

Abstract
B7‐H3 is highly overexpressed in a variety of human clinical tumors, and its expression is significantly associated with poor outcomes. In our study, we aimed to develop new antitumor mAbs by employing cancer cell immunization, and succeeded in generating a mouse anti‐human B7‐H3 antibody (M30) that shows antitumor activity. M30 was humanized (Hu‐M30), and an afucosylated Hu‐M30 (DS‐5573a) was also generated. To assess the potency of DS‐5573a as a therapeutic mAb, we characterized this mAb and evaluated its antitumor activity in vitro and in vivo. Flow cytometry analysis showed that B7‐H3 proteins were expressed on various types of cancer cell lines broadly, and DS‐5573a binds to IgC1 and IgC2 domains of human B7‐H3. Antibody‐dependent cellular cytotoxicity activity of DS‐5573a was drastically enhanced against medium to high B7‐H3‐expressing cancer cell lines MDA‐MB‐231 and NCI‐H322. DS‐5573a also induced high antibody‐dependent cellular cytotoxicity activity against low B7‐H3‐expressing cancer cell line COLO205, whereas Hu‐M30 induced little activity against it. In addition, DS‐5573a was found to be a novel anti‐B7‐H3 antibody which showed antibody‐dependent cellular phagocytosis activity. Furthermore, DS‐5573a showed dose‐dependent and significant antitumor efficacy (0.03–3 mg/kg) in MDA‐MB‐231‐bearing SCID mice (which have functional natural killer cells and macrophages), but little antitumor efficacy in NOG mice (which lack natural killer cells and have reduced macrophage function). These results suggest that antitumor activity of DS‐5573a is mediated by effector cells, and this mAb could be a promising antitumor therapy for patients with a wide range of B7‐H3‐expressing tumors.
Keywords: Antibody‐dependent cellular cytotoxicity, antibody‐dependent cellular phagocytosis, B7‐H3, DS‐5573a, therapeutic antibody
With approximately 14.1 million new cases and 8.2 million cancer‐related deaths in 2012,1 cancer figures among the leading causes of morbidity and mortality worldwide, and the number of cancer patients has been increasing steadily every year. Mabs have been a standard component of cancer therapy for over 15 years. At present, approximately 30 mAbs have been approved for use in clinical practice, and many more are currently being tested in clinical trials.2 Unmodified mAbs such as trastuzumab and rituximab, known as first‐generation mAbs with signal blocking or antibody‐dependent cellular cytotoxicity (ADCC) functions, have shown clinical benefits, but the responses are often limited to tumors with highly overexpressed antigens, and drug resistance is acquired as time proceeds.3, 4 To address these problems, next‐generation mAbs such as ADCC enhanced antibodies created by Fc engineering technologies have been developed. They have shown significant therapeutic advantages in preclinical and clinical studies.5, 6, 7, 8 However, almost all such antibodies under development are targeted for limited functional molecules, and there is still a strong need for new therapeutic mAbs against novel target antigens that have the potential to treat refractory cancer.
B7‐H3 is a member of the B7 family and 20–27% of its amino acids are identical to those of other members.9 Several clinical studies have reported that B7‐H3 is overexpressed in a broad spectrum of tumor tissues,10, 11, 12, 13, 14, 15 and higher B7‐H3 expression in tumor tissue is significantly associated with lymph node metastasis,13, 15 worse event‐free survival, 11, 12 , 14, 15 advanced pathological stage,10, 14 and decreased infiltration of T cells in the tumor.10, 13, 14 Preclinical studies in vitro have shown that knockdown of B7‐H3 expression reduced cell adhesion, migration, or invasion in hepatocellular, breast, and prostate cancer cells.15, 16, 17 Expression of B7‐H3 mRNA has been reported to occur in various normal tissues,9 whereas B7‐H3 protein expression is more limited.10, 15, 18 Based on these findings, B7‐H3 has the potential of being a promising target for safe cancer therapy.
In our study, we aimed to develop new antitumor mAbs by employing cancer cell immunization methods. Many antitumor mAbs were obtained, but we focused specifically on a mouse anti‐human B7‐H3 mAb (M30), because of the correlation between its expression and tumor progression, and the suitable expression profiles. To generate a therapeutic mAb, M30 was humanized (Hu‐M30), and an afucosylated humanized anti‐B7‐H3 IgG1 antibody, designated DS‐5573a, was generated from Hu‐M30. In this article, we characterize DS‐5573a and show its remarkable antitumor activity against B7‐H3‐expressing cells in vitro and in vivo.
Materials and Methods
Cells
Human cancer cell lines NCI‐H1975 (non‐small‐cell lung cancer), 786‐O (renal cell adenocarcinoma), NCI‐N87 (gastric carcinoma), DU145 and PC‐3 (prostate carcinoma), RL95‐2 (endometrial carcinoma), BxPC‐3 (pancreatic adenocarcinoma), TF‐1α (erythroleukemia), MDA‐MB‐231 and MCF‐7 (breast adenocarcinoma), COLO205 (colorectal adenocarcinoma), CCRF‐CEM (acute lymphoblastic leukemia), CHO‐K1, and the murine myeloma cell line P3X63Ag8U.1 were purchased from ATCC (Manassas, VA, USA). NCI‐H322 (non‐small‐cell lung cancer) was purchased from European Collection of Cell Cultures (Salisbury, UK). HEC‐1 (endometrial carcinoma) was purchased from Japanese Collection of Research Bioresources (Osaka, Japan).
Generation of M30
Female transgenic mice with the GANP gene (Transgenic, Fukuoka, Japan)19 were s.c. immunized with MCF‐7, and mice splenocytes were fused with P3X63Ag8U.1 cells using PEG 4000 (Immuno‐Biological Laboratories, Gunma, Japan). One of the hybridoma that produced a mouse IgG2a mAb (M30) was selected because M30 showed antitumor activity against NCI‐H322‐bearing nude mice. The antigen of M30 was identified as B7‐H3 by mass spectrometry.20
Establishment of Hu‐M30 and DS‐5573a
cDNAs of the heavy‐ and light‐chain variable regions of M30 were obtained by RT‐PCR. Hu‐M30 was generated using the CDR grafting method. Expression vectors of Hu‐M30 were transfected into 293‐F cells (Life Technologies, Tokyo, Japan), and Hu‐M30 was purified from the supernatant.20 Afucosylated Hu‐M30, DS‐5573a, was produced by the POTELLIGENT® CHOK1SV expression system (BioWa, La Jolla, CA, USA, and Lonza, Allendale, NJ, USA).
Epitope mapping of DS‐5573a
Full‐length human B7‐H3 (NCBI Reference Sequence, NP_001019907.1: a.a. 27–534), and IgV1 (a.a. 27–139), IgC1 (a.a. 140–244), IgV2 (a.a. 245–357), or IgC2 (a.a. 358–456) domain expression vectors, each with transmembrane/intracellular domain (a.a. 457–534), were transfected into CHO‐K1 cells. Each vector had FLAG‐tag at the N‐terminus. They were then treated with 1 μg/mL DS‐5573a or human IgG1 isotype control (Enzo Life Sciences, New York, NY, USA), and were stained with FITC‐conjugated anti‐human IgG (Jackson ImmunoResearch, West Grove, PA, USA). To detect the expressed B7‐H3 proteins on the cell surface, cells were treated with 1 μg/mL anti‐FLAG antibody (Sigma‐Aldrich, Tokyo, Japan) or mouse IgG1 isotype control (BD, Tokyo, Japan), and were stained with FITC‐conjugated anti‐mouse IgG (Cappel, Aurora, OH, USA). Samples were analyzed by Cytomics FC500 MPL (Beckman Coulter, Tokyo, Japan).
Biacore assay
The binding affinity of DS‐5573a or Hu‐M30 against recombinant human B7‐H3 protein (4IgB7‐H3 or 2IgB7‐H3) (R&D Systems, Minneapolis, MN, USA) was analyzed by surface plasmon resonance using Biacore 3000 or 4000 (GE Healthcare, Tokyo, Japan). DS‐5573a or Hu‐M30 was immobilized to sensor chips using a human antibody capture kit (GE Healthcare), and then each recombinant human B7‐H3 protein was injected. The K D values were calculated using Biacore 3000 or 4000 Evaluation Software (version 4.1.1 or version 1.0; GE Healthcare).
Antigen expression analysis by flow cytometry
Quantification of cell surface antigen was carried out using QIFIKIT (Dako, Tokyo, Japan). Each cell line was treated with a saturating concentration of M30 or mouse IgG2a isotype control (BD or eBioscience, San Diego, CA, USA) (>25 μg/mL), and was stained with FITC‐conjugated anti‐mouse IgG (Dako). Each sample was analyzed by Cytomics FC500 MPL, and the number of binding sites per cell was calculated according to the manufacturer's instructions.
Blood donors
Blood donors were randomly selected from healthy volunteers registered at Shinagawa R&D Center, Daiichi Sankyo, Co., Ltd (Tokyo, Japan). All donors provided informed consent in accordance with the Daiichi Sankyo Ethics Committee.
ADCC assay
Chromium‐51 (51Cr) labeled target cells (1 × 104 cells) were treated with serial dilutions of antibodies and added to PMBCs (3 × 105 cells) obtained from the donors. After 4 h of incubation at 37°C, radioactivity in the supernatant was measured by TopCount‐NXT (PerkinElmer, Yokohama, Japan). 51Cr release was considered to be at maximum after incubation of the cells in 1% Triton‐X solution. All experiments were done in triplicate. ADCC activity (%) was determined according to the following formula:
| (1) |
where S, M, and B represent 51Cr release of each sample, maximum 51Cr release, and background 51Cr release, respectively.
Antibody‐dependent cellular phagocytosis (ADCP) assay
Human macrophages were derived from PBMCs by treatment with 10 ng/mL recombinant human granulocyte M‐CSF (PeproTech, Rocky Hill, NJ, USA) and recombinant human M‐CSF (PeproTech) for 13 days. The day before the assay, macrophages were treated with 250 U/mL recombinant human interferon‐γ (PeproTech), and 10 ng/mL M‐CSF. On the assay day, PKH26‐labeled target cells (5 × 104 cells) treated with serial dilutions of antibodies were mixed with macrophages (1 × 105 cells). After 3 h of incubation at 37°C, each sample was stained with APC‐labeled anti‐human CD11b mAb (BD), and was measured by FACSCanto II (BD) after fixation with 1% paraformaldehyde. All experiments were carried out in triplicate. ADCP activity (%) was calculated using the following formula:
In vivo evaluation
All experimental procedures were carried out according to the in‐house guidelines of the Institutional Animal Care and Use Committee of Daiichi Sankyo Co., Ltd. To confirm the efficacy of DS‐5573a in vivo, MDA‐MB‐231 cells suspended in Matrigel (BD) (5 × 106 cells) were s.c. inoculated into the right flank of female SCID mice (CB17/Icr‐Prkdc scid/CrlCrlj; Charles River, Yokohama, Japan), and DS‐5573a at doses of 0.003–3 mg/kg or vehicle (PBS) was injected i.p. once a week for 5 weeks. To confirm whether the efficacy of DS‐5573a is mediated from effector cells, 3 mg/kg DS‐5573a, human IgG1 isotype control (Eureka Therapeutics, Emeryville, CA, USA), or vehicle was injected i.p. into MDA‐MB‐231‐bearing SCID mice (5 × 106 cells) or NOG mice (NOD/Shi‐scid, IL‐2RγKO; In‐vivo Science, Kawasaki, Japan) (1 × 106 cells) once a week for 5 weeks. As positive control, irinotecan hydrochloride hydrate (Daiichi Sankyo) was injected i.v. twice a week for 2 weeks. Estimated tumor volumes were calculated according to the following formula:
Statistical analysis
In the dose‐response study, estimated tumor volumes at 7 days after the final treatment were compared by Dunnett's test between vehicle‐treated group and DS‐5573a‐treated groups. In the comparison between SCID and NOG mice, estimated tumor volumes at 7 days after the final treatment were compared by Student's t‐test between the irinotecan‐treated group and vehicle‐treated group, and between the DS‐5573a‐treated group and isotype control‐treated group. All statistical analyses were carried out using SAS System Release 9.2 (SAS Institute Inc., Cary, NC, USA).
Results
Generation of M30, Hu‐M30, and DS‐5573a
First, we generated a mouse anti‐human B7‐H3 mAb (M30), which has antitumor efficacy against nude mice bearing the NCI‐H322 cell line with high B7‐H3 expression.20 M30 was then humanized, and designated Hu‐M30. We found that M30 and Hu‐M30 showed antitumor activity in vivo, but the proliferation of cells expressing B7‐H3 was not affected by treatment with either mAb in vitro, suggesting that M30 and Hu‐M30 do not have direct killing activity (data not shown). We also found that Hu‐M30 showed ADCC activity against several B7‐H3‐expressing cell lines, suggesting that ADCC activity may be one of the modes of action of Hu‐M30. Enhancement of the ADCC activity of Hu‐M30 would be expected to result in a more effective therapeutic mAb, thus afucosylated Hu‐M30, DS‐5573a, was generated.
Characterization of DS‐5573a
Next, we evaluated the binding site and affinity of DS‐5573a to human B7‐H3. To analyze the binding site, a series of extracellular domain expressing vectors were constructed (Fig. 1), and transiently transfected into CHO‐K1 cells. Flow cytometry analysis revealed that DS‐5573a was bound to CHO‐K1 cells expressing either the full‐length human B7‐H3 (4IgB7‐H3), IgC1 or IgC2 domains, but not to cells expressing either the IgV1 or IgV2 domain (Fig. 2a). Cell surface expression of each B7‐H3 protein was confirmed by measuring the binding of the anti‐FLAG antibody (Fig. 2b). Based on these results, we concluded that DS‐5573a binds to the IgC1 and IgC2 domains of human B7‐H3. To evaluate the effect of afucosylation of the Fc portion of Hu‐M30 on its binding to B7‐H3, the K D values of DS‐5573a and Hu‐M30 were assessed using the Biacore assay. The affinity of DS‐5573a and Hu‐M30 to human 4IgB7‐H3 was similar, with K D values of 1.8 nM and 1.6 nM, respectively, suggesting that afucosylation has little effect on binding activity. We also found that DS‐5573a and Hu‐M30 bound to 2IgB7‐H3, another isoform of human B7‐H3 that has the IgV1–IgC2 extracellular domain,9 with approximately one order of magnitude lower affinity (K D values of 11 nM and 16 nM, respectively) than those to 4IgB7‐H3.
Figure 1.
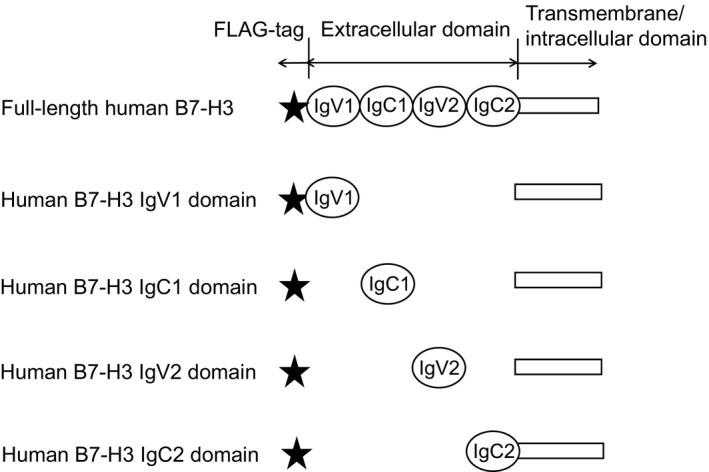
Full‐length human B7‐H3, and IgV1, IgC1, IgV2, or IgC2 domain expression vectors, each with transmembrane/intracellular domain, were constructed. Each vector had FLAG‐tag at the N‐terminus.
Figure 2.
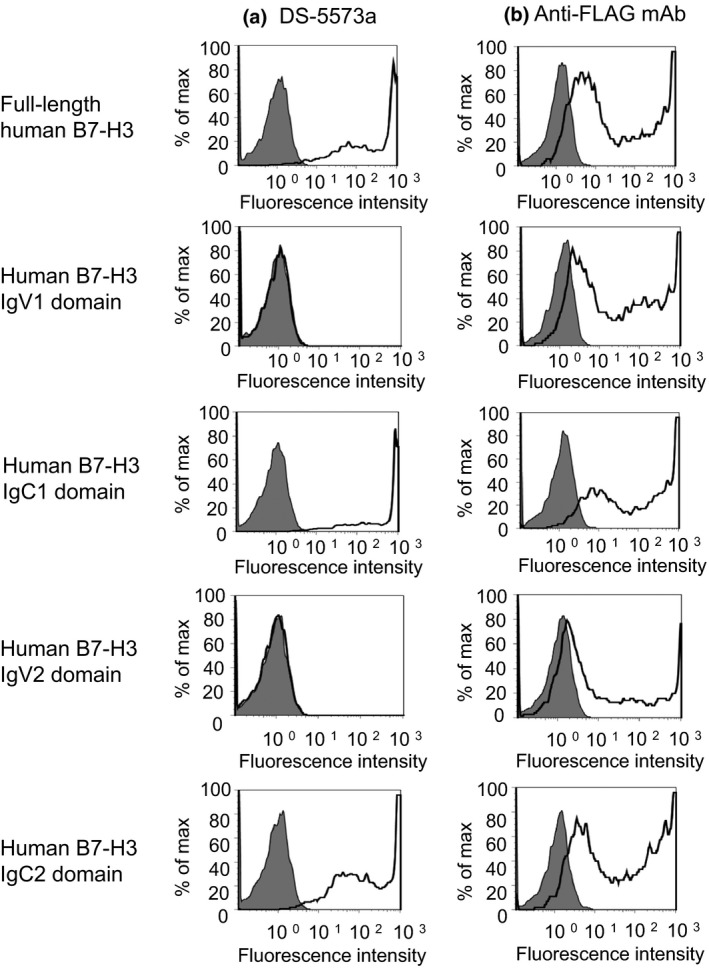
DS‐5573a binds to IgC1 and IgC2 domains of human B7‐H3. (a) Expression vector‐transfected CHO‐K1 cells were treated with 1 μg/mL DS‐5573a (open histogram) or isotype control (gray histogram) and were stained with FITC‐conjugated anti‐human antibody. (b) Cells were treated with 1 μg/mL anti‐FLAG mAb (open histogram) or isotype control (gray histogram) and were stained with FITC‐conjugated anti‐mouse antibody.
B7‐H3 proteins are widely expressed in cancer cell lines
To investigate whether B7‐H3 proteins are expressed not only in the clinical tumors10, 11, 12, 13, 14, 15 but also on the surface of cancer cell lines, quantitative flow cytometry analysis using M30 was carried out in various cancer cell lines. Results showed that B7‐H3 proteins were expressed on various types of cancer cell lines broadly (Table 1). Among the cell lines, we selected NCI‐H322 as a cell line with high B7‐H3 expression (>1 × 105 per cell), MDA‐MB‐231 as a cell line with medium B7‐H3 expression (approximately 5 × 104 per cell), COLO205 as a cell line with low B7‐H3 expression (approximately 1 × 104 per cell), and CCRF‐CEM as a cell line that is negative for B7‐H3 expression, and used them for in vitro assays.
Table 1.
Expression level of B7‐H3 in each cancer cell line
| Origin | Cell line | B7‐H3 expression, per cell |
|---|---|---|
| Lung | NCI‐H322 | 3.4E+05 |
| NCI‐H1975 | 1.9E+05 | |
| Kidney | 786‐O | 1.5E+05 |
| Stomach | NCI‐N87 | 1.4E+05 |
| Prostate | DU145 | 1.2E+05 |
| PC‐3 | 7.1E+04 | |
| Endometrium | HEC‐1 | 1.3E+05 |
| RL95‐2 | 6.2E+04 | |
| Pancreas | BxPC‐3 | 5.0E+04 |
| Breast | MDA‐MB‐231 | 4.8E+04 |
| Erythroleukemia | TF‐1α | 3.7E+04 |
| Colon | COLO205 | 1.7E+04 |
| ALL | CCRF‐CEM | Not detectable |
Quantification of cell surface antigen was carried out using QIFIKIT. Underlined cell lines were used in in vitro assays. ALL, acute lymphoblastic leukemia.
DS‐5573a exerts much more potent human PBMC‐mediated ADCC than Hu‐M30
Next, to confirm whether DS‐5573a exerts enhanced ADCC activity as expected, we compared the activity of DS‐5573a and Hu‐M30 against cancer cell lines with various levels of B7‐H3 expression in the presence of human PBMCs. As shown in Figure 3, notably the ADCC activity of DS‐5573a and Hu‐M30 was found to fit to a sigmoid curve with different maximal levels of cytotoxicity against B7‐H3‐expressing cell lines. DS‐5573a showed enhanced ADCC activity against NCI‐H322 and MDA‐MB‐231 cells in a dose‐dependent manner at concentrations of 0.1–100 ng/mL, whereas Hu‐M30 was effective at concentrations of 10–1000 ng/mL. Of note, DS‐5573a could induce high ADCC activity against COLO205, whereas Hu‐M30 induced little ADCC activity. The high ADCC activity of DS‐5573a was also observed against various other B7‐H3‐expressing cancer cell lines (data not shown). In contrast, minimal cytotoxicity was observed against a B7‐H3‐negative cancer cell line, CCRF‐CEM. These results suggest that DS‐5573a exerts much more potent B7‐H3‐dependent ADCC activity than Hu‐M30.
Figure 3.
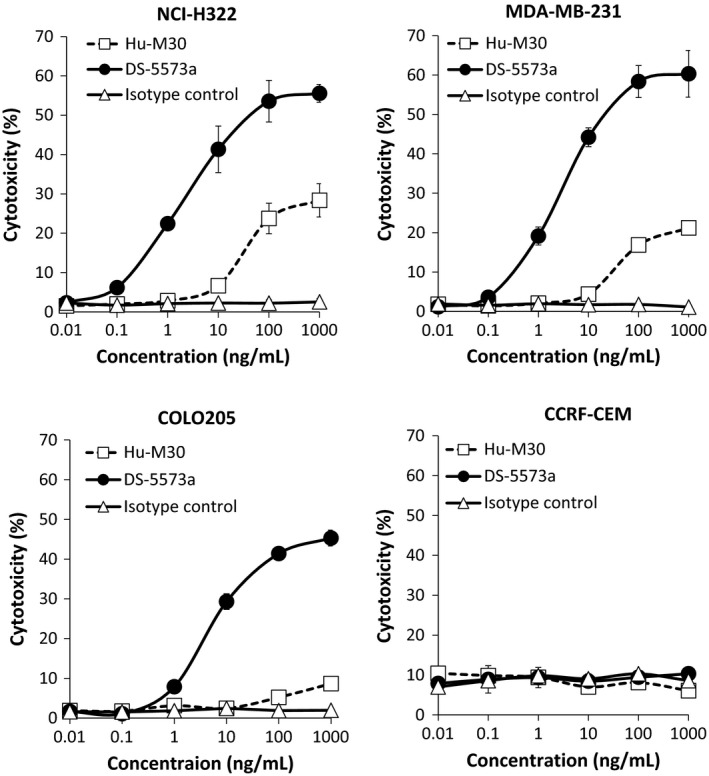
Human PBMC‐mediated antibody‐dependent cellular cytotoxicity (ADCC) activity of DS‐5573a and Hu‐M30 against cancer cell lines with various levels of B7‐H3 expression. ADCC activity was measured by 4‐h 51Cr release assay, in the presence of DS‐5573a or Hu‐M30 (effector/target = 30/1). Isotype control was also evaluated as negative control. Mean ± SD of triplicates are shown.
DS‐5573a exerts human macrophage‐mediated ADCP
Recently, it was shown that macrophages serve as critical immune effectors of therapeutic mAbs.21 Although ADCP activity has been a focus of attention, there is no report on an anti‐B7‐H3 antibody with ADCP activity. To further evaluate the potential of DS‐5573a in vitro, we evaluated the human macrophage‐mediated ADCP activity of DS‐5573a and Hu‐M30 against cancer cell lines with various B7‐H3 expression levels. We found that antibody dose‐dependent ADCP activity of DS‐5573a was detected against NCI‐H322, MDA‐MB‐231, COLO205 (Fig. 4), and other B7‐H3‐expressing cancer cell lines (data not shown). In contrast, DS‐5573a did not mediate ADCP activity against B7‐H3‐negative CCRF‐CEM cells. In addition, it was revealed that afucosylation of Hu‐M30 did not impair the ADCP activity. These results suggest that DS‐5573a also has the potency of B7‐H3‐dependent ADCP activity.
Figure 4.
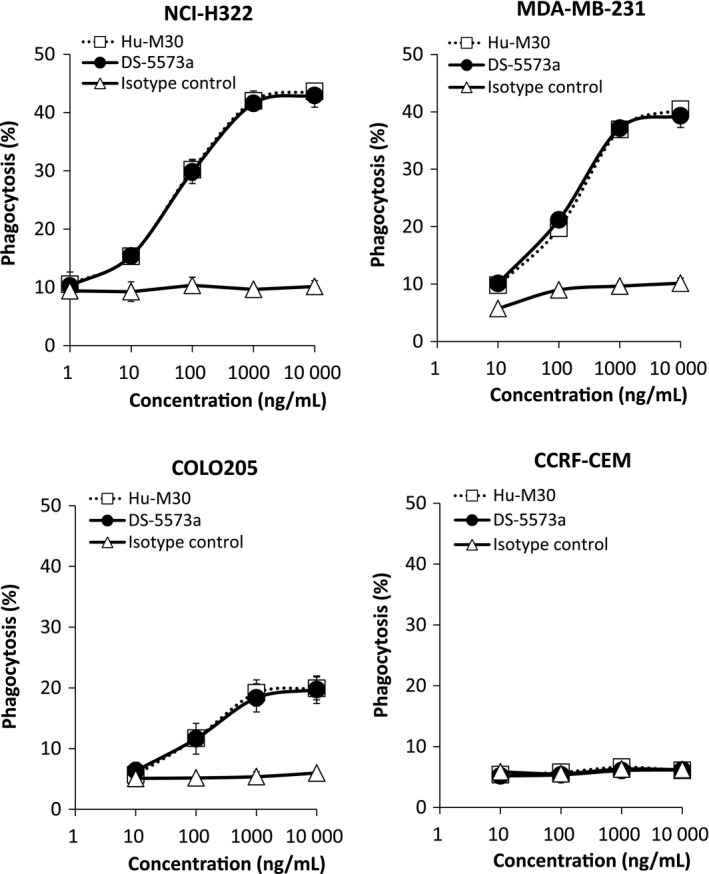
Human macrophage‐mediated antibody‐dependent cellular phagocytosis (ADCP) activity of DS‐5573a and Hu‐M30 against cancer cell lines with various level of B7‐H3 expression. ADCP activity against PKH26‐labeled target cells was measured in the presence of DS‐5573a or Hu‐M30 (effector/target = 2/1). Isotype control was also evaluated as negative control. After 3 h of incubation, each sample was stained with APC‐labeled anti‐human CD11b mAb and measured by flow cytometry. Mean ± SD of triplicates are shown.
DS‐5573a shows effector cell‐mediated antitumor activity in vivo
To further evaluate the therapeutic potential of DS‐5573a, its antitumor activity was evaluated using MDA‐MB‐231‐bearing SCID mice, in which the tumors are poorly differentiated. At first, DS‐5573a at doses of 0.003–3 mg/kg or vehicle was injected i.p. once a week for 5 weeks. As shown in Figure 5, dose‐dependent and statistically significant antitumor efficacy was observed compared with the vehicle‐treated group (0.03 mg/kg, P = 0.043; 0.3 and 3 mg/kg, P < 0.001). Next, to confirm whether the efficacy of DS‐5573a is mediated from effector cells, not only in vitro but also in vivo, the efficacy of DS‐5573a was compared in MDA‐MB‐231‐bearing SCID mice (which have functional NK cells and macrophages) and NOG mice (which lack NK cells and have reduced macrophage function). DS‐5573a, human IgG1 isotype control (both 3 mg/kg), or vehicle was injected i.p. once a week for 5 weeks. As positive control, irinotecan (60 mg/kg), which is a standard chemotherapy for metastatic breast cancer, was injected i.v. twice a week for 2 weeks. As shown in Figure 6, statistically significant suppression of tumor growth was observed in the irinotecan‐treated group in both mouse models (both P < 0.001). Statistically significant suppression of tumor growth was also observed in the DS‐5573a‐treated group in SCID mice compared with the isotype control‐treated group (P < 0.001; Fig. 6a), but no statistically significant difference was observed in NOG mice (Fig. 6b). These results suggest that the antitumor activity of DS‐5573a is mediated by effector cells.
Figure 5.
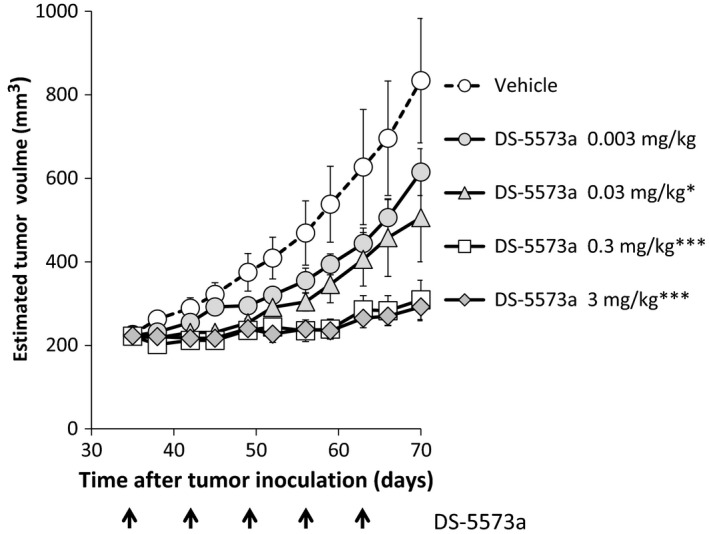
Antitumor activity of DS‐5573a in MDA‐MB‐231‐bearing SCID mice. DS‐5573a at doses of 0.003–3 mg/kg or vehicle (PBS) was injected i.p. into MDA‐MB‐231‐bearing SCID mice once a week for 5 weeks (n = 10). The arrow shows the timing of DS‐5573a or vehicle injection. Data represent mean ± SEM. Estimated tumor volumes at 7 days after the final treatment were compared by Dunnett's test between the vehicle‐treated group and DS‐5573a‐treated groups. *P < 0.05, ***P < 0.001.
Figure 6.
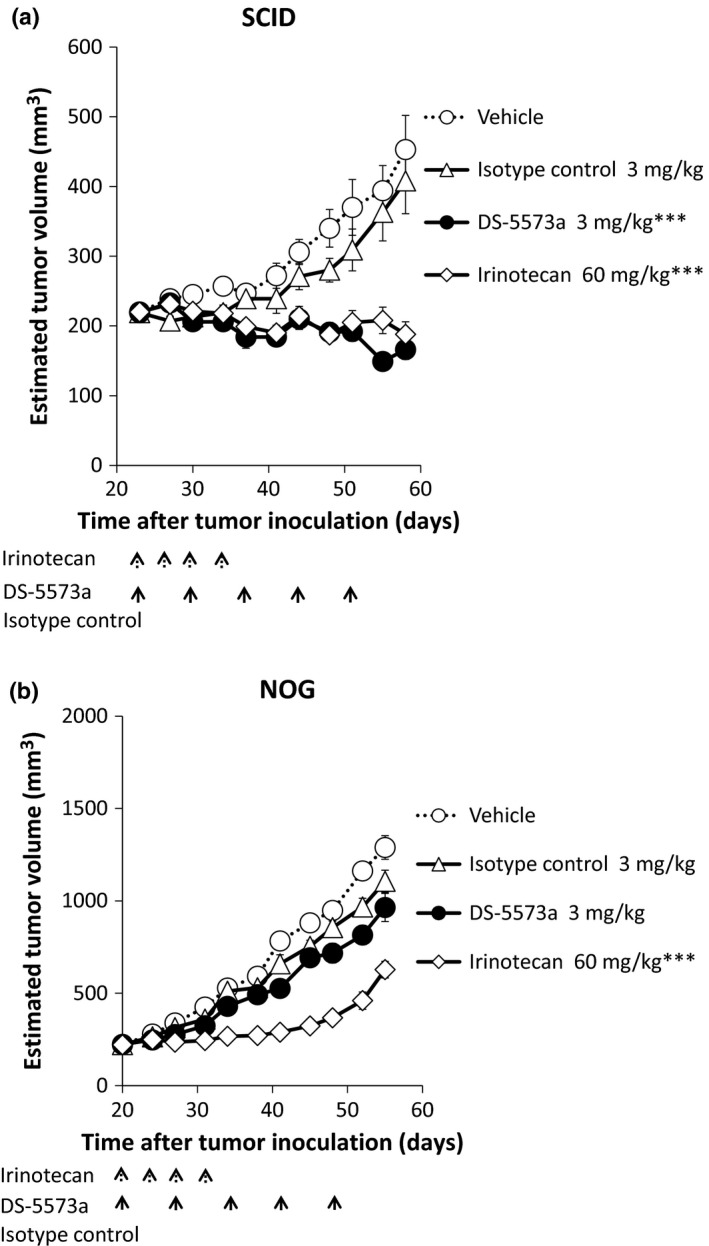
Antitumor activity of DS‐5573a in MDA‐MB‐231‐bearing SCID mice (a) and NOG mice (b). DS‐5573a, isotype control (both 3 mg/kg), or vehicle was injected i.p. once a week for 5 weeks (SCID mice, n = 10; NOG mice, n = 6). As positive control, 60 mg/kg irinotecan was injected i.v. twice a week for 2 weeks. The dashed arrows show the timing of irinotecan injection, and the solid arrows show the timing of DS‐5573a, isotype control, or vehicle injection. Data represent mean ± SEM. Estimated tumor volumes were compared by Student's t‐test between irinotecan‐treated group and vehicle‐treated group, and between DS‐5573a‐treated group and isotype control‐treated group at 7 days after final treatment. ***P < 0.001.
Discussion
In our study, we aimed to develop new antitumor mAbs, and succeeded in generating an afucosylated humanized anti‐human B7‐H3 mAb, DS‐5573a. DS‐5573a induced drastically enhanced ADCC activity against medium to high B7‐H3‐expressing cancer cell lines MDA‐MB‐231 and NCI‐H322 at approximately 100‐fold lower mAb concentrations compared to Hu‐M30. In addition, DS‐5573a induced high ADCC activity against low B7‐H3‐expressing cancer cell line COLO205, whereas Hu‐M30 induced little ADCC activity against it.
It is known that ADCC plays a key role in mAb therapy, but some cancer patients could be resistant to the ADCC of current therapeutic mAbs due to shortage of antigen expression, and individual heterogeneity of functional polymorphism of FcγRIIIa. Based on published reports and our data, DS‐5573a is expected to be effective against a wide range of B7‐H3‐expressing tumors at lower mAb concentrations, regardless of the single nucleotide polymorphism in the FCGR3A gene. Specifically, it has been clearly shown that human IgG1 binds more strongly to homozygous FcγRIIIa‐158V NK cells than to homozygous FcγRIIIa‐158F or heterozygous NK cells,22 and FCGR3A gene allelic polymorphism affects response rates and progression‐free survival in cancer patients treated with ADCC‐mediated mAbs such as rituximab, trastuzumab, and cetuximab.23, 24, 25 However, the FCGR3A‐158F allele is more prevalent than the FCGR3A‐158V allele.22, 26, 27 These reports suggest that conventional ADCC therapy with first‐generation mAbs would be less effective against clinical tumors. In contrast, a low‐fucose version of rituximab showed significantly enhanced binding activity against not only FcγRIIIa‐158V but also FcγRIIIa‐158F, and its ADCC activity was increased 10‐ to 100‐fold compared with rituximab, regardless of FCGR3A genotypes.28 Quantitative analysis revealed that the amount of antigen on target cells required for an equivalent degree of ADCC induction was 3‐ to 10‐fold lower for the low‐fucose IgG1 than that for the high‐fucose IgG1.7
One of the most important findings in this study is that DS‐5573a has ADCP activity against B7‐H3‐expressing cancer cells. As far as we know, this is the first report about an anti‐B7‐H3 mAb that has not only ADCC but also ADCP activity. It is suggested that macrophages are commonly found in tumors in high numbers, and play a critical function as effectors of mAb therapies.21 For example, the novel drugs anti‐CD47 mAb29 and high‐affinity SIRPα‐Fc fusion proteins30 enhanced phagocytosis by macrophages in rituximab‐treated non‐Hodgkin's lymphoma cells and trastuzumab‐treated breast cancer cells in vitro. Also these treatments showed remarkable synergistic antitumor effects in vivo. DS‐5573a might be more effective for enhancement of ADCP activity if used in combination with these biologics. We also showed that ADCP activity of DS‐5573a against B7‐H3‐expressing cells is almost the same as that of Hu‐M30. It has been revealed that FcγRIIa plays the most influential role for macrophages,31 and the binding affinity of low‐fucose IgG1 was greatly increased against FcγRIIIa, but was slightly increased against FcγRIIa.28 Another group suggested that ADCC activity of an afucosylated mAb was increased more than 20‐fold compared with a fucosylated version of the mAb, with no impairment of ADCP activity,32 which is consistent with our data.
Furthermore, we found that DS‐5573a has dose‐dependent antitumor efficacy in MDA‐MB‐231‐bearing SCID mice; in a comparison between SCID and NOG mice (Fig. 6), the efficacy of DS‐5573a depended largely on effector cells. Our in vitro and in vivo data suggested that DS‐5573a may be effective against B7‐H3‐expressing tumors containing NK cells and macrophages. One report revealed that the strength of ADCC activity of the low‐fucose version of rituximab is correlated with the percentage of NK cells in PBMCs,28 so the NK portion may be an important factor for DS‐5573a to exert its antitumor activity efficiently.
In conclusion, DS‐5573a has a unique dual mode of action, effector functions of ADCC and ADCP, and shows strong antitumor activity against B7‐H3‐expressing cancer cell lines in vitro and in vivo. A high level of B7‐H3 protein expression was detected in various types of cancer cell lines in this study, and several immunohistochemistry studies have revealed that moderate to high B7‐H3 protein expression was detected in more than 50% of various types of tumors.10, 11, 14, 15 We also confirmed that repeated administration of DS‐5573a in cynomolgus monkeys was well tolerated (unpublished observation). These reports and our data suggest that DS‐5573a has potential as a novel therapeutic mAb to address unmet medical needs in B7‐H3‐positive cancer patients. A phase I clinical trial in patients with advanced solid malignant tumors is underway (ClinicalTrials.gov identifier: NCT02192567).
Disclosure Statement
All authors are employees of Daiichi Sankyo Co., Ltd.
Abbreviations
- a.a.
amino acid
- ADCC
antibody‐dependent cellular cytotoxicity
- ADCP
antibody‐dependent cellular phagocytosis
- APC
allophycocyanin
- KD
dissociation constant
- M30
mouse anti‐human B7‐H3 mAb
- Hu‐M30
humanized M30
- DS‐5573a
afucosylated Hu‐M30
- M‐CSF
macrophage colony‐stimulating factor
- NK
natural killer
Acknowledgments
The authors thank Yoko Oda, Haruyuki Nishigori, and Atsushi Urano for generating and screening mouse mAbs, Kazuishi Kubota and Rika Nakano for antigen identification of M30, Kenji Murakami, Takeshi Takizawa, and Tatsuji Matsuoka for generating Hu‐M30, members of Biologics Technology Research Laboratories for purification of M30, Hu‐M30, and DS‐5573a, Michiko Kitamura for establishment of the in vivo evaluation model of DS‐5573a, Akira Okubo for technical support, and Yuki Abe, Kenji Iida, and Donald J. Hinman and all project members for helpful discussions.
Cancer Sci 107 (2016) 674–681
Funding Information
Daiichi Sankyo Co., Ltd.
References
- 1. Stewart BW, Wild CP. World Cancer Report 2014.
- 2. Liu JK. The history of monoclonal antibody development – Progress, remaining challenges and future innovations. Ann Med Surg 2014; 3: 113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Slamon DJ, Leyland‐Jones B, Shak S et al Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–92. [DOI] [PubMed] [Google Scholar]
- 4. Coiffier B, Lepage E, Briere J et al CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large‐B‐cell lymphoma. N Engl J Med 2002; 346: 235–42. [DOI] [PubMed] [Google Scholar]
- 5. Mössner E, Brünker P, Moser S et al Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti‐CD20 antibody with enhanced direct and immune effector cell‐mediated B‐cell cytotoxicity. Blood 2010; 115: 4393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goede V, Fischer K, Busch R et al Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014; 370: 1101–10. [DOI] [PubMed] [Google Scholar]
- 7. Niwa R, Sakurada M, Kobayashi Y et al Enhanced natural killer cell binding and activation by low‐fucose IgG1 antibody results in potent antibody‐dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res 2005; 11: 2327–36. [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto K, Utsunomiya A, Tobinai K et al Phase I study of KW‐0761, a defucosylated humanized anti‐CCR4 antibody, in relapsed patients with adult T‐cell leukemia–lymphoma and peripheral T‐cell lymphoma. J Clin Oncol 2010; 28: 1591–8. [DOI] [PubMed] [Google Scholar]
- 9. Chapoval AI, Ni J, Lau JS et al B7‐H3: a costimulatory molecule for T cell activation and IFN‐gamma production. Nat Immunol 2001; 2: 269–74. [DOI] [PubMed] [Google Scholar]
- 10. Sun J, Chen LJ, Zhang GB et al Clinical significance and regulation of the costimulatory molecule B7‐H3 in human colorectal carcinoma. Cancer Immunol Immunother 2010; 59: 1163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roth TJ, Sheinin Y, Lohse CM et al B7‐H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res 2007; 67: 7893–900. [DOI] [PubMed] [Google Scholar]
- 12. Crispen PL, Sheinin Y, Roth TJ et al Tumor cell and tumor vasculature expression of B7‐H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res 2008; 14: 5150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun Y, Wang Y, Zhao J et al B7‐H3 and B7‐H4 expression in non‐small‐cell lung cancer. Lung Cancer 2006; 53: 143–51. [DOI] [PubMed] [Google Scholar]
- 14. Brunner A, Hinterholzer S, Riss P, Henize G, Brustmann H. Immunoexpression of B7‐H3 in endometrial cancer: relation to tumor T‐cell infiltration and prognosis. Gynecol Oncol 2012; 124: 105–11. [DOI] [PubMed] [Google Scholar]
- 15. Kang FB, Wang L, Jia HC et al B7‐H3 promotes aggression and invasion of hepatocellular carcinoma by targeting epithelial‐to‐mesenchymal transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int 2015; 15: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen YW, Tekle C, Fodstad O. The immunoregulatory protein human B7H3 is a tumor‐associated antigen that regulates tumor cell migration and invasion. Curr Cancer Drug Targets 2008; 8: 404–13. [DOI] [PubMed] [Google Scholar]
- 17. Yuan H, Wei X, Zhang G, Li C, Zhang X, Hou J. B7‐H3 over expression in prostate cancer promotes tumor cell progression. J Urol 2011; 186: 1093–9. [DOI] [PubMed] [Google Scholar]
- 18. Xu H, Cheung IY, Guo HF, Cheung NK. MicroRNA miR‐29 modulates expression of immunoinhibitory molecule B7‐H3: potential implications for immune based therapy of human solid tumors. Cancer Res 2009; 69: 6275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sakaguchi N, Kimura T, Matsushita S et al Generation of high‐affinity antibody against T cell‐dependent antigen in the Ganp gene‐transgenic mouse. J Immunol 2005; 174: 4485–94. [DOI] [PubMed] [Google Scholar]
- 20. Takahashi S, Matsuoka T, Murakami K et al ANTI‐B7‐H3 ANTIBODY. WO 2012/147713.
- 21. Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. Mabs 2015; 7: 303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa‐158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa‐48L/R/H phenotype. Blood 1997; 90: 1109–14. [PubMed] [Google Scholar]
- 23. Cartron G, Dacheux L, Salles G et al Therapeutic activity of humanized anti‐CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002; 99: 754–8. [DOI] [PubMed] [Google Scholar]
- 24. Musolino A, Naldi N, Bortesi B et al Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab‐based therapy in patients with HER‐2⁄neu‐positive metastatic breast cancer. J Clin Oncol 2008; 26: 1789–96. [DOI] [PubMed] [Google Scholar]
- 25. Zhang W, Gordon M, Schultheis AM et al FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single‐agent cetuximab. J Clin Oncol 2007; 25: 3712–8. [DOI] [PubMed] [Google Scholar]
- 26. Leppers‐van de Straat FG, van der Pol WL, Jansen MD et al A novel PCR‐based method for direct Fc gamma receptor IIIa (CD16) allotyping. J Immunol Methods 2000; 242: 127–32. [DOI] [PubMed] [Google Scholar]
- 27. Lehrnbecher T, Foster CB, Zhu S et al Variant genotypes of the low‐affinity Fcgamma receptors in two control populations and a review of low‐affinity Fcgamma receptor polymorphisms in control and disease populations. Blood 1999; 94: 4220–32. [PubMed] [Google Scholar]
- 28. Niwa R, Hatanaka S, Shoji‐Hosaka E et al Enhancement of the antibody‐dependent cellular cytotoxicity of low‐fucose IgG1 Is independent of FcgammaRIIIa functional polymorphism. Clin Cancer Res 2004; 10: 6248–55. [DOI] [PubMed] [Google Scholar]
- 29. Chao MP, Alizadeh AA, Tang C et al Anti‐CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non‐Hodgkin lymphoma. Cell 2010; 142: 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiskopf K, Ring AM, Ho CC et al Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013; 341: 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008; 7: 2517–27. [DOI] [PubMed] [Google Scholar]
- 32. Silence K, Dreier T, Moshir M et al ARGX‐110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. Mabs 2014; 6: 523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]