS (*P < 0.01).
The effects of VASH1, secreted by SHIN‐3/VASH1 and KOC‐2S/VASH1 cells, on HUVECs were investigated. The HUVEC count was significantly higher at 48 h after the addition of SHIN‐3/LUC culture supernatant when compared to base levels before addition, but was not increased by the addition of the SHIN‐3/VASH1 culture supernatant, as shown in Figure 3(c). The HUVEC count was also increased 48 h after the addition of KOC‐2S/LUC culture supernatant. Likewise, the addition of the KOC‐2S/VASH1 culture supernatant resulted in increased HUVEC count; however, this increase was significantly smaller than that observed for the KOC‐2S/LUC culture supernatant‐treated cells (Fig. 3d), showing that VASH1 secreted by both SHIN‐3/VASH1 and KOC‐2S/VASH1 cells inhibited vascular endothelial cell growth in vitro.
Inhibition of tumor growth and vascularization by sFlt‐1 and VASH1
The tumor growth‐ and vascularization‐inhibitory effects of sFlt‐1 expression on SHIN‐3 cells have been reported previously.7 The growth curves of s.c. KOC‐2S/LUC and KOC‐2S/sFlt‐1 tumors are shown in Figure 4(a). No significant difference was observed in the tumor growth between the KOC‐2S/LUC and KOC‐2S/sFlt‐1 groups, indicating that sFlt‐1 expression did not influence the growth of s.c. PDGF‐producing KOC‐2S tumors in vivo.
Figure 4.
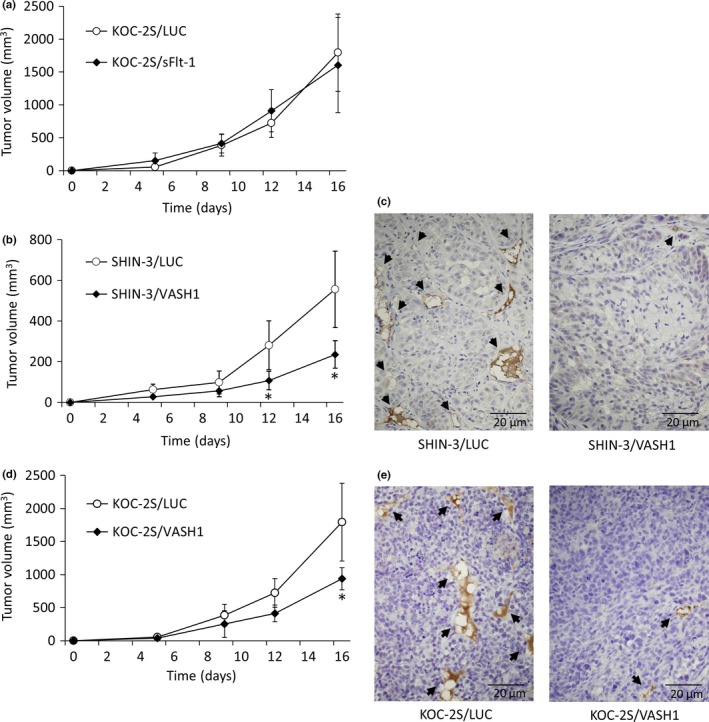
Inhibition of tumor growth and vascularization by soluble vascular endothelial growth factor receptor‐1 (sFlt‐1) and vasohibin‐1 (VASH1). (a) Growth curves of s.c. KOC‐2S/LUC and KOC‐2S/sFlt‐1 tumors. No significant differences in tumor growth were noted between the KOC‐2S/LUC and KOC‐2S/sFlt‐1 groups. (b) Growth curves of s.c. SHIN‐3/LUC and SHIN‐3/VASH1 tumors. The tumor volume 12 days after inoculation and thereafter was significantly smaller in the SHIN‐3/VASH1 group compared to the SHIN‐3/LUC group. (c) CD31 immunohistochemical staining, developed with diaminobenzidine, of new blood vessels (black arrows) in SHIN‐3/LUC and SHIN‐3/VASH1 tumor tissues. (d) Growth curves of s.c. KOC‐2S/LUC and KOC‐2S/VASH1 tumors. Tumor volume 16 days after inoculation was significantly lower in the KOC‐2S/VASH1 group than the KOC‐2S/LUC group. (e) CD31 immunohistochemical staining, developed with diaminobenzidine, of new blood vessels (black arrows) in KOC‐2S/LUC and KOC‐2S/VASH1 tumor tissues. Data points for the graphs are represented as mean ± SD. *P < 0.01.
The growth curves of s.c. SHIN‐3/LUC and SHIN‐3/VASH1 tumors are shown in Figure 4(b). Tumor growth was slower in the SHIN‐3/VASH1 group than SHIN‐3/LUC group. New blood vessels in excised SHIN‐3/LUC and SHIN‐3/VASH1 tumor tissue were evaluated using immunohistochemical staining (Fig. 4c). The number of blood vessels per visual field at a high magnification was significantly lower in SHIN‐3/VASH1 tumors than in SHIN‐3/LUC tumors (1.6 ± 0.9 and 9.8 ± 2.5, respectively) (n = 3 per group, P < 0.01), thus showing that the expression of VASH1 resulted in the inhibition of SHIN‐3 tumor vascularization and growth.
The growth curves for s.c. KOC‐2S/LUC and KOC‐2S/VASH1 tumors are shown in Figure 4(d). Tumor growth was slower in the KOC‐2S/VASH1 group than in the KOC‐2S/LUC group. New blood vessels in excised KOC‐2S/LUC and KOC‐2S/VASH1 tumor specimens were evaluated using immunohistochemical staining (Fig. 4e). The number of blood vessels per visual field at a high magnification was significantly lower in KOC‐2S/VASH1 tumors than in KOC‐2S/LUC tumors (1.6 ± 0.5 and 8.6 ± 0.9, respectively) (n = 3 per group, P < 0.01), revealing that the expression of VASH1 led to the inhibition of KOC‐2S tumor vascularization and growth.
Inhibition of peritoneal dissemination by sFlt‐1 and VASH1
The inhibition of peritoneal dissemination and ascites development, and prolongation of the survival of the host, as an effect of sFlt‐1 expression in SHIN‐3 cells has been previously reported.7 The survival curves of the mice that underwent i.p. transplantation of KOC‐2S/LUC and KOC‐2S/sFlt‐1 cells are shown in Figure 5(a). No significant difference was seen between these two groups (n = 5 per group), showing that the expression of sFlt‐1 did not influence the survival time of mice with peritoneal dissemination of PDGF‐producing KOC‐2S cells.
Figure 5.
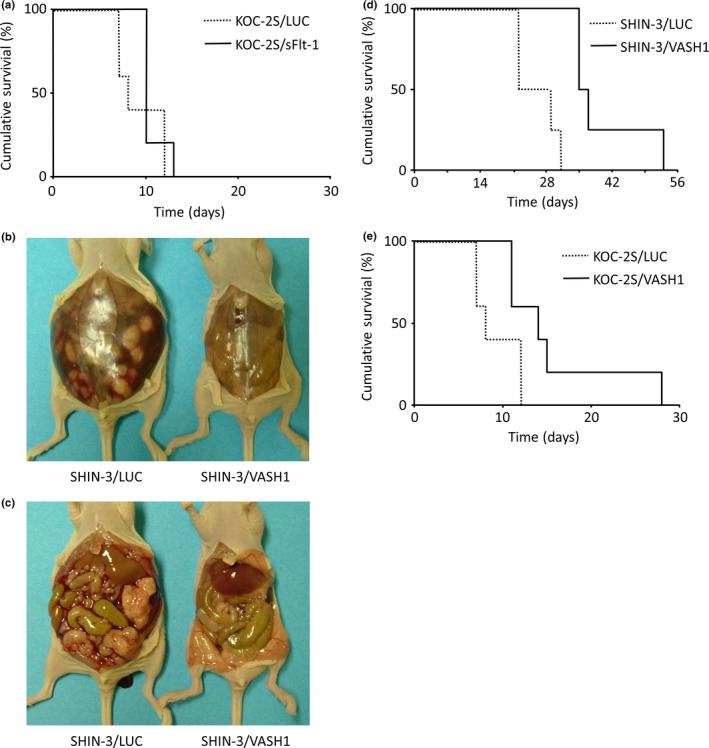
Inhibition of peritoneal dissemination by soluble vascular endothelial growth factor receptor‐1 (sFlt‐1) and vasohibin‐1 (VASH1). (a) Survival curves for mice i.p. transplanted with either KOC‐2S/LUC or KOC‐2S/sFlt‐1 cells. No significant difference was noted between the two groups. (b, c) Representative images of the laparotomic findings, carried out 21 days after i.p. SHIN‐3/LUC or SHIN‐3/VASH1 transplantation into mice. A large volume of ascites (b, left) and many peritoneal disseminated lesions (c, left) were observed in SHIN‐3/LUC‐transplanted mice. In contrast, no ascites (b, right) or peritoneal dissemination (c, right) was noted in SHIN‐3/VASH1‐transplanted mice. (d) Survival curves of mice i.p. transplanted with SHIN‐3/LUC or SHIN‐3/VASH1 cells. The survival time was significantly longer in the SHIN‐3/VASH1 group than in the SHIN‐3/LUC group (P < 0.01). (e) Survival curves of mice i.p. transplanted with KOC‐2S/LUC or KOC‐2S/VASH1 cells. The survival time was significantly longer in the KOC‐2S/VASH1 group than in the KOC‐2S/LUC group (P < 0.01).
SHIN‐3/LUC and SHIN‐3/VASH1 cells were i.p. transplanted into nude mice, and laparotomy was carried out after 21 days. A large volume of ascites (Fig. 5b, left) and many peritoneal disseminated lesions (Fig. 5c, left) were observed in SHIN‐3/LUC‐transplanted mice, but these were absent in SHIN‐3/VASH1‐transplanted mice (Fig. 5b,c right), illustrating that the expression of VASH1 inhibited the peritoneal dissemination of SHIN‐3 cancer cells.
The survival curves of mice transplanted i.p. with SHIN‐3/LUC and SHIN‐3/VASH1 cells are shown in Figure 5(d). All mice in the SHIN‐3/LUC group died within 31 days of transplantation, whereas the SHIN‐3/VASH1 group survived significantly longer (n = 4 per group, P < 0.01), showing that the expression of VASH1 inhibited the peritoneal dissemination of VEGF‐producing ovarian cancer cells, SHIN‐3, and prolonged the survival time of these mice.
The survival curves of mice i.p. transplanted with KOC‐2S/LUC and KOC‐2S/VASH1 are shown in Figure 5(e). All mice in the KOC‐2S/LUC group died from peritoneal dissemination and ascites within 13 days of transplantation, whereas the KOC‐2S/VASH1 group survived significantly longer (n = 5 per group, P < 0.01), elucidating that the expression of VASH1 inhibited the peritoneal dissemination of PDGF‐producing ovarian cancer cells, KOC‐2S, and prolonged the survival time of the KOC‐2S mice.
Discussion
We investigated the efficacy of VASH1 in treating ovarian cancer by using high VEGF‐producing and high PDGF‐producing ovarian cancer cells. Expression of the VEGF‐antagonist, sFlt‐1, inhibits the tumor vascularization and growth of high VEGF‐producing ovarian cancer cells, reduces peritoneal dissemination and ascites development, and prolongs the survival time of the host.7 However, in the present study, these effects were not observed in high PDGF‐producing ovarian cancer cells. In contrast, the expression of VASH1 inhibited tumor vascularization and growth of not only high VEGF‐producing cells, but also high PDGF‐producing cells, reduced peritoneal dissemination and ascites development, and prolonged the survival time of the host. sFlt‐1 specifically inhibits VEGF, similar to the anti‐VEGF mAb bevacizumab, whereas VASH1 inhibits various angiogenic factors in addition to VEGF, including FGF and PDGF.9, 13 This multitarget inhibition ability of VASH1 was reflected in the results of our study.
In previous studies involving ovarian cancer patients, the levels of angiogenic factors other than VEGF, that is, FGF and PDGF, were high (79–94% for FGF;18, 19 73–88% for PDGF20, 21) demonstrating that various angiogenic factors are involved in tumor vascularization. Therefore, a treatment targeting only a single angiogenic factor would likely be ineffective, and it may be necessary to simultaneously target many angiogenic factors for improving patient survival. In this regard, VASH1 may be useful in treating ovarian cancer therapy.
With the growing use of anti‐VEGF therapy, including the use of bevacizumab, a newly arisen resistance phenomenon, termed evasive resistance and seen in tumors following anti‐VEGF therapy, has been attracting attention.8 The acquisition mechanism of this resistance is speculated to involve the mobilization of bone marrow‐derived endothelial precursor cells, the coverage of tumor vascular endothelial cells by pericytes, and the increased infiltrating ability of tumor cells, in addition to the hypothesis that angiogenic factors other than VEGF are induced.8 An animal study in which pancreatic cancer was treated with an anti‐VEGF receptor‐2 (VEGFR‐2) antibody showed that after the initial vascular regression and tumor size reduction, tumor vascularization was reinduced, leading to recommencement of tumor growth. In the same study, when the mRNA expression in these regrown tumors was analyzed, the expression of angiogenic factors other than VEGF, such as FGF, ephrin, and angiopoietin, was found to be enhanced.22 In an animal study documenting the treatment of colorectal cancer, melanoma, and pancreatic cancer by using an anti‐VEGFR‐2 antibody, the production of an angiogenic factor, placenta growth factor (PlGF), was promoted in all tumors after treatment.23 Furthermore, an increase in serum FGF levels after VEGFR tyrosine kinase inhibitor treatment was observed in glioma patients in a clinical study.24 Based on these findings, it can be inferred that tumors resist treatments that target a single angiogenic factor by producing other angiogenic factors, that is, single‐target treatments may induce resistance. To prevent the induction of resistance and to develop an effective treatment, it may be necessary to simultaneously target many angiogenic factors. With this in mind, researchers combined an anti‐VEGFR‐2 antibody with an FGF antagonist, soluble FGF receptor, or anti‐PlGF antibody in an animal study, and a stronger treatment effect was observed.22, 23 Vasohibin‐1, which inhibits several angiogenic factors, is also likely to be a superior alternative to the use of agents inhibiting only VEGF, with regard to overcoming evasive resistance.
Various pro‐angiogenesis factors, such as VEGF, generally contribute to the survival of endothelial cells. However, anti‐angiogenic factors, such as thrombospondins,25 generally cause endothelial cell death and vascular regression. The inhibitory action of VASH1 on angiogenesis has been reported by various researchers.9, 11, 13, 26 It was recently reported that VASH1 protects endothelial cells from premature senescence and stress‐induced cell death through the induction of superoxide dismutase 2 and sirtuin 1.27 Superoxide dismutase 2 plays a critical role in protecting cells against oxidative stress,28 and sirtuin 1 is considered to be responsible for the stress resistance of endothelial cells. Accordingly, VASH1 has the unique characteristics of not only inhibiting angiogenesis but increasing the stress tolerance and survival of endothelial cells as well.29 We are trying to elucidate the mechanism of these unique activities of VASH1, including the VASH1 receptor and its signaling pathway.
In the current study, we showed that VASH1 inhibited tumor vascularization and peritoneal dissemination of not only VEGF‐producing, but also PDGF‐producing ovarian cancer cells, and prolonged the survival time of mice in an ovarian cancer model. These results suggest that treatment with VASH1 represents a novel and very useful therapeutic strategy for ovarian cancer.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgment
This work was supported by a Japan Society for the Promotion of Science Kakenhi grant (Grant‐in‐Aid for Scientific Research no. 25462606) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Cancer Sci 107 (2016) 629–637
Funding Information
Japan Society for the Promotion of Science Kakenhi grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan (25462606)
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 2. Heintz AP. Surgery in advanced ovarian carcinoma: is there proof to show the benefit? Eur J Surg Oncol 1988; 14: 91–9. [PubMed] [Google Scholar]
- 3. McGuire WP, Hoskins WJ, Brady MF et al Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med 1996; 334: 1–6. [DOI] [PubMed] [Google Scholar]
- 4. Takei Y, Suzuki M, Ohwada M et al A feasibility study of paclitaxel and carboplatin therapy in Japanese patients with epithelial ovarian cancer. Oncol Rep 2003; 10: 951–5. [PubMed] [Google Scholar]
- 5. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–6. [DOI] [PubMed] [Google Scholar]
- 6. Kohno T, Mizukami H, Suzuki M et al Interleukin‐10‐mediated inhibition of angiogenesis and tumor growth in mice bearing VEGF‐producing ovarian cancer. Cancer Res 2003; 63: 5091–4. [PubMed] [Google Scholar]
- 7. Takei Y, Mizukami H, Saga Y et al Suppression of ovarian cancer by muscle‐mediated expression of soluble VEGFR‐1/Flt‐1 using adeno‐associated virus serotype 1‐derived vector. Int J Cancer 2007; 120: 278–84. [DOI] [PubMed] [Google Scholar]
- 8. Bergers G, Hanahan D. Modes of resistance to anti‐angiogenic therapy. Nat Rev Cancer 2008; 8: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watanabe K, Hasegawa Y, Yamashita H et al Vasohibin as an endothelium‐derived negative feedback regulator of angiogenesis. J Clin Invest 2004; 114: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shimizu K, Watanabe K, Yamashita H et al Gene regulation of a novel angiogenesis inhibitor, vasohibin, in endothelial cells. Biochem Biophys Res Commun 2005; 327: 700–6. [DOI] [PubMed] [Google Scholar]
- 11. Takahashi Y, Saga Y, Koyanagi T et al The angiogenesis regulator vasohibin‐1 inhibits ovarian cancer growth and peritoneal dissemination and prolongs host survival. Int J Oncol 2015; 47: 2057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiener JR, Nakano K, Kruzelock RP et al Decreased Src tyrosine kinase activity inhibits malignant human ovarian cancer tumor growth in a nude mouse model. Clin Cancer Res 1999; 5: 2164–70. [PubMed] [Google Scholar]
- 13. Heishi T, Hosaka T, Suzuki Y et al Endogenous angiogenesis inhibitor vasohibin1 exhibits broad‐spectrum antilymphangiogenic activity and suppresses lymph node metastasis. Am J Pathol 2010; 176: 1950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Imai S, Kiyozuka Y, Maeda H, Noda T, Hosick HL. Establishment and characterization of a human ovarian serous cystadenocarcinoma cell line that produces the tumor markers CA‐125 and tissue polypeptide antigen. Oncology 1990; 47: 177–84. [DOI] [PubMed] [Google Scholar]
- 15. Kataoka A, Nishida T, Hirai N, Tomioka Y, Sugiyama T, Yakushiji M. Induction of apoptosis in ovarian carcinoma cell line by glucocorticoids, and sex steroid hormones. Oncol Rep 1997; 4: 1249–53. [DOI] [PubMed] [Google Scholar]
- 16. Machida S, Saga Y, Takei Y et al Inhibition of peritoneal dissemination of ovarian cancer by tyrosine kinase receptor inhibitor SU6668 (TSU‐68). Int J Cancer 2005; 114: 224–9. [DOI] [PubMed] [Google Scholar]
- 17. Urabe M, Hasumi Y, Ogasawara Y et al A novel dicistronic AAV vector using a short IRES segment derived from hepatitis C virus genome. Gene 1997; 200: 157–62. [DOI] [PubMed] [Google Scholar]
- 18. Le Page C, Ouellet V, Madore J et al From gene profiling to diagnostic markers: IL‐18 and FGF‐2 complement CA125 as serum‐based markers in epithelial ovarian cancer. Int J Cancer 2006; 118: 1750–8. [DOI] [PubMed] [Google Scholar]
- 19. Gan Y, Wientjes MG, Au JL. Expression of basic fibroblast growth factor correlates with resistance to paclitaxel in human patient tumors. Pharm Res 2006; 23: 1324–31. [DOI] [PubMed] [Google Scholar]
- 20. Henriksen R, Funa K, Wilander E, Bäckström T, Ridderheim M, Oberg K. Expression and prognostic significance of platelet‐derived growth factor and its receptors in epithelial ovarian neoplasms. Cancer Res 1993; 53: 4550–4. [PubMed] [Google Scholar]
- 21. Schilder RJ, Sill MW, Lee RB et al Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol 2008; 26: 3418–25. [DOI] [PubMed] [Google Scholar]
- 22. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late‐stage pancreatic islet tumors. Cancer Cell 2005; 8: 299–309. [DOI] [PubMed] [Google Scholar]
- 23. Fischer C, Jonckx B, Mazzone M et al Anti‐PlGF inhibits growth of VEGF(R)‐inhibitor‐resistant tumors without affecting healthy vessels. Cell 2007; 131: 463–75. [DOI] [PubMed] [Google Scholar]
- 24. Batchelor TT, Sorensen AG, di Tomaso E et al AZD2171, a pan‐VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007; 11: 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo N, Krutzsch HC, Inman JK, Roberts DD. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res 1997; 57: 1735–42. [PubMed] [Google Scholar]
- 26. Hosaka T, Kimura H, Heishi T et al Vasohibin‐1 expression in endothelium of tumor blood vessels regulates angiogenesis. Am J Pathol 2009; 175: 430–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyashita H, Watanabe T, Hayashi H et al Angiogenesis inhibitor vasohibin‐1 enhances stress resistance of endothelial cells via induction of SOD2 and SIRT1. PLoS ONE 2012; 7: e46459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fukai T, Ushio‐Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 2011; 15: 1583–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato Y. Novel link between inhibition of angiogenesis and tolerance to vascular stress. J Atheroscler Thromb 2015; 22: 327–34. [DOI] [PubMed] [Google Scholar]