

Abstract
In the FIRST trial (MM‐020), lenalidomide plus low‐dose dexamethasone (Rd) reduced the risk of disease progression or death compared with combination melphalan–prednisone–thalidomide. As the FIRST trial did not include any Japanese patients, the efficacy and safety of continuous treatment with Rd was evaluated in 26 Japanese patients with newly diagnosed multiple myeloma (NDMM) in a single‐arm, multicenter, open‐label phase II trial (MM‐025). Patients received lenalidomide on days 1–21 of each 28‐day cycle, with a starting dose of 25 mg/day (dose adjusted for renal impairment), and 40 mg/day dexamethasone (dose adjusted for age) on days 1, 8, 15 and 22 of each 28‐day cycle until disease progression or discontinuation for any reason. In the efficacy evaluable population, overall response rate was 87.5%, including 29.2% of patients who achieved a complete response/very good partial response. Median durations of response, progression‐free survival and overall survival have not been reached. The most common grade 3–4 adverse events were neutropenia (23%) and anemia (19%). The efficacy and safety of Rd were consistent with data from larger studies, including the FIRST trial, thereby supporting the use of Rd continuous in Japanese patients with NDMM who are ineligible for stem cell transplantation.
Keywords: Dexamethasone, Japan, Japanese, lenalidomide, multiple myeloma
Currently in Japan, the recommended therapy for patients with newly diagnosed multiple myeloma (NDMM) who are ineligible for stem cell transplantation (SCT) is melphalan plus prednisone (MP) combined with either bortezomib (MPV) or thalidomide (MPT).1, 2 Bortezomib was first approved in Japan in 2006 for the treatment of patients with relapsed and refractory multiple myeloma (RRMM).
In the phase III VISTA trial, MPV was shown to significantly improve median overall survival (OS) compared with MP (56.4 vs 43.1 months, respectively; P < 0.001), with a 31% risk reduction of death (hazard ratio [HR] 0.695).3, 4, 5 A meta‐analysis of data from six randomized trials in transplant‐ineligible NDMM patients showed that the MPT regimen, compared with MP, significantly reduced the risk of death (HR 0.83; P < 0.0001) or disease progression and death (HR 0.68; P < 0.004).6 MPT is recommended as first‐line therapy for transplant‐ineligible NDMM patients in the USA and the European Union.2, 7 Although MPT is not yet approved in Japan, the Japanese Society of Hematology has given the regimen a Category 1 recommendation, based on high‐level evidence and uniform consensus.1
Both MPV and MPT are associated with adverse events (AE) that may impact on quality of life and treatment outcome.8 In addition, prolonged treatment with melphalan‐containing regimens, including those combined with lenalidomide,9 may increase the risk of second primary malignancies (SPMs), such as myelodysplastic syndromes and acute myeloid leukemia. Thus, limiting the duration of melphalan‐containing regimens to 6–18 months is generally recommended.10, 11, 12 In the VISTA trial, grade ≥3 peripheral neuropathy was reported in 13% of patients treated with MPV, with 12% of patients discontinuing study treatment due to peripheral neuropathy.3 In trials evaluating MPT, up to 20% of patients developed grade ≥2 peripheral neuropathy during MPT therapy.13 These findings suggest the need for a combination therapy that is effective and less toxic for long‐term use in transplant‐ineligible NDMM patients.
Lenalidomide is an oral immunomodulatory agent which, when given in combination with dexamethasone, is a standard therapy option for the treatment of patients with RRMM.14, 15 A randomized study comparing lenalidomide plus high‐dose dexamethasone (RD) with lenalidomide plus low‐dose dexamethasone (Rd) in patients with NDMM found that the 1‐year OS rate was significantly higher with Rd vs RD (96% vs 87%, respectively; P = 0.0002); Rd was also associated with fewer AE.16
In the pivotal, global, randomized phase III FIRST trial (MM‐020, the largest global registration study of transplant‐ineligible NDMM patients conducted to date), continuous treatment with Rd was compared with fixed‐duration treatment with MPT.17 The Rd regimen, compared with MPT, significantly prolonged progression‐free survival (PFS) (HR 0.72, 95% confidence interval [CI] 0.61–0.85; P < 0.001), yielded favorable interim OS results (HR 0.78, 95% CI 0.64–0.96; P = 0.02), and showed a higher overall response rate (ORR) (P < 0.001). Results from the Asian subpopulation in the FIRST trial were generally consistent with the findings in the overall population (PFS: HR 0.61, 95% CI 0.33–1.14; and OS: HR 0.52, 95% CI 0.24–1.13;18 ORR: P = 0.06 [Celgene data on file]). However, only approximately 8% of the patients in the FIRST trial were Asian (enrolled in China, South Korea and Taiwan),18 and the Rd regimen has not been studied specifically in Japanese patients. Although the safety and efficacy of lenalidomide in combination with high‐dose dexamethasone has been investigated in Japanese RRMM patients,19 there has been no such study in the Japanese NDMM patient population using low‐dose dexamethasone. In response to this gap in knowledge, an original phase II study was initiated, in reference to the FIRST trial, to confirm the efficacy and safety of Rd in Japanese patients with NDMM who are ineligible for SCT.
Materials and Methods
Study design and treatment
MM‐025 was a phase II, multicenter, open‐label, single‐arm study conducted at 24 treatment centers in Japan (ClinicalTrials.gov Identifier: NCT01698801). The study consisted of three periods: screening, treatment and follow‐up. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice and applicable regulations. Written informed consent was obtained from each patient before any study procedure was undertaken. The Institutional Review Board at each study site approved the study and the contents of the informed consent document.
Potential eligible patients underwent screening within 28 days before receiving study treatment. Eligible patients had previously untreated, symptomatic and measurable multiple myeloma (MM), were aged ≥65 years (patients aged <65 years were included if they were considered ineligible for SCT, refused SCT or did not have access to SCT), and had an Eastern Cooperative Oncology Group (ECOG) performance status score of 0–2. Patients were excluded if they had: received prior antimyeloma therapy; significant laboratory abnormalities; severe renal impairment requiring dialysis; grade ≥2 peripheral neuropathy; or a contraindication for dexamethasone. Patients also had to be able and willing to undergo antithrombotic therapy.
Patients who met the eligibility criteria entered the treatment period and received continuous Rd until documentation of disease progression or discontinuation of study treatment for any reason. Lenalidomide was given on days 1–21 of each 28‐day cycle. The starting dose of lenalidomide was adjusted based on patient renal function: 25 mg/day for normal renal function/mild renal impairment (creatinine clearance [CrCl] ≥60 mL/min); 10 mg/day for moderate renal impairment (CrCl ≥30 to <60 mL/min); and 15 mg once every other day for severe renal impairment (CrCl <30 mL/min). Patients requiring dialysis were excluded from the study. Dexamethasone was given weekly (i.e. on days 1, 8, 15 and 22 of each 28‐day cycle) and the starting dose was adjusted based on patient age: 40 mg/day for patients aged ≤75 years and 20 mg/day for patients aged >75 years. For patients who experienced a treatment‐emergent AE (TEAE), doses of lenalidomide and/or dexamethasone were adjusted according to dose modification guidelines specified in the protocol. The dose and schedule of lenalidomide and dexamethasone selected for the current study were based on those used in the FIRST trial and in a previous phase III trial conducted by ECOG in previously untreated MM subjects,16 which demonstrated favorable efficacy and safety profiles. The pharmacokinetics of lenalidomide have been shown to be similar in Japanese and white patients,20 and the dose and schedule of lenalidomide used in the current study are consistent with those approved for the treatment of RRMM in Japan and beyond.
Patients who had experienced deep‐vein thrombosis (DVT) or pulmonary embolism (PE) within 5 years of starting the study treatment received prophylactic anticoagulation with heparin, a low‐molecular‐weight heparin, or warfarin for at least the first 16 weeks of the study. Thereafter, they either continued anticoagulation therapy or received low‐dose aspirin, at the investigator's discretion, for the remainder of the study treatment period. Patients who had not experienced DVT or PE within 5 years before starting treatment received low‐dose aspirin or other prophylactic antithrombotic treatment during the study treatment period at the discretion of the investigator. Bisphosphonates and other supportive therapies were allowed at the investigator's discretion. Other antimyeloma therapies, however, were not allowed during the study.
Patients who discontinued the study treatment entered the follow‐up period and were monitored every 2 months for survival, disease progression (for those who did not progress) and SPM for ≥5 years from the start of Rd treatment until the last patient enrolled in the study (unless the patient withdrew consent, was lost to follow‐up or died).
Endpoints and assessments
The primary endpoint was ORR (defined as complete response [CR], very good partial response [VGPR] or partial response [PR]), based on the International Myeloma Working Group (IMWG) criteria.21 Secondary endpoints included: time to response (TTR), duration of response (DOR), PFS, OS and safety (type, frequency and severity of AE, and their relationship to study drug). Disease progression data were reviewed by the Efficacy and Safety Evaluation Committee using IMWG criteria.21 The severity of AE was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.0. The intent‐to‐treat (ITT) population comprised all enrolled patients regardless of whether they received the study drug. The safety population included all patients who received ≥1 dose of the study drug, and the efficacy evaluable (EE) population included patients who met the protocol requirements (all eligibility criteria) and were evaluated for efficacy after receiving ≥1 dose of the study drug. The primary efficacy population was the EE population. The efficacy analysis was performed after all patients had completed ≥24 weeks of study treatment or discontinued for any reason.
Statistical considerations
The required sample size was calculated using the expected ORR of 60%. Assuming an ORR of 60%, 20 patients would be required to reject the null hypothesis (H0: ORR 30%) with a probability of ≥80%, an α error of 0.05 and β error of 0.20. The threshold ORR (30%) was based on the ORR achieved with MP in the VISTA trial (39%),3 indicating that an ORR of 30% was the minimum requirement to continue the development of Rd in this population. A one‐sample binomial test was performed for the dichotomized ORR proportion against the null hypothesis to provide the P‐value for the ORR. The treatment was considered effective if the lower limit of the CI was >30% in the EE population.
Results
Patients and treatment
The primary analysis was conducted after all patients completed 24 weeks of treatment or discontinued the study treatment prior to 24 weeks for any reason. The data cutoff date was 15 July 2014.
A total of 26 patients were enrolled in the study and received Rd continuous (Fig. 1). As of the data cutoff date, 15 patients were receiving treatment and 11 patients had discontinued. The primary reasons for discontinuation were AE (n = 4), withdrawal of consent (n = 1), progressive disease (n = 1), protocol violation (n = 1) and “other” (n = 4). Of the 10 patients who entered the follow‐up phase following treatment discontinuation, six remained in follow‐up and four discontinued (due to death).
Figure 1.
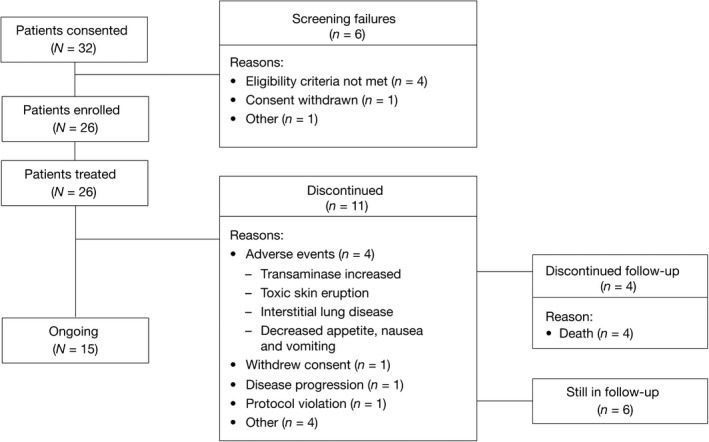
Disposition of study patients.
Patient baseline characteristics for the ITT population are presented in Table 1. The median age was 75 years (range 60–85 years), and 46.2% (n = 12) of patients were aged >75 years. Half of all patients were male (n = 13) and 19.2% (n = 5) had International Staging System (ISS) stage III disease. The ECOG performance status score was 0–1 in 76.9% (n = 20) of patients and was 2 in 23.1% (n = 6). The baseline CrCl level was <30 mL/min in 7.7% (n = 2) of patients and ≥30 to <60 mL/min in 53.8% (n = 14). Baseline characteristics of patients in the MM‐025 trial were generally similar to those in the FIRST trial in terms of age, sex and disease type; however, the MM‐025 patients generally appeared to have less severe MM. The majority of MM‐025 patients (80.8%) had ISS stage I or II disease compared with 59.4% of patients in the FIRST trial (in the FIRST trial Rd continuous arm, 59.6% of patients had ISS stage I or II disease).
Table 1.
Patient baseline characteristics (intent‐to‐treat population)
| Characteristic | N = 26 |
|---|---|
| Age, median (range), years | 75 (60–85) |
| Age distribution, n (%) | |
| ≤75 years | 14 (53.8) |
| >75 years | 12 (46.2) |
| <65 years | 2 (7.7) |
| ≥65 years | 24 (92.3) |
| Sex, n (%) | |
| Male | 13 (50.0) |
| Female | 13 (50.0) |
| ECOG performance status score, n (%) | |
| 0 | 13 (50.0) |
| 1 | 7 (26.9) |
| 2 | 6 (23.1) |
| International Staging System stage, n (%) | |
| I | 7 (26.9) |
| II | 14 (53.8) |
| III | 5 (19.2) |
| Creatinine clearance, n (%) | |
| ≥60 mL/min | 10 (38.5) |
| ≥30 to <60 mL/min | 14 (53.8) |
| <30 mL/min | 2 (7.7) |
| Albumin level, n (%) | |
| ≤3.5 g/dL | 20 (76.9) |
| >3.5 g/dL | 6 (23.1) |
| β2‐microglobulin level, n (%) | |
| ≤5.5 mg/L | 21 (80.8) |
| >5.5 mg/L | 5 (19.2) |
| Multiple myeloma subtype, n (%) | |
| IgG | 18 (69.2) |
| IgA | 6 (23.1) |
| Bence–Jones protein | 2 (7.7) |
ECOG, Eastern Cooperative Oncology Group; Ig, immunoglobulin.
Efficacy
The ORR, the primary endpoint, is shown in Table 2. In the EE population (n = 24), the ORR was 87.5% (95% CI 74–100; P < 0.0001). Two patients (8.3%) achieved CR, 5 (20.8%) achieved VGPR and 14 (58.3%) had PR. In patients who achieved at least PR, the median TTR was 1.97 months (range 0.9–13.8 months). The median DOR based on Kaplan–Meier estimate has not been reached. The ORR was 83% in the 12 patients aged >75 years and 92% in the 12 patients aged ≤75 years. Responses were observed regardless of renal impairment status; at least PR was reported in 100% (2 of 2) of patients with severe renal impairment (CrCl <30 mL/min), 84.6% (11 of 13) with moderate renal impairment (CrCl ≥30 to <60 mL/min) and 88.9% (8 of 9) with normal renal function/mild renal impairment (CrCl ≥60 mL/min).
Table 2.
Response rate and TTR (efficacy evaluable population)
| Variable | N = 24 |
|---|---|
| ORR (CR + VGPR + PR), n (%) | 21 (87.5) |
| CR | 2 (8.3) |
| VGPR | 5 (20.8) |
| PR | 14 (58.3) |
| Stable disease, n (%) | 2 (8.3) |
| Progressive disease, n (%) | 0 |
| Not evaluable, n (%) | 1 (4.2) |
| TTR, median (range), † months | 1.97 (0.9–13.8) |
†In patients who achieved at least PR. CR, complete response; ORR, overall response rate; PR, partial response; TTR, time to response; VGPR, very good PR.
Of the 24 patients in the EE population, three had progressed as of the data cutoff date. With a median follow‐up of 14.2 months, the median PFS based on the Kaplan–Meier estimates had not been reached (Fig. 2). The interim OS data, with a data cutoff of 15 July 2014, revealed that there were three deaths during the follow‐up period.
Figure 2.
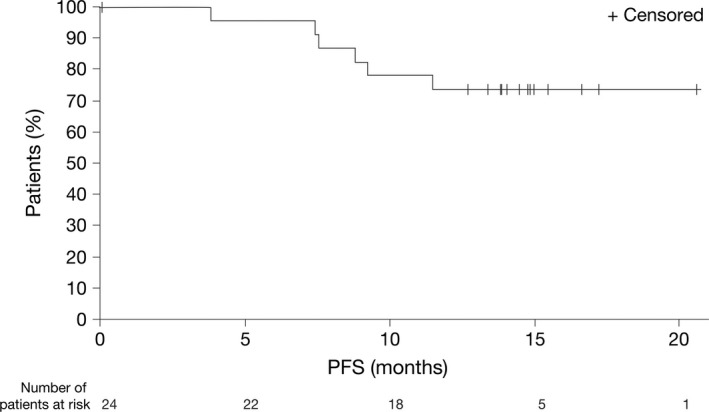
Interim progression‐free survival (PFS) in the efficacy evaluable population (N = 24).
Safety
All patients experienced ≥1 TEAE. The most common TEAEs were rash (50.0%; n = 13), nasopharyngitis (42.3%; n = 11), constipation (30.8%; n = 8), anemia (30.8%; n = 8), neutropenia (26.9%; n = 7), thrombocytopenia (23.1%; n = 6), leukopenia (23.1%; n = 6), insomnia (23.1%; n = 6) and peripheral edema (23.1%; n = 6). There was one reported incidence of grade 2 DVT.
Eighteen patients (69.2%) reported ≥1 grade 3–4 TEAE (Table 3). The most commonly reported grade 3–4 TEAEs were neutropenia (23.1%; n = 6), anemia (19.2%; n = 5) and thrombocytopenia (15.4%; n = 4). Serious TEAEs were reported in 42.3% (n = 11) of patients and included the following: abnormal hepatic function, acute cardiac failure, coronary artery stenosis, interstitial lung disease, lower gastrointestinal hemorrhage, pneumonia, aspiration pneumonia, presyncope, tumor lysis syndrome, thrombocytopenia, toxic skin eruption and urinary tract infection (all single cases). No deaths due to treatment‐related AE, thromboembolic events or SPM were reported as of the data cutoff. The deaths that did occur were due to MM disease progression and related complications (n = 3) or pneumonia (n = 1), neither of which were related to study drug.
Table 3.
Grade 3–4 adverse events occurring in >5% of the safety population
| n (%) | N = 26 |
|---|---|
| Any grade 3–4 treatment‐emergent adverse events | 18 (69.2) |
| Neutropenia | 6 (23.1) |
| Anemia | 5 (19.2) |
| Thrombocytopenia | 4 (15.4) |
| Leukopenia | 3 (11.5) |
| Lymphopenia | 3 (11.5) |
| Rash | 3 (11.5) |
| Hypoalbuminemia | 2 (7.7) |
| Hyponatremia | 2 (7.7) |
| Hypophosphatemia | 2 (7.7) |
| Pneumonia | 2 (7.7) |
| Hypertension | 2 (7.7) |
Study treatment and dosing are shown in Table 4. The median duration of treatment was 13.8 months (range 0.2–20.6 months). Median relative dose intensity for all patients was 0.87 (range 0.2–1.3) for lenalidomide and 0.93 (range 0.3–1.0) for dexamethasone. Relative dose intensity was lower in patients aged >75 years (0.72 for lenalidomide and 0.80 for dexamethasone). TEAEs led to dose reductions in 13 (50%) patients for lenalidomide and 9 (34.6%) patients for dexamethasone. The median time to first lenalidomide dose reduction was 1.9 months. TEAEs led to dose interruption in 18 patients (69.2%) for lenalidomide and 14 patients (53.8%) for dexamethasone. Four patients (15.4%) discontinued the study treatment because of the following TEAEs: increased transaminase level; interstitial lung disease; toxic skin eruption; and decreased appetite, nausea and vomiting (all single cases).
Table 4.
Study drug treatment and dosing
| Lenalidomide | Dexamethasone | |
|---|---|---|
| Duration of treatment, median (range), months | 13.8 (0.2–20.6) | |
| Cumulative dose, median (range), † mg | 3057.5 (25–9075) | 1058.0 (40–2760) |
| Average daily dose, median (range), ‡ mg/day | 11.9 (8.2–25.0) | 20.0 (12.9–40.0) |
| Dose intensity, median (range), § mg/week | 51.7 (14.0–131.3) | 19.7 (6.6–40.0) |
| Relative dose intensity, median (range) ¶ | 0.87 (0.2–1.3) | 0.93 (0.3–1.0) |
†Cumulative dose is total dose taken across the treatment period. ‡Average daily dose is calculated as cumulative dose divided by dose exposure (in days). §Dose intensity is calculated as cumulative dose divided by treatment duration for lenalidomide (in weeks). ¶Relative dose intensity is calculated as dose intensity divided by planned dose intensity.
Discussion
In this Japanese cohort of patients with NDMM, continuous treatment with Rd achieved an ORR of 87.5%, including a CR + VGPR rate of 29.2%. Responses were rapid (median TTR was 1.97 months), and the median DOR and PFS have not been reached. Interim OS data show only three deaths (during the follow‐up phase) at the data cutoff. Despite differences in the MM‐025 trial in terms of patient number and population, these findings are consistent with previously published results from the FIRST trial, where the continuous Rd 4‐year OS was 59% and the median PFS was 25.5 months.15 In the FIRST trial, Rd continuous achieved an ORR of 75% and a CR + VGPR rate of 43.6%.17 Median PFS was significantly longer with Rd continuous (25.5 months) compared with MPT (21.2 months; HR 0.72; P < 0.001) or Rd given form 18 cycles (20.7 months; HR 0.70; P < 0.001).17 Findings from the FIRST trial Asian subpopulation were similar to those from the MM‐025 study in terms of ORR (the Rd continuous arm showed a response rate of 78%17 vs 87.5%, respectively); however, the CR + VGPR rate was lower in the MM‐025 study (29.5% vs 50%) (Celgene data on file). This difference in depth of response between the two trials may be due to the shorter follow‐up for the MM‐025 study compared with the FIRST trial (median follow‐up: 14.2 vs 37.0 months, respectively).17
As in the FIRST trial, the starting dose of lenalidomide in the MM‐025 study was adjusted according to patient renal function. Compared with the Rd continuous arm in the FIRST trial, there were fewer patients with normal/mild renal impairment (CrCl ≥60 mL/min) in the MM‐025 study (MM‐025, 38.5% and FIRST, 50.1%); similar numbers of patients had severe renal impairment in both studies (MM‐025, 7.7% and FIRST, 8.4%). Overall, the majority of patients in the MM‐025 study had moderate renal impairment, which led to >60% of MM‐025 patients receiving a lenalidomide dose of either 10 mg/day or 15 mg every other day, on days 1–21 of each 28‐day cycle. In the FIRST trial, just over 50% of patients had normal/mild renal impairment, which meant that a higher percentage of patients received a lenalidomide dose of 25 mg/day compared with the MM‐025 study. Despite this difference in dosing, the ORR from the MM‐025 trial was comparable with that in the Rd continuous arm of the FIRST trial, and the Asian subpopulation of the FIRST trial (87.5% vs 75%17 and 78%,18 respectively). Of the 10 patients in the MM‐025 study who initially received the full 25 mg/day dose of lenalidomide, four patients remained at this dosing level at the data cutoff. After Rd continuous treatment, the majority of patients (78.3%; 18 of 23) had similar renal function to the baseline. The adjustments in lenalidomide dose were based on those used in the US Revlimid® prescribing information; further research and patient monitoring will be necessary to determine whether these doses are appropriate for Japanese patients with NDMM.
Median relative dose intensity of lenalidomide was lower for patients aged >75 years compared with all patients (0.72 vs 0.8). This was not unexpected as elderly patients are more likely to have decreased renal function, and generally lower levels of physiological function. However, this does indicate that elderly patients should be carefully monitored when treated with lenalidomide.22
The safety profile of Rd continuous in the MM‐025 study was consistent with the known safety profile of lenalidomide. Rd continuous was well tolerated despite the fact that the median age of patients was 75 years (46.2% of patients were aged >75 years), 19.2% had ISS stage III disease and 7.7% had severe renal impairment (CrCl <30 mL/min) not requiring dialysis. The most common grade 3–4 AE were hematological, including neutropenia, anemia and thrombocytopenia, and were generally manageable with dose modifications and standard interventions. The frequency of hematological AE was similar to that reported in the continuous Rd arm of the FIRST trial, where the most common grade 3–4 AE were infection (29%), neutropenia (28%), anemia (18%) and cardiac disorder (12%).17 In the present study, 11.5% (n = 3) of patients developed grade 3–4 rash compared with 6% in the FIRST trial. Grade 3–4 infection developed in only 7.7% (n = 2) of patients in the present study, which is lower than the rate of 29% reported in the FIRST trial; however, the incidence of grade 3–4 pneumonia (7.7%) was similar to that found in the FIRST trial (8.0%). There were no reports of SPM, and no deaths were attributed to TEAEs. There was one incident of grade 2 DVT, contrasting with the FIRST trial, in which 8% of continuous Rd patients experienced grade 3–4 DVT.17 This is not an unexpected finding, given the lower prevalence of venous thromboembolism in Asian patients compared with white patients.23 The efficacy and safety results from this study suggest that Rd continuous has a favorable benefit–risk profile in Japanese patients with NDMM. One limitation of the MM‐025 study, given the increasing importance of cytogenetics in predicting patient outcomes from treatment,24 is that cytogenetic information was not collected from the patients.
Current treatment options for transplant‐ineligible patients consist primarily of melphalan‐containing regimens (MPV or MPT).1, 2, 7 Rd continuous was shown to be superior to fixed‐duration MPT in the FIRST trial, and results in the MPT control group were consistent with outcomes achieved with MPT in previous studies.6, 17 No trials have directly compared Rd continuous with MPV. In the VISTA trial, MPV produced a response rate of 71%, and a CR + VGPR rate of 41%, including a high CR rate of 33% (International Uniform Response Criteria analysis).3 Median TTP was 24.0 months and, with extended follow‐up, median OS was 56.4 months.5 Notably, as in the FIRST trial, <10% of patients in the VISTA trial were Asian.3 Differences in terms of patient demographics and disease characteristics preclude meaningful comparisons between the VISTA trial and trials of Rd continuous. Nevertheless, there is clearly a need for less‐intensive therapies for older and frailer patients, who may have difficulty tolerating melphalan‐based triplet regimens. Myeloma is generally a disease of the elderly, and the presence of advanced age, frailty and comorbidity can increase the risk of AE and treatment modifications that may compromise efficacy.25, 26, 27 The regimen of Rd, which can be given continuously with a manageable and predictable safety profile, fulfills an unmet need in the treatment of elderly patients with MM and lends itself to the development of risk‐adapted strategies for the management of MM.10
In conclusion, the MM‐025 study demonstrated that Rd continuous was effective and well tolerated over the study period in Japanese patients with NDMM who are ineligible for SCT. Response rates and safety data were consistent with what has been reported in the pivotal FIRST trial and other studies evaluating the Rd continuous regimen. Evidence regarding the long‐term treatment of Japanese NDMM patients with lenalidomide is extremely limited; therefore, longer follow‐up of safety and efficacy data will be needed, along with studies involving larger patient numbers. Further research into the most appropriate dosing for patients with renal impairment is also required. However, the MM‐025 data presented here support the potential role of Rd continuous as a first‐line treatment for Japanese patients with NDMM who are ineligible for SCT.
Disclosure Statement
M.T. received research funding from Chugai Pharmaceutical, Bristol‐Myers Squibb and Kyowa Hakko Kirin. K.I. received research funding from Celgene K.K., GSK, Janssen Pharmaceutical, Kyowa Hakko Kirin and Takeda Pharmaceutical, and received honoraria from Celgene K.K., Chugai Pharmaceutical, Kyowa Hakko Kirin and Takeda Pharmaceutical. K.M. received honoraria from Celgene K.K. Y. Ogawa received honoraria from Janssen Pharmaceutical. M.M. received honoraria from Celgene K.K., Janssen Pharmaceutical and Ono Pharmaceutical. S.I. received honoraria from Celgene K.K., Ono Pharmaceutical, Takeda Pharmaceutical, Bristol Myers Squib and Janssen Pharmaceutical, and received research funding from Celgene K.K., Chugai Pharmaceutical, Eli Lilly Japan K.K., Janssen Pharmaceutical, Kyowa Hakko Kirin, Astellas, Toyama Chemical Co., Ltd. and Ono Pharmaceutical. I.M. received honoraria from Celgene K.K., and received research funding from Celgene K.K. Y. Ogaki is a Celgene K.K. employee with stock ownership. S.M. is a Celgene K.K. employee with stock ownership. V.H. is a Celgene Corporation employee with stock ownership. A.E.‐H. is a Celgene Corporation employee with stock ownership. T.C. received honoraria from Bristol‐Myers Squibb, Celgene K.K., Janssen Pharmaceutical, Novartis Pharma K.K. and Takeda Pharmaceutical. The remaining authors have no conflict of interest to declare. The authors acknowledge financial support for this study from Celgene K.K. Lenalidomide was provided by Celgene Corporation. The authors received editorial assistance from Keisha Peters, MSc of Excerpta Medica, sponsored by Celgene Corporation. All authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication.
Acknowledgments
The authors would like to thank the patients, nurses and study personnel who were involved in this clinical trial.
Cancer Sci 107 (2016) 653–658
Funding Information The authors received financial support for this study from Celgene K.K.
ClinicalTrials.gov Identifier: NCT01698801.
References
- 1. Japanese Society of Hematology . Guideline for multiple myeloma. Rinsho Ketsueki 2013; 54(10): 1850–5. (Article in Japanese). [PubMed] [Google Scholar]
- 2. Moreau P, San Miguel J, Ludwig H et al Multiple myeloma: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2013; 24(Suppl 6): vi133–7. [DOI] [PubMed] [Google Scholar]
- 3. San Miguel JF, Schlag R, Khuageva NK et al Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008; 359: 906–17. [DOI] [PubMed] [Google Scholar]
- 4. Mateos MV, Richardson PG, Schlag R et al Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow‐up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol 2010; 28: 2259–66. [DOI] [PubMed] [Google Scholar]
- 5. San Miguel JF, Schlag R, Khuageva NK et al Persistent overall survival benefit and no increased risk of second malignancies with bortezomib–melphalan–prednisone versus melphalan–prednisone in patients with previously untreated multiple myeloma. J Clin Oncol 2013; 31: 448–55. [DOI] [PubMed] [Google Scholar]
- 6. Fayers PM, Palumbo A, Hulin C et al Thalidomide for previously untreated elderly patients with multiple myeloma: meta‐analysis of 1685 individual patient data from 6 randomized clinical trials. Blood 2011; 118: 1239–47. [DOI] [PubMed] [Google Scholar]
- 7. National Comprehensive Cancer Network . NCCN Clinical Practice Guidelines: Multiple myeloma. Version 2.2015. [Cited 9 September 2015.] Available from URL: http://www.nccn.org.
- 8. Palumbo A, Mateos MV, Bringhen S, San Miguel JF. Practical management of adverse events in multiple myeloma: can therapy be attenuated in older patients? Blood Rev 2011; 25: 181–91. [DOI] [PubMed] [Google Scholar]
- 9. Palumbo A, Bringhen S, Kumar SK et al Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: a meta‐analysis of individual patient data. Lancet Oncol 2014; 15: 333–42. [DOI] [PubMed] [Google Scholar]
- 10. Gertz MA, Lacy MQ, Lust JA, Greipp PR, Witzig TE, Kyle RA. Long‐term risk of myelodysplasia in melphalan‐treated patients with immunoglobulin light‐chain amyloidosis. Haematologica 2008; 93: 1402–6. [DOI] [PubMed] [Google Scholar]
- 11. Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med 2004; 351: 1860–73. [DOI] [PubMed] [Google Scholar]
- 12. Mikhael JR, Dingli D, Roy V et al Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk‐Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc 2013; 88: 360–76. [DOI] [PubMed] [Google Scholar]
- 13. Palumbo A, Facon T, Sonneveld P et al Thalidomide for treatment of multiple myeloma: 10 years later. Blood 2008; 111: 3968–77. [DOI] [PubMed] [Google Scholar]
- 14. Dimopoulos MA, Spencer A, Attal M et al Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 2007; 357: 2123–32. [DOI] [PubMed] [Google Scholar]
- 15. Weber DM, Chen C, Niesvizky R et al Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med 2007; 357: 2133–42. [DOI] [PubMed] [Google Scholar]
- 16. Rajkumar SV, Jacobus S, Callander NS et al Lenalidomide plus high‐dose dexamethasone versus lenalidomide plus low‐dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open‐label randomised controlled trial. Lancet Oncol 2010; 11: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benboubker L, Dimopoulos MA, Dispenzieri A et al Lenalidomide and dexamethasone in transplant‐ineligible patients with myeloma. N Engl J Med 2014; 371: 906–17. [DOI] [PubMed] [Google Scholar]
- 18. Lu J, Lee JH, Huang S‐Y et al Continuous treatment with lenalidomide and low‐dose dexamethasone in transplant‐ineligible patients with newly diagnosed multiple myeloma in Asia: subanalysis of the First Trial [abstract]. Blood 2015; 126: Abstract 4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iida S, Chou T, Okamoto S et al Lenalidomide plus dexamethasone treatment in Japanese patients with relapsed/refractory multiple myeloma. Int J Hematol 2010; 92: 118–26. [DOI] [PubMed] [Google Scholar]
- 20. Chen N, Kasserra C, Reyes J, Liu L, Lau H. Single‐dose pharmacokinetics of lenalidomide in healthy volunteers: dose proportionality, food effect, and racial sensitivity. Cancer Chemother Pharmacol 2012; 70: 717–25. [DOI] [PubMed] [Google Scholar]
- 21. Durie BG, Harousseau JL, Miguel JS et al International uniform response criteria for multiple myeloma. Leukemia 2006; 20: 1467–73. [DOI] [PubMed] [Google Scholar]
- 22. Revlimid® prescribing information. [Cited 27 December 2015.] US available from URL: http://www.revlimid.com/wp-content/uploads/full-prescribing-information.pdf. Japan. [Cited 27 December 2015] available from URL: http://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/380809_4291024M1024_1_21. (Article in Japanese.)
- 23. White RH. The epidemiology of venous thromboembolism. Circulation 2003; 107(23 Suppl 1): I4–8. [DOI] [PubMed] [Google Scholar]
- 24. Bergsagel PL, Mateos MV, Gutierrez NC, Rajkumar SV, San Miguel JF. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high‐risk multiple myeloma. Blood 2013; 121: 884–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Palumbo A, Hajek R, Delforge M et al Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med 2012; 366: 1759–69. [DOI] [PubMed] [Google Scholar]
- 26. Larocca A, Bringhen S, Evangelista A et al A simple score, based on geriatric assessment, improves prediction of survival, and risk of serious adverse events in elderly newly diagnosed multiple myeloma patients [abstract]. Blood 2013; 122: Abstract 687. [Google Scholar]
- 27. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011; 364: 1046–60. [DOI] [PubMed] [Google Scholar]