

Abstract
TAS‐102, a novel oral antitumor agent, consists of trifluridine and tipiracil hydrochloride (molar ratio, 1:0.5). We investigated the effects of food on trifluridine and tipiracil hydrochloride. The efficacy and safety of TAS‐102 were evaluated in patients with advanced solid tumors. We analyzed drug pharmacokinetics using a randomized, single‐dose, two‐treatment (fed versus fasting), two‐period, two‐sequence cross‐over design, followed by repeated administration. Patients were given single doses of TAS‐102 (35 mg/m2) in the pharmacokinetic phase and received twice‐daily doses of TAS‐102 in 28‐day cycles in the repeated administration phase for evaluating efficacy and safety. Food showed no effect on the area under the curve from 0 to 12 h or 0 h–infinity values of trifluridine following administration of TAS‐102 under fasting and fed conditions, whereas those of tipiracil hydrochloride decreased by approximately 40%. Maximum concentrations of both drugs decreased by approximately 40%, indicating that food influenced the absorption and bioavailability of trifluridine and tipiracil hydrochloride, respectively. During the repeated administration, stable disease was observed in nine patients with rectal, small‐cell lung, breast, thymic, duodenal, and prostate cancers. Major adverse events were neutropenia, leukopenia, anemia, and nausea. Postprandial administration was optimal for TAS‐102 because trifluridine's area under the curve was not changed by food, indicating that its clinical efficacy would not be affected. Additionally, postprandial administration was reasonable because the maximum concentration of trifluridine decreased in neutrophils, which correlated with previous studies. These results suggest that TAS‐102 would be an effective treatment for small‐cell lung, thymic, and colorectal cancers. This trial is registered with the Japan Pharmaceutical Information Center (no. JapicCTI‐111482).
Keywords: Effect of food, pharmacokinetics, TAS‐102, tipiracil hydrochloride, trifluridine
Trifluridine (FTD) showed excellent anticancer activity in non‐clinical studies.1, 2, 3 Trifluridine suppresses cancer cell growth by incorporation into DNA and inhibition of thymidylate synthase.4, 5 Trifluridine incorporation into DNA is the primary mechanism underlying its antitumor activity because inhibition of thymidylate synthase was rapidly abrogated following the removal of FTD from the culture medium of cancer cells in an in vitro study.6 Plasma FTD concentration decreased rapidly with an extremely short half‐life (approximately 12 min) following i.v. administration in humans and, subsequently, plasma 5‐trifluoromethyluracil (FTY) and 5‐carboxyuracil (5‐CU) concentrations increased.7 Trifluridine is degraded by thymidine phosphorylase (TPase) to the inactive FTY and 5‐CU.
Tipiracil hydrochloride (TPI) is a competitive inhibitor of TPase and inhibits FTD degradation in extracts of rodent, monkey, and human tissue.8 Monkey studies have shown that plasma FTD concentrations were considerably increased after oral administration of FTD with TPI compared to administration of FTD alone, indicating that the oral bioavailability of FTD is low in the absence of TPI. The low bioavailability of FTD is attributable to the first‐pass effect mediated mainly by TPase.9 Therefore, TPI increases FTD concentration by TPase inhibition, and this discovery led to the development of the novel combination drug TAS‐102.8
In a phase 1 clinical study in Japanese patients with solid tumors, TAS‐102 (containing FTD at a dose of 15–35 mg/m2) was given within 1 h after morning and evening meals on days 1–5 and 8–12, followed by a 14‐day recovery period in 28‐day cycles. Plasma concentrations were measured following the morning dose on day 1 and 12. After FTD had reached the maximum drug concentration (C max) on day 1 and 12 of repeated TAS‐102 administration, it decreased with an elimination half‐life of 1.17–1.88 and 1.52–2.44 h, respectively. The concentration of FTD in plasma increased 2.6 times on day 12 (compared with day 1).10 In contrast, the pharmacokinetic parameters of TPI on day 12 showed little change from those on day 1, indicating a lack of TPI accumulation. The concentration of FTD and TPI on day 1 increased dose‐dependently. The common grade 3 and 4 adverse events were hematological toxicities. Dose‐dependency was observed in the severity of hematological toxicity following repeated administration of TAS‐102. Significant negative correlations were observed between the reduction in the neutrophil count and each of the C max and area under the curve at 0–10 h (AUC0–10) of FTD after repeated administration of TAS‐102. No significant safety issues were noted when TAS‐102 was continuously administered at a dose of 35 mg/m2 b.i.d.10 Based on these results, the 70‐mg/m2/day dosing regimen (35 mg/m2/dose) was selected as the recommended phase 2 regimen and dosage for Japanese patients with solid tumors.
As TAS‐102 is given orally, assessment of the effect of food on the pharmacokinetics of its constituents is required. Therefore, the primary objective of this study was to assess the effect of food on the pharmacokinetics of FTD and TPI after a single oral administration of TAS‐102. The secondary objective was to investigate the efficacy and safety of repeated TAS‐102 administration. A high‐fat, high‐calorie meal was used to evaluate the maximal effect of food intake on pharmacokinetics. The nutritional value of the meal was adjusted base on body weight of Japanese patients according to the US FDA standard guidance.
Material and Methods
Patients
The major inclusion criteria for the study were: (i) provision of written consent to participate; (ii) a solid tumor confirmed histologically or cytologically; (iii) unresponsiveness to the standard therapy or unavailability of suitable therapy; (iv) acceptance of oral administration of the study drug and the high‐fat, high‐calorie meal specified for the study that contained 110, 180, and 360–430 kcal of protein, carbohydrates, and fat, respectively (adjustment by patients' body weight in accordance with the FDA standard guidance); (v) aged 20 years or older at the time of informed consent; (vi) measurement of the bone marrow, liver, and kidney function parameters within 15 days prior to enrolment (hemoglobin concentration, ≥8.0 g/dL; neutrophil count, ≥1500/mm3; platelet count, ≥7.5 × 104/mm3; serum total bilirubin, ≤1.5 mg/dL; aspartate aminotransferase, glutamate oxaloacetate transaminase/alanine aminotransferase, glutamate pyruvate transaminase, ≤2.5 times the upper limit of the institutional normal range; and serum creatinine, ≤1.5 mg/dL); and (vii) performance status of 0–1.
The major exclusion criteria were: (i) co‐existing gastric cancer or post‐gastrectomy; (ii) anticancer or extensive radiation therapy during the 22 or 43 days, respectively, prior to TAS‐102 administration; (iii) presence or suspicion of extensive bone metastasis; (iv) serious complications; and (v) continuous systemic steroid administration.
Ethics and good clinical practice
This study was carried out at the National Cancer Center Hospital East (Kashiwa, Japan) between May 2011 and December 2013. The study was carried out after review and approval by the Institutional Review Board of the National Cancer Center (Tokyo, Japan). The protocol was prepared based on the ethical principles delineated in the Declaration of Helsinki, the Pharmaceutical Affairs Law, the Ordinance for Enforcement of the Pharmaceutical Affairs Act, Ordinances Concerning Good Clinical Practice (GCP), Implementation of the GCP, Operation of Implementation Standards for Clinical Drug Studies, and FDA standards. The study was carried out in compliance with the approved protocol and GCP. The investigator or co‐investigator provided an explanation of the study details to the patients before enrolment based on the informed consent document. The patients were given sufficient time to consider participating in the study and voluntary written consent was obtained.
Methodology
The subjects were first enrolled in the pharmacokinetic phase to investigate the effect of food, and then in the repeated administration phase. In addition, there was no washout period prior to the repeated administration phase (Japan Pharmaceutical Information Center registration no. JapicCTI‐111482).
Pharmacokinetic phase
The effects of food on the pharmacokinetics of FTD and TPI, the active constituents of TAS‐102, were investigated using a randomized cross‐over design. Sixteen patients were randomly assigned to two groups, Group A and Group B, with eight patients each. The investigational drug was TAS‐102 tablets (15 or 20 mg) containing FTD (15 or 20 mg, respectively) and TPI (7.065 or 9.42 mg, respectively). TAS‐102 was given orally once daily at 35 mg/m2 (dosage of FTD) to patients in Group A (first under fed and then fasting conditions) and Group B (first under fasting and then fed conditions). This was followed by a treatment‐free period of at least 4 days between administration under fed and fasting conditions. For the drug administration under fasting conditions, patients fasted for 10 and 4 h before and after TAS‐102 administration, respectively. For administration under fed conditions, patients fasted for 10 h before administration and completely ingested a meal within 0.5 h. The study drug was ingested with 240 mL water and for 1 h before and after TAS‐102 administrations under both conditions, ingestion of water was prohibited except for that taken with the medication. Food intake was prohibited for 4 h after TAS‐102 administration.
Four milliliters of blood was collected at the predetermined time points (within 0.5 h before and 0.25, 0.5, 1, 2, 4, 6, 8, 10, and 12 h after TAS‐102 administration under fasting or fed conditions) and the plasma concentrations of FTD, major FTD metabolites FTY and 5‐CU, and TPI were determined using validated liquid chromatography MS/MS. FTD, FTY, and TPI were measured at Toray Research Center, Inc. (Kanagawa, Japan); 5‐CU was measured at the Tokushima Research Center of Taiho Pharmaceutical Co., Ltd. (Tokushima, Japan).
Repeated administration phase
Following a 28‐day cycle (5 and 2 days on and off, respectively, repeated for 2 weeks, followed by a 14‐day recovery), TAS‐102 containing 35 mg/m2 FTD was given orally twice daily (after morning and evening meals). TAS‐102 administration was repeated until completion of the pharmacokinetic study unless the patient met any of the discontinuation criteria for repeated administration: (i) obvious growth of tumor or clinical progression; (ii) an adverse event that made continued administration difficult; (iii) a washout period exceeding 30 consecutive days; and (iv) difficulty continuing visits to the institution. The efficacy and safety of TAS‐102 were evaluated for each patient. The efficacy endpoints were the best overall response and progression‐free survival (PFS) based on the Response Evaluation Criteria in Solid Tumors version 1.1 guideline. In the study, image evaluation was carried out once every 4–6 weeks. Adverse events that occurred during the period from the start of the study to completion of the post‐dose observation period (30 days after final administration of the study drug) or during implementation of the next treatment (whichever was earlier), were recorded according to the Common Terminology Criteria for Adverse Events version 4.0.
Statistical analyses
In this study, the sample size was determined based on the FDA's guidance that specifies the minimum number of patients required for evaluation of food effect as 12, instead of the statistical setting of the sample size. Each of the two groups will consist of eight patients, that is, six patients plus two more patients to be considered for possible dropouts.
Pharmacokinetic phase
The pharmacokinetic parameters were calculated from the plasma concentrations. The C max and time to achieve C max (t max) were based on observed maximum concentrations, whereas AUC0–12, AUC at 0 h–infinity (AUC0–inf), elimination half‐life, oral clearance, and apparent volume of distribution were calculated using non‐compartmental analysis with WinNonlin pharmacokinetic analysis software (Pharsight, Certara L.P., Princeton, NJ, USA).
The statistical analysis was carried out using ANOVA with the common logarithms of the C max, AUC0–12, and AUC0–inf of FTD and TPI. The geometric mean ratios of the parameters under fed to fasting conditions and the 90% confidence intervals (CIs) of the geometric mean ratios were calculated using SAS statistical analysis software (SAS Institute Inc., Cary, NC, USA).
Repeated administration period
Regarding the efficacy of TAS‐102, the survival curves were estimated using the Kaplan–Meier method. Median survival was calculated based on the PFS of all 16 patients and the PFS of cancers afflicting three or more patients. Progression‐free survival was defined as the period from the registration day in the repeated administration phase up to the assessment day of disease progression or the day of death from all causes (whichever was earlier). Stable disease (SD) was confirmed if the criteria were met ≥6 weeks after the enrolment day, which was defined as the commencement date for the repeated administration phase. The actual dose intensity and relative dose intensity were calculated for each patient, and the dose intensity of TAS‐102 was determined.
Results
Patients
Sixteen patients (eight each in Groups A and B) were enrolled from May through December 2011. All enrolled patients completed the pharmacokinetics and repeated administration phases. The baseline patient characteristics for the pharmacokinetic phase are presented in Table 1. There were two patients and one patient who took proton pump inhibitors during the pharmacokinetic phase in Group A and Group B, respectively.
Table 1.
Baseline characteristics of patients with advanced solid tumors treated with TAS‐102 under fasting and fed conditions
| n | % | ||
|---|---|---|---|
| Gender | Male | 9 | 56.3 |
| Female | 7 | 43.8 | |
| Age, years | Mean ± SD | 58.5 ± 12.8 | |
| Median | 62.0 | ||
| Range (min–max) | (37–73) | ||
| <65 | 10 | 62.5 | |
| ≥65 | 6 | 37.5 | |
| Height, cm | Mean ± SD | 160.25 ± 8.31 | |
| Median | 159.95 | ||
| Range (min–max) | (148.0–175.3) | ||
| Weight, kg | Mean ± SD | 58.49 ± 9.36 | |
| Median | 55.55 | ||
| Range (min–max) | (46.4–74.2) | ||
| BSA, m2 | Mean ± SD | 1.5631 ± 0.1495 | |
| Median | 1.5555 | ||
| Range (min–max) | (1.357–1.827) | ||
| 1.250–1.500 | 7 | 43.8 | |
| >1.500 | 9 | 56.3 | |
| Primary lesion | Rectum | 5 | 31.3 |
| Lung | 5 | 31.3 | |
| Breast | 2 | 12.5 | |
| Thymus | 1 | 6.3 | |
| Duodenum | 1 | 6.3 | |
| Prostate | 1 | 6.3 | |
| Unknown primary | 1 | 6.3 | |
| Histological type | Well‐differentiated adenocarcinoma | 2 | 12.5 |
| Moderately differentiated adenocarcinoma | 4 | 25.0 | |
| Poorly differentiated adenocarcinoma | 3 | 18.8 | |
| Squamous cell carcinoma | 2 | 12.5 | |
| Infiltrating duct carcinoma | 2 | 12.5 | |
| Well‐differentiated neuroendocrine carcinoma | 1 | 6.3 | |
| Small‐cell carcinoma | 2 | 12.5 | |
| Concomitant medication with TAS‐102 | Proton pump inhibitors | 3 | 18.8 |
Analysis set: evaluable patients in the pharmacokinetic phase of the study. BSA, body surface area.
All 16 patients were suitable for evaluation of the pharmacokinetics of FTD and TPI, but two in Group B violated the protocol‐specified prohibition of food intake during the 4‐h fasting period after TAS‐102 administration. Therefore, the pharmacokinetic analysis was carried out on data of 14 patients.
Pharmacokinetics of FTD and TPI
Figure 1 presents the plasma concentration–time profiles of FTD and TPI in 14 patients included in the analysis following administration of TAS‐102 under fasting and fed conditions. The mean plasma concentration–time profiles showed that the C max of FTD was slightly lower under fed conditions than it was under fasting conditions. In contrast, the mean FTD plasma concentrations at time points after C max were slightly higher under fed conditions than they were under fasting conditions. The mean TPI plasma concentration remained lower under fed conditions than it was under fasting conditions. The geometric mean ratios (fed/fasting) and 90% CIs of C max, AUC0–12, and AUC0–inf are presented in Tables 2 and 3. The point estimates of the geometric mean ratios of the C max, AUC0–12, and AUC0–inf of FTD were 0.6074, 0.9560, and 0.9559, respectively (Table 2). The 90% CI of the C max was outside the bioequivalence range (0.80–1.25), but those of the AUC0–12, AUC0–inf, and AUC0–t were within the bioequivalence limits. The difference in the C max of FTD was statistically significant (P = 0.0005) but the differences in FTD's AUCs were not. These results indicate that food intake reduced the C max of FTD by approximately 40% but did not affect its AUC0–12 or AUC0–inf. The point estimates of the geometric mean ratios of the C max, AUC0–12, and AUC0–inf of TPI were 0.5578, 0.5526, and 0.5581, respectively. The 90% CIs of the C max and AUC of TPI were outside the bioequivalence range (0.80–1.25). The differences in the C max, AUC0–12, and AUC0–inf of TPI were statistically significant (<0.0001 for every parameter). The t max values of FTD and TPI under fed and fasting conditions were similar. These results showed that food intake reduced the C max, AUC0–12, and AUC0–inf of TPI by approximately 40%.
Figure 1.
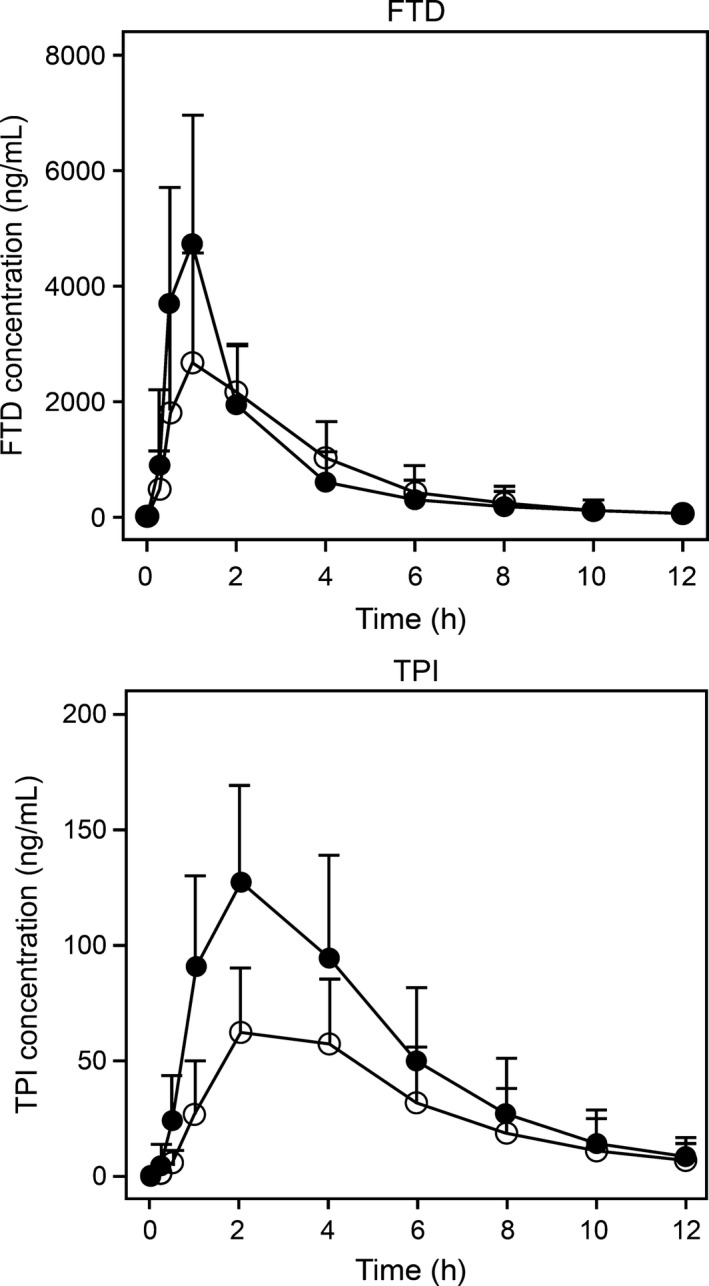
Plasma concentration–time profiles (Mean ± SD) of trifluridine (FTD, top panel) and tipiracil hydrochloride (TPI, bottom panel) in 14 patients with solid tumors following treatment with TAS‐102 at 35 mg/m2 under fasting (Closed circles) and fed (open circles) conditions.
Table 2.
Effect of food on pharmacokinetics of trifluridine after oral administration of TAS‐102 in patients with solid tumors (n = 14)
| State | Statistic | C max, ng/mL | t max, h | AUC0–12, ng h/mL | AUC0–inf, ng h/mL | t 1/2, h | CL/F, L/h/kg | Vd/F, L/kg |
|---|---|---|---|---|---|---|---|---|
| Fasting | Mean ± SD | 5630 ± 1840 | 0.88 ± 0.42 | 10648 ± 5011 | 10943 ± 5581 | 2.13 ± 0.76 | 0.106 ± 0.056 | 0.310 ± 0.181 |
| Fed | Mean ± SD | 3510 ± 1380 | 1.32 ± 0.93 | 9840 ± 4247 | 10082 ± 4593 | 1.72 ± 0.58 | 0.115 ± 0.060 | 0.260 ± 0.102 |
| Fed/fasting | Geometric mean | 0.6074 | – | 0.9560 | 0.9559 | – | – | – |
| 90% CI | 0.5037–0.7323 | – | 0.8566–1.0670 | 0.8556–1.0680 | – | – | – | |
| P‐value† | 0.0005 | – | 0.4791 | 0.4825 | – | – | – |
Under fasting conditions, patients fasted for 10 and 4 h before and after TAS‐102 administration, respectively. Under fed conditions, patients fasted for 10 h before administration and completely ingested a meal within 0.5 h. The study drug was ingested with 240 mL water and for 1 h before and after TAS‐102 administrations under both conditions; ingestion of water was prohibited except for that taken with the medication. †Estimated by ANOVA. –, Not calculated. AUC0–12, area under the curve at 0–12 h; AUC0–inf, AUC at 0 h–infinity; CI, confidence interval; CL/F, oral clearance; C max, maximum plasma drug concentration; t max, time to achieve C max; t 1/2, elimination half‐life; Vd/F, apparent volume of distribution.
Table 3.
Effect of food on pharmacokinetics of tipiracil hydrochloride after oral administration of TAS‐102 in patients with solid tumors (n = 14)
| State | Statistic | C max, ng/mL | t max, h | AUC0–12, ng h/mL | AUC0–inf, ng h/mL | t 1/2, h | CL/F, L/h/kg | Vd/F, L/kg |
|---|---|---|---|---|---|---|---|---|
| Fasting | Mean ± SD | 135 ± 39 | 2.07 ± 0.92 | 647 ± 281 | 677 ± 309 | 2.19 ± 0.66 | 0.775 ± 0.320 | 2.42 ± 1.25 |
| Fed | Mean ± SD | 76.8 ± 26.3 | 2.79 ± 1.37 | 361 ± 160 | 384 ± 189 | 2.22 ± 0.45 | 1.34 ± 0.45 | 4.10 ± 1.24 |
| Fed/fasting | Geometric mean | 0.5578 | – | 0.5526 | 0.5581 | – | – | – |
| 90% CI | 0.4732–0.6576 | – | 0.4802–0.6358 | 0.4872–0.6392 | – | – | – | |
| P‐value† | <0.0001 | – | <0.0001 | <0.0001 | – | – | – |
Under fasting conditions, patients fasted for 10 and 4 h before and after TAS‐102 administration, respectively. Under fed conditions, patients fasted for 10 h before administration and completely ingested a meal within 0.5 h. The study drug was ingested with 240 mL water and for 1 h before and after TAS‐102 administrations under both conditions; ingestion of water was prohibited except for that taken with the medication. †Estimated by ANOVA. –, Not calculated. AUC0–12, area under the curve at 0–12 h; AUC0–inf, AUC at 0 h–infinity; CI, confidence interval; CL/F, oral clearance; C max, maximum plasma drug concentration; t max, time to achieve C max; t 1/2, elimination half‐life; Vd/F, apparent volume of distribution.
Table 4 presents the pharmacokinetic parameters of FTY and 5‐CU, the metabolites of FTD. The plasma concentration–time profiles of 5‐CU under fasting and fed conditions were similar. The C max of FTY was slightly higher under fasting than it was under fed conditions, but no significant difference was observed in AUC between fasting and fed conditions. There was no marked difference in parameters of 5‐CU between fasting and fed conditions.
Table 4.
Pharmacokinetic parameters of trifluridine metabolites 5‐trifluoromethyluracil (FTY) and 5‐carboxyuracil (5‐CU) after oral administration of TAS‐102 in patients with solid tumors (n = 14)
| State | Metabolite | C max, ng/mL | t max, h | AUC0–12, ng h/mL | AUC0–inf, ng h/mL | t 1/2, h |
|---|---|---|---|---|---|---|
| Fasting | FTY | 860 ± 207 | 1.43 ± 0.51 | 2900 ± 837 | 2972 ± 868 | 2.41 ± 0.61 |
| 5‐CU | 2.93 ± 1.69 | 4.43 ± 1.79 | 17 ± 8 | 32 ± 8† | 3.91 ± 0.60† | |
| Fed | FTY | 728 ± 186 | 1.96 ± 1.01 | 3011 ± 855 | 3121 ± 941 | 2.08 ± 0.69 |
| 5‐CU | 2.32 ± 0.57 | 4.86 ± 1.29 | 15 ± 7 | 29 ± 10‡ | 4.84 ± 1.18‡ |
Under fasting conditions, patients fasted for 10 and 4 h before and after TAS‐102 administration, respectively. Under fed conditions, patients fasted for 10 h before administration and completely ingested a meal within 0.5 h. The study drug was ingested with 240 mL water and for 1 h before and after TAS‐102 administrations under both conditions; ingestion of water was prohibited except for that taken with the medication. †n = 4. ‡n = 5. Data are shown as mean ± standard deviation. AUC0–12, area under the curve at 0–12 h; AUC0–inf, AUC at 0 h–infinity; C max, maximum plasma drug concentration; t max, time to achieve C max; t 1/2, elimination half‐life.
Efficacy and safety
Sixteen patients completed the pharmacokinetic and repeated administration phases of the study. One patient with non‐small‐cell lung cancer discontinued the treatment after the first dosing following the withdrawal of informed consent.
The median cycle of TAS‐102 treatment was 3.5, with medians of 32.5 total dosing days, 3115.0 mg total dose (range, 120–20100 mg), and 83.5 treatment period days. The mean actual dose intensity and relative dose intensity of TAS‐102 were 132.28 mg/m2/week and 77.65%, respectively.
No patient treated with TAS‐102 achieved a complete response or partial response, whereas SD was observed in nine patients. Stable disease was observed in three of five patients with rectal cancer, both patients with small‐cell lung cancer, one of two patients with breast cancer, and in the only patients with thymic, duodenal, and prostate cancers. Six patients were assessed as having progressive disease, while one patient with non‐small‐cell lung cancer was assessed as not evaluable. The individual PFS of each patient is shown in Figure 2. The median PFS of the 16 patients was 3.7 months. For each cancer subtype with three or more patients, the median PFS was 7.1 months for rectal cancer (n = 5) and 1.1 months for non‐small‐cell lung cancer (n = 3).
Figure 2.
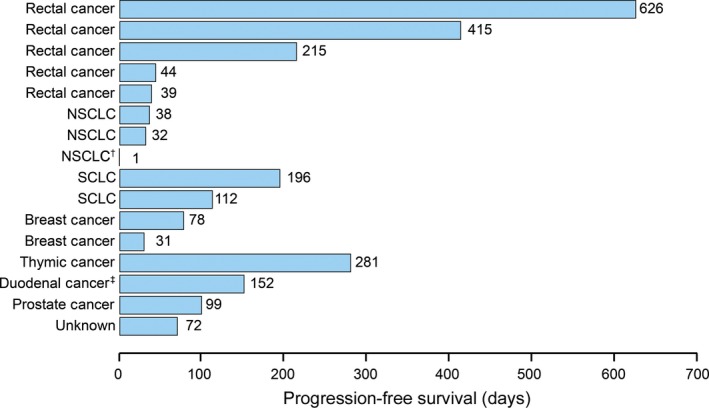
Progression‐free survival of 16 patients with advanced solid tumors following treatment with single doses of TAS‐102 (35 mg/m2) (pharmacokinetic phase) and twice‐daily doses of TAS‐102 in 28‐day cycles (repeated administration phase) under fed and fasting conditions. Stable disease was observed in nine patients. †Discontinue due to refusal of informed consent;. ‡Discontinue due to failure to meet conditions required to begin treatment. NSCLC, non‐small‐cell lung carcinoma; SCLC, small‐cell lung carcinoma.
Adverse events were reported in 16 patients. The incidence of major adverse events as tabulated using the System Organ Class and Preferred Term of the MedDRA/J (version 16.0) are shown in Table 5. Adverse events reported by Preferred Term that occurred in at least 20% of patients were decreased neutrophil and white blood cell counts in 10 patients each (62.5%), anemia and nausea in nine each (56.3%), fatigue in six (37.5%), decreased lymphocyte count and decreased appetite in five each (31.3%), and diarrhea in 4 (25.0%). No treatment‐related death was observed. Decreased appetite occurred as a serious adverse event in one patient, but the event was assessed as unrelated to the study drug, and the patient recovered from decreased appetite on day 6 after the onset. There were no adverse events that required discontinuation of the study treatment; however, decreased neutrophil counts required a reduction of the TAS‐102 dose in five patients (six events). Nine events of grade 4 decreased neutrophil count, as specified by Common Terminology Criteria for Adverse Events version 4.0, were noted in five patients. All occurrences of grade 4 decreased neutrophil count were resolved by prolongation of the washout period (n = 5/5 patients), and reduction of TAS‐102 dose (n = 4/5 patients) or administration of granulocyte colony‐stimulating factor or both (n = 3/5 patients). No febrile neutropenia was observed in any patient in the study.
Table 5.
Classification and incidence of adverse events of any grade observed in more than 10% of patients with solid tumors in the repeated administration phase of a study of TAS‐102 (n = 16)
| MedDRA version 16.0 System Organ Class/preferred term | G1 | G2 | G3 | G4 | Any grade† | G3–4 |
|---|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Adverse events | 2 (12.5) | 4 (25.0) | 5 (31.3) | 5 (31.3) | 16 (100.0) | 10 (62.5) |
| Blood and lymphatic system disorders | 2 (12.5) | 4 (25.0) | 3 (18.8) | 0 (0.0) | 9 (56.3) | 3 (18.8) |
| Anaemia | 2 (12.5) | 4 (25.0) | 3 (18.8) | 0 (0.0) | 9 (56.3) | 3 (18.8) |
| Gastrointestinal disorders | 9 (56.3) | 3 (18.8) | 0 (0.0) | 0 (0.0) | 12 (75.0) | 0 (0.0) |
| Abdominal pain upper | 3 (18.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (18.8) | 0 (0.0) |
| Constipation | 3 (18.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (18.8) | 0 (0.0) |
| Diarrhoea | 4 (25.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (25.0) | 0 (0.0) |
| Nausea | 7 (43.8) | 2 (12.5) | 0 (0.0) | 0 (0.0) | 9 (56.3) | 0 (0.0) |
| Stomatitis | 2 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (12.5) | 0 (0.0) |
| Vomiting | 2 (12.5) | 1 (6.3) | 0 (0.0) | 0 (0.0) | 3 (18.8) | 0 (0.0) |
| General disorders and administration site conditions | 7 (43.8) | 2 (12.5) | 0 (0.0) | 0 (0.0) | 9 (56.3) | 0 (0.0) |
| Fatigue | 4 (25.0) | 2 (12.5) | 0 (0.0) | 0 (0.0) | 6 (37.5) | 0 (0.0) |
| Influenza like illness | 3 (18.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (18.8) | 0 (0.0) |
| Malaise | 2 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (12.5) | 0 (0.0) |
| Investigations | 2 (12.5) | 1 (6.3) | 4 (25.0) | 5 (31.3) | 12 (75.0) | 9 (56.3) |
| Lymphocyte count decreased | 2 (12.5) | 1 (6.3) | 2 (12.5) | 0 (0.0) | 5 (31.3) | 2 (12.5) |
| Neutrophil count decreased | 0 (0.0) | 3 (18.8) | 2 (12.5) | 5 (31.3) | 10 (62.5) | 7 (43.8) |
| Platelet count decreased | 0 (0.0) | 1 (6.3) | 2 (12.5) | 0 (0.0) | 3 (18.8) | 2 (12.5) |
| White blood cell count decreased | 1 (6.3) | 2 (12.5) | 7 (43.8) | 0 (0.0) | 10 (62.5) | 7 (43.8) |
| Metabolism and nutrition disorders | 3 (18.8) | 1 (6.3) | 1 (6.3) | 0 (0.0) | 5 (31.3) | 1 (6.3) |
| Decreased appetite | 3 (18.8) | 1 (6.3) | 1 (6.3) | 0 (0.0) | 5 (31.3) | 1 (6.3) |
| Musculoskeletal and connective tissue disorders | 1 (6.3) | 1 (6.3) | 0 (0.0) | 0 (0.0) | 2 (12.5) | 0 (0.0) |
| Respiratory, thoracic, and mediastinal disorders | 7 (43.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 7 (43.8) | 0 (0.0) |
| Cough | 2 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (12.5) | 0 (0.0) |
| Laryngeal pain | 2 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (12.5) | 0 (0.0) |
| Rhinitis allergic | 2 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (12.5) | 0 (0.0) |
†Any grade (%) = (number of patients who experienced adverse events in each category [preferred term, system organ class, or any events])/(number of all patients) × 100. If a patient was reported to have the same toxicity more than once, then that patient was only counted once for the summary of that toxicity, using the most severe intensity.
During the study period, no clinically relevant abnormalities were noted in vital signs, electrocardiography, or laboratory values other than those related to hematological toxicities.
Discussion
We investigated the effects of food on the pharmacokinetics of FTD and TPI, the active ingredients of TAS‐102, after a single oral administration of TAS‐102 under fasting and fed conditions in patients with advanced solid tumors. Three patients took proton pump inhibitors during the pharmacokinetic phase; however, the effect of the inhibitors on the absorption of FTD and TPI was considered limited because the dissolution of the formulation is rapid (85% or more in 15 min) in a buffered media at pH 1.2–6.8. The absorption rate of FTD was not considered to have been changed by food intake although the FTD C max decreased by 40% in the fed condition because no differences were noted in the AUC between fasting and fed conditions. Similarly, the FTY C max was higher in fasting conditions while the FTY AUC was similar in both conditions, indicating that the pharmacokinetics of FTY reflected those of FTD. Therefore, the inhibitory activity of TPI was considered similar between fasting and fed conditions, although the TPI C max and AUC decreased in the fed condition. These results suggest that TPI substantially inhibited the metabolism of FTD8 even in the fed condition when patients were given a dose of 35 mg/m2. Based on these results, food would slow down the absorption of FTD with a similar absorption rate occurring in both conditions. In addition, the anticancer activity of TAS‐102 was not considered to be affected notably by food intake because the AUC of FTD, the anticancer component, was similar between fasting and fed conditions. However, postprandial administration, which led to a lower C max, was reasonable because a previous phase 1 clinical study of TAS‐102 in Japanese patients with solid tumors showed a significant correlation between decreased neutrophil counts and the C max and AUC of FTD.10 These results were accepted by the FDA for implementation of a large‐scale global phase 3 clinical study of TAS‐102.
After patients underwent pharmacokinetic evaluations in the pharmacokinetic phase, they then proceeded to the repeated administration phase in which they received 70 mg/m2 under fed conditions. This study did not provide data on repeated administration under fasting conditions, therefore, it is not within the scope of the results to discuss the effect of food in terms of the manifestation frequency or severity of adverse effects. Furthermore, in a phase 2 clinical study in Japanese patients with colorectal cancer, the dosing regimen of TAS‐102 that used as the same postprandial administration as that in our repeated administration phase resulted in a significantly lower risk of death in the TAS‐102‐treated group than in the placebo group, without significant safety issues.11 Based on these results, postprandial administration is considered acceptable and applicable to therapy with TAS‐102.
No complete response or partial response was observed in the 16 patients who received TAS‐102 in the repeated administration study, whereas SD was noted in nine patients. Stable disease was observed in three of five patients with rectal cancer, which was a ratio similar to that of patients showing SD in a phase 2 study of 112 Japanese patients with colon and rectal cancer (54%).11 The PFS times in two patients with small‐cell lung cancer were 6.4 and 3.7 months, which were similar to the PFS time of 3.5 months observed in the amrubicin group in a previous phase 2 study in patients with small‐cell lung cancer.12 The present study also enrolled one patient with thymic cancer who had previously received combination therapy with cisplatin and gemcitabine, as well as treatment with docetaxel. Following the administration of TAS‐102 on consecutive days, pleural dissemination was markedly improved on day 41 of treatment. The patient continued to show a marked improvement in pleural dissemination until the drug was discontinued due to exacerbation of hepatic metastasis (non‐target lesion) on day 280 after initiating daily administration (Fig. 3). Although this study enrolled a limited number of patients, and the results need to be investigated in a larger sample size, these findings suggest that TAS‐102 may exert its inhibitory effect on lung cancer (small‐cell) and thymic cancer, in addition to colorectal cancer.
Figure 3.
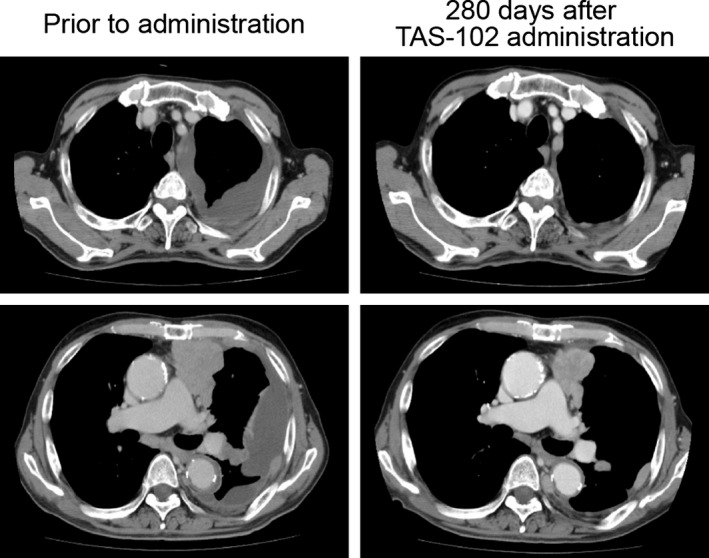
Computed tomographic images of pleural dissemination in a patient with thymic cancer treated with TAS‐102 at 35 mg/m2, twice daily. The patient showed marked improvement until the drug was discontinued due to exacerbation of hepatic metastasis (non‐target lesion) on day 280.
All 16 patients treated with TAS‐102 on consecutive days experienced adverse events. Frequently observed adverse events included hematological toxicities, gastrointestinal symptoms, and fatigue. Adverse events of grade 3 or higher mainly consisted of hematological toxicities. The observed adverse events were consistent with those previously reported in phase 1 and phase 2 studies.10, 11 No new adverse events, including those of grade 2 or below, were reported in the present study.
This study shows that postprandial administration after morning and evening meals is reasonable. Furthermore, the results suggest that TAS‐102 would be an effective therapeutic agent for small‐cell lung, thymic, and colorectal cancers.
Disclosure Statement
All authors received research funding from Taiho Pharmaceutical. Takashi Kojima received funding from Merck Serono; Hideaki Bando received funding from AstraZeneca; Takayuki Yoshino and Koichi Goto received honoraria from Taiho Pharmaceutical; Atsushi Ohtsu has an immediate family member who is affiliated with Celgene. This study was designed under the responsibility of Taiho Pharmaceutical in collaboration with all authors. The study was funded by Taiho Pharmaceutical. Study drug was provided by Taiho Pharmaceutical. Taiho Pharmaceutical collected and analyzed the data, and contributed the interpretation of the study. Data were recorded at the clinical center maintained by Taiho Pharmaceutical.
Acknowledgment
All authors received research funding from Taiho Pharmaceutical.
Cancer Sci 107 (2016) 659–665
Funding Information
Taiho Pharmaceutical.
References
- 1. Heidelberger C, Parsons DG, Remy DC. Syntheses of 5‐trifluoromethyluracil and 5‐trifluoromethyl‐2′‐deoxyuridine. J Med Chem 1963; 7: 1–5. [DOI] [PubMed] [Google Scholar]
- 2. Heidelberger C, Anderson SW. Fluorinated pyrimidines. XXI. The tumor‐inhibitory activity of 5‐trifluoromethyl‐2ifdeoxyuridine. Cancer Res 1964; 24: 1979–85. [PubMed] [Google Scholar]
- 3. Heidelberger C, Boohar J, Kampschroer B. Fluorinated pyrimidines: XXIV. In vivo metabolism of 5‐trifluoromethyluracil‐2‐C14 and 5‐trifluoromethyl‐2‐deoxyuridine‐2‐C14. Cancer Res 1965; 25: 377–81. [PubMed] [Google Scholar]
- 4. Fujiwara Y, Heidelberger C. Fluorinated pyrimidines VIII. The incorporation of 5‐trifluoromethyl‐2′‐deoxyuridine into the deoxyribonucleic acid of vaccinia virus. Mol Pharmacol 1970; 6: 281–91. [PubMed] [Google Scholar]
- 5. Fujiwara Y, Oki T, Heidelberger C. Fluorinated pyrimidines, VII. Effects of 5‐trifluoromethyl‐2′‐deoxyuridine on the synthesis of deoxyribonucleic acid of mammalian cells in culture. Mol Pharmacol 1970; 6: 273–80. [PubMed] [Google Scholar]
- 6. Tanaka N, Sakamoto K, Okabe H et al Repeated oral dosing of TAS‐102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep 2014; 32: 2319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dexter DL, Wolberg WH, Ansfield FJ, Helson L, Heidelberger C. The clinical pharmacology of 5‐trifluoromethyl‐2′‐deoxyuridine. Cancer Res 1972; 32: 247–53. [PubMed] [Google Scholar]
- 8. Fukushima M, Suzuki N, Emura T et al Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′‐deoxyribonucleosides. Biochem Pharmacol 2000; 59: 1227–36. [DOI] [PubMed] [Google Scholar]
- 9. Emura T, Suzuki N, Fujioka A, Ohshimo H, Fukushima M. Potentiation of the antitumor activity of alpha, alpha, alpha‐trifluorothymidine by the co‐administration of an inhibitor of thymidine phosphorylase at a suitable molar ratio in vivo. Int J Oncol 2005; 27: 449–55. [PubMed] [Google Scholar]
- 10. Doi T, Ohtsu A, Yoshino T et al Phase I study of TAS‐102 treatment in Japanese patients with advanced solid tumors. Br J Cancer 2012; 107: 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshino T, Mizunuma N, Yamazaki K et al TAS‐102 monotherapy for pretreated metastatic colorectal cancer: a double‐blind randomized, placebo‐controlled phase 2 trial. Lancet Oncol 2012; 13: 993–1001. [DOI] [PubMed] [Google Scholar]
- 12. Inoue A, Sugawara S, Yamazaki K et al Randomized phase II trial comparing amrubicin with topotecan in patients with previously treated small‐cell lung cancer: North Japan Lung Cancer Study Group Trial 0402. J Clin Oncol 2008; 26: 5401–6. [DOI] [PubMed] [Google Scholar]