

Abstract
Although rituximab, a chimeric monoclonal antibody that specifically binds to CD20, has significantly improved the prognosis for diffuse large B cell lymphoma (DLBCL), one‐third of DLBCL patients demonstrate resistance to rituximab or relapse after rituximab treatment. Thus, a novel approach to rituximab‐based treatment is likely to be required to improve the efficacy of DLBCL treatment. As complement dependent cytotoxicity (CDC) is a key mechanism mediating rituximab's tumoricidal activity, rituximab binding to CD20 on tumor cells is a critical factor for effective rituximab‐based treatments against DLBCL. We found that gemcitabine (GEM), but not lenalidomide (LEN) or azacitidine (AZA), can upregulate CD20 expression in TK and KML‐1 cells, two human DLBCL cell lines. Treatment of TK and KML‐1 cells with GEM enhanced CD20 expression at both the mRNA and protein levels. CD20 upregulation by GEM treatment was accompanied by increased rituximab binding to CD20. In TK cells, GEM treatment synergistically increased rituximab‐mediated CDC activity in a dose‐dependent manner. In KML cells, GEM treatment also induced upregulation of complement regulatory proteins, possibly leading to resistance to CDC. Treatment with LEN, a drug that did not upregulate CD20, did not enhance rituximab‐mediated CDC activity. GEM treatment activated nuclear factor‐kappa B (NF‐kB) signaling in these cells. Furthermore, a specific inhibitor to NF‐kB suppressed GEM‐induced CD20 upregulation, indicating that GEM‐induced NF‐kB activation is closely associated with CD20 upregulation. These results suggest that when used in combination, GEM might enhance the antitumor efficacy of rituximab against DLBCL due to its unique ability to upregulate CD20.
Keywords: CD20, complement dependent cytotoxicity, diffuse B cell lymphoma, gemcitabine, rituximab
Non‐Hodgkin lymphoma (NHL) is a common hematological cancer in adults. As the majority of lymphomas are of B‐cell origin and 80% of B‐cell lymphomas express CD20 on the cell surface, rituximab, an anti‐CD20 monoclonal antibody, has become a standard drug for NHL treatment.1, 2, 3 Rituximab has been used to treat hematological cancers such as high‐grade diffuse large B cell lymphoma (DLBCL), low‐grade follicular lymphoma and chronic lymphocytic leukemia, as well as non‐hematological diseases such as rheumatoid arthritis, granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).3, 4, 5, 6, 7, 8 The antitumor effect of rituximab is mediated by antibody‐dependent cell‐mediated cytotoxicity (ADCC), complement‐dependent cytotoxicity (CDC), antibody‐mediated phagocytosis of tumor cells and rituximab‐induced apoptosis.9, 10 Several trials have demonstrated that the addition of rituximab to conventional chemotherapy improved the response rate and prolonged the survival of DLBCL patients.11, 12 Therefore, rituximab is included in the R‐CHOP, R‐EPOCH, R‐ESHAP and R‐GEMOX regimens recommended for DLBCL treatment.11, 13, 14, 15 Two‐year overall survival was achieved in 79% of the patients treated with R‐CHOP, compared with 70% in patients treated with CHOP alone.12
However, despite the high response rate of rituximab‐containing chemotherapy, approximately 50% of DLBCL patients experience recurrent disease within 5 years.16 Furthermore, DLBCL patients who are resistant to rituximab‐containing chemotherapy or those who relapse after treatment have a very low survival rate. The mechanism of rituximab resistance is largely unknown, but potential mechanisms include downregulation or loss of CD20 expression, the formation of soluble CD20 molecules or inhibition of ADCC and CDC.17, 18 A recent study reported that downregulation of CD20 expression decreased rituximab‐mediated CDC activity against B cell lymphomas.19
Some therapeutic agents, such as farnesyltransferase inhibitors, bryostatin‐1, histone deacetylase inhibitors20, 21, 22 and some cytokines,23 can enhance CD20 expression in lymphoma, and upregulation of CD20 expression on B cell lymphoma cells increases the cytotoxic activity of rituximab.21, 22 In the present study, we demonstrate that gemcitabine (GEM), which is conventionally used for DLBCL treatment,24, 25 augments CD20 expression on DLBCL cells. Upregulation of CD20 on DLBCL cells by GEM treatment enhances rituximab‐mediated CDC activity, suggesting that combined treatment with GEM and rituximab might improve outcomes in DLBCL treatment.
Materials and Methods
Cell culture
Human diffuse large B‐cell lymphoma cell lines (TK:JCRB0157 and KML‐1:JCRB1347) were purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). TK cells were cultured in alpha‐MEM (Wako, Osaka, Japan), and KML‐1 cells were cultured in RPMI 1640 (Wako) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), penicillin (100 U/mL) and streptomycin (100 μg/mL). Cells were cultured at 37°C in a fully humidified atmosphere of 5% CO2 and passaged every other day.
Reagents
Gemcitabine (GEM) was purchased from Eli Lilly Japan (Kobe, Japan). Lenalidomide (LEN) was purchased from Toronto Research Chemicals (Toronto, Canada). Azacitidine (AZA) was purchased from Sigma‐Aldrich (St. Louis, MO, USA). BAY 11‐7082, a nuclear factor‐kappa B (NF‐kB) inhibitor, was purchased from Wako Pure Chemical Industries. Rituximab was a gift from Zenyaku Kogyo (Tokyo, Japan).
Flow cytometry
Cell surface CD20 expression was examined by flow cytometry. A suspension of untreated or agent‐treated cells was stained with phycoerythrin (PE)‐conjugated anti‐human CD20 (clone 2H7; BioLegend, CA, USA) anti‐human CD46 (clone TRA‐2‐10; BioLegend), anti‐human CD55 (clone JS11; BioLegend), anti‐human CD59 (clone p282; BioLegend) and the appropriate isotype controls (BioLegend) for 30 min at 4°C in the dark. Propidium iodide (PI, 1 μg/mL final concentration) (Sigma‐Aldrich) staining was performed to identify dead cells to exclude from flow cytometry analysis. Cells were analyzed on a MACSQuantAnalyzer (Milteny Biotec, Bergisch Gladbach, Germany) using MACSQuantify Software Version 2.0.
Quantitative reverse transcription PCR
Total RNA was extracted from untreated or agent‐treated TK and KML‐1 cells using the RNeasy kit (QIAGEN GmbH, Hilden, Germany). Complementary DNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). qRT‐PCR was carried out in a 7300 real‐time PCR system (Applied Biosystems). TaqMan primers for the CD20 (Assay ID: Hs0.0144819_m1) CD46 (Assay ID: Hs00611257_m1), CD55 (Assay ID: Hs00892618_m1), CD59 (Assay ID: Hs00174141_m1) and 18S ribosomal RNA (Assay ID: Hs99999901_s1) genes were purchased from Applied Biosystems. Relative expression is calculated using the delta‐Ct method.
Complement dependent cytotoxicity assays
TK and KML‐1 cells were incubated with GEM (0–1000 ng/mL), rituximab (1–100 μg/mL) and 10% human AB serum as a complement source for 48 h (h). Cell viability in the CDC assay was determined using propidium iodide (PI) staining and the MACSQuantAnalyzer and was expressed as the relative viability of tumor cells compared with control cultures incubated with medium and human AB serum.26
Immunofluorescent microscopy
TK cells were left untreated or were treated with GEM for 48 h. The cells were then pasted to a prepared slide (Mastsunami, Osaka, Japan) by the Shandon cytospin 3 centrifuge (Thermo Fisher Scientific). The cells were fixed with 4% formaldehyde for 15 min at room temperature and methanol for 10 min at −20°C. Each slide was treated with 100 μL blocking solution (PBS/0.3% Triton/5% BSA) for 1 h at room temperature. The slides were subsequently incubated with primary antibody (CD20, clone 2H7; BioLegend) diluted in antibody solution (PBS/0.3% Triton/1%BSA) overnight at 4°C. The slides were incubated with secondary antibody (Alexa488 anti‐rabbi IgG; Thermo Fisher Scientific) for 1 h at room temperature and mounted with 30 μL of VECTASHIELD mounting medium containing DAPI (Vector, CA, USA). Each slide was observed under DeltaVision (GE Healthcare UK, Buckinghamshire, England).
Western blot
For analyses on the cell signaling, cells were incubated with GEM (1000 ng/mL) for 6 or 12 h and lysed in RIPA buffer (Nacalai tesque, Kyoto, Japan) supplemented with Phosphatase Inhibitor Cocktail (Nacalai tesque). Protein concentration was measured using the Bio‐Rad protein assay (Bio‐Rad, California, USA). Then, 20 μg protein was separated on 5–20% gradient e‐PAGEL gels (ATTO, Tokyo, Japan). The proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane using Invitrogen's iBlot transfer system (Applied BioSystems). The membrane was blocked with Blocking One‐P buffer (Nacalai tesque) for 30 min at room temperature. The following antibodies were purchased from Cell Signaling Technology (Danvers, MO, USA): NF‐kB p65, phospho‐NF‐kb (p‐NF‐kB) (Ser536), AKT, phospho‐AKT (p‐AKT) (Ser473), ERK1/2, phospho‐ERK1/2 (p‐ERK1/2) (Thr202/Tyr204), STAT3, phospho‐STAT3 (p‐STAT3) (Tyr705) and beta‐actin.
For analyses on CD20 expression, cells were incubated with GEM (1000 ng/mL) for 24, 36 or 48 h and lysed in the same way. The membrane was incubated with anti‐CD20 antibody (Spring Bioscience, Pleasanton, CA, USA) in TBST (0.01 M Tris‐HCl and 0.15 M NaCl with 0.01% Tween20) buffer with gentle agitation overnight at 4°C and incubated for 1 h at room temperature with HRP‐conjugated anti‐rabbit IgG. The signal was visualized using Chemi‐Lumi One Super (Nacalai tesque).
Rituximab binding assay
Cells were left untreated or were treated with both GEM (1000 ng/mL) and rituximab (0–100 μg/mL) for 48 h. Untreated or agent‐treated cells were stained with PE‐conjugated anti‐human IgG1 antibodies (clone IS11‐12E4.23.20) purchased from Milteny Biotec or with an appropriate isotype control (BioLegend) for 30 min at 4°C in the dark. The cells were analyzed on a MACSQuantAnalyzer (Milteny Biotec) using MACSQuantify Software Version 2.0.
Inhibition of nuclear factor kappa B
TK and KML‐1 cells were pre‐treated with BAY 11‐7082 (0.3, 1 μM) (Wako) for 1 h, and subsequently treated with GEM (1000 ng/mL) and BAY 11‐7082 for 48 h. Expression of CD20mRNA and CD20 protein was assayed by qRT‐PCR and flow cytometry, respectively, as described above.
Results
Gemcitabine induced upregulation of CD20 expression in diffuse large B cell lymphoma cells in vitro
Chemotherapeutic agents for DLBCL treatment were examined for their ability to upregulate CD20 expression in TK and KML‐1 cells. The cell‐surface expression of the CD20 protein was significantly increased by GEM treatment (Fig. 1a,b). In TK cells, CD20 expression increased gradually after 12 h GEM treatment. However, in KML‐1 cells, CD20 upregulation was observed after 48 h GEM treatment (Fig. 1c). The concentrations of GEM (100–1000 ng/mL) in this study are similar to those in patients’ plasma.
Figure 1.
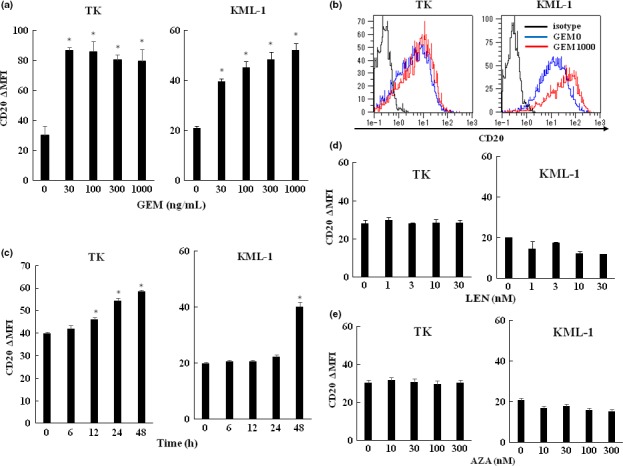
Effects of various chemotherapeutic agents on surface CD20 expression in TK and KML‐1 cells. (a) TK and KML‐1 cells were incubated with gemcitabine (GEM) (0–1000 ng/mL). (b) Histograms of CD20 expression in TK and KML‐1 cells untreated or treated with GEM. The black line indicates the isotype control, the blue line indicates untreated cells and the red line indicates cells treated with 1000 ng/mL of GEM. (c) TK and KML‐1 cells were incubated with GEM (1000 ng/mL) for 0, 6, 12, 24 and 48 h. (d) LEN (0–30 nM) and (e) AZA (0–300 nM). After a 48‐h incubation, the cells were analyzed for CD20 expression by flow cytometry (n = 3). The results were presented as CD20∆MFI (mean fluorescence intensity [MFI] of CD20 subtracting MFI of isotype control). P‐values were calculated by Student's t‐test (*P < 0.01). Error bars represent SD.
In contrast, LEN treatment did not induce CD20 upregulation, nor did AZA treatment. (Fig. 1d,e).
Enhanced expression of CD20 protein in TK cells was demonstrated by western blot analysis (Fig. 2a). Immuno‐fluorescent microscopy also demonstrated CD20 upregulation in TK cells by GEM treatment (Fig. 2b). CD20 mRNA expression was significantly upregulated after 12 h treatment with GEM (1000 ng/mL) (Fig. 2c) but decreased following GEM treatment for longer than 12 h.
Figure 2.
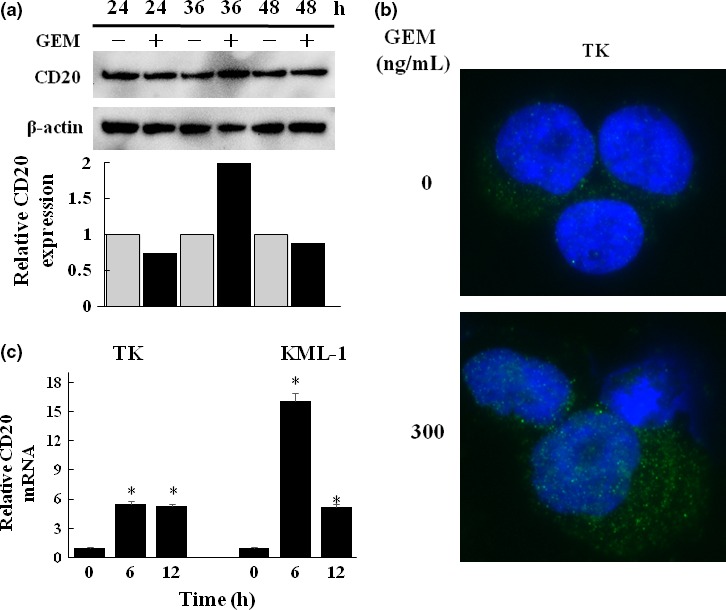
Effect of gemcitabine (GEM) on the expression of CD20 mRNA and protein. (a) TK cells were untreated or treated with GEM (1000 ng/mL) for 24, 36 and 48 h and CD20 expression was examined by western blot at each time point. Analysis of the relative CD20 expression was performed using Image J. (b) TK cells were untreated or treated with GEM (300 ng/mL) for 48 h. They were subsequently stained with a FITC‐conjugated anti‐CD20 mAb (green) and DAPI (blue). The cells were observed under immuno‐fluorescent microscopy. (c) TK and KML‐1 cells were incubated with GEM (1000 ng/mL) for 6 and 12 h, and CD20mRNA expression was examined by qRT‐PCR. The results were quantified by the delta‐Ct method using 18S as a reference (*P < 0.01).
Rituximab binding level on TK and KML‐1 cells was enhanced by CD20 upregulation by gemcitabine treatment
We next examined whether upregulation of surface CD20 expression enhanced the level of rituximab binding in TK and KML cells. Rituximab binding to surface CD20 in GEM‐treated TK and KML‐1 cells was increased compared with untreated cells (Fig. 3a,b).
Figure 3.
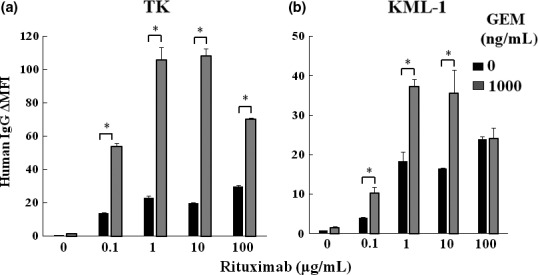
Gemcitabine (GEM) treatment increased rituximab binding to CD20 on DLBCL cells. (a) TK and (b) KML‐1 cells were incubated with GEM (1000 ng/mL) and rituximab (0–100 μg/mL) for 48 h. The binding of rituximab (human IgG1) was examined using flow cytometry (n = 3). The results were presented as human IgG∆MFI (MFI of human IgG subtracting MFI of isotype control). P‐values were calculated by Student's t‐test (*P < 0.01). Error bars represent SD (*P < 0.01).
Gemcitabine enhanced rituximab‐mediated complement dependent cytotoxicity
We investigated whether the GEM‐induced upregulation of surface CD20 leads to enhanced rituximab‐mediated CDC activity. GEM treatment of TK cells synergistically enhanced rituximab‐mediated CDC activity in a dose‐dependent manner (Fig. 4a). In KML‐1 cells, however, a synergistic effect of GEM and rituximab was observed only in cells treated with 1000 ng/mL of GEM and 100 μg/mL of rituximab (Fig. 4b). In contrast, treatment of TK and KML‐1 cells with LEN, a drug that did not increase CD20 expression, did not enhance rituximab‐mediated‐CDC activity (Fig. 4c,d).
Figure 4.
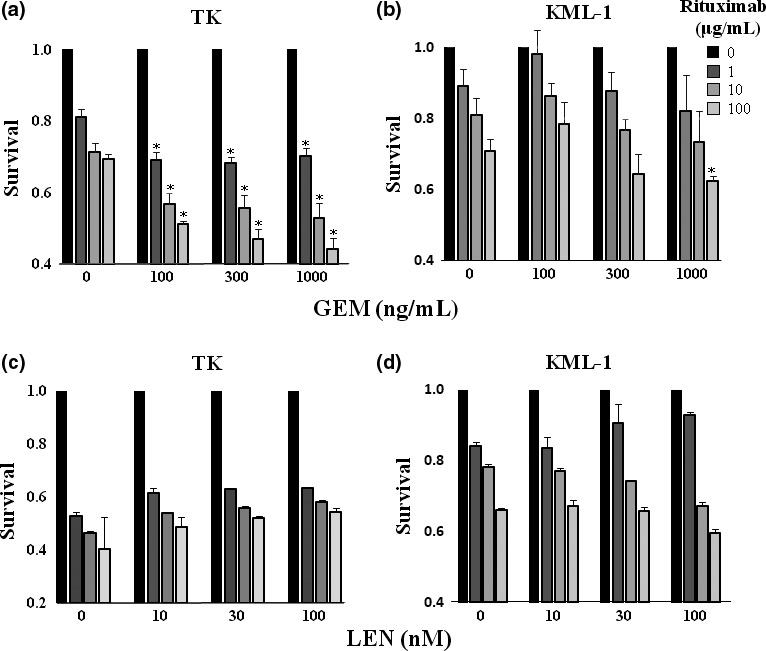
Gemcitabine (GEM) treatment enhanced rituximab‐mediated complement dependent cytotoxicity (CDC) activity. (a) TK cells and (b) KML cells were incubated with GEM (0–1000 ng/mL) and rituximab (0–100 μg/mL) with 10% human AB serum for 48 h. Following staining with propidium iodide (PI), cells were examined for apoptosis by flow cytometry (n = 3). The results were presented as a ratio of the viability of untreated cells to cells treated with GEM. Statistical analysis was performed between the cells treated with both rituximab and GEM and those treated with rituximab alone. P‐values were calculated by Student's t‐test (*P < 0.01). Error bars represent SD. (c) TK cells and (d) KML cells were incubated with LEN (0–100 nM) and rituximab (0–100 μg/mL) with 10% human AB serum for 48 h. Following staining with PI, cells were examined for apoptosis by flow cytometry (n = 3). The results were presented as a ratio of the viability of untreated cells to cells treated with LEN. Statistical analysis was performed between the cells treated with both rituximab and LEN and those treated with rituximab alone. P‐values were calculated by Student's t‐test (*P < 0.01). Error bars represent SD.
Gemcitabine treatment enhanced expression levels of the complement regulatory proteins
We examined expression levels of complement regulatory proteins which work as complement inhibitors, CD46, CD55 and CD59, in GEM‐treated TK and KML‐1 cells. GEM treatment significantly enhanced CD46 and CD59 protein expressions in KML‐1 cells, but not in TK cells (Fig. 5a,c). In KML‐1 cells, expression of CD46, CD55 and CD59 mRNA was enhanced by GEM treatment (Fig. 5d), while in TK cells, only CD55 mRNA expression was enhanced by GEM treatment (Fig. 5b), but CD55 protein expression was not (Fig. 5a).
Figure 5.
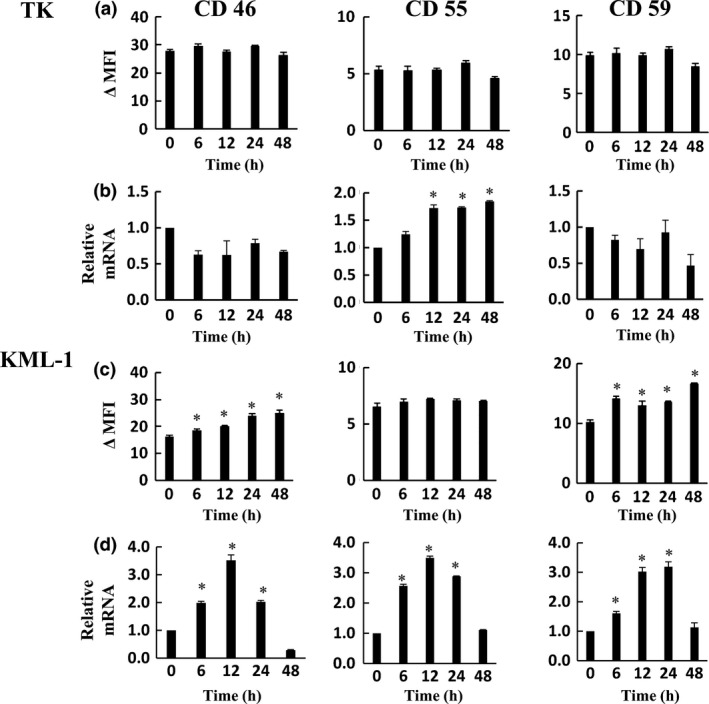
Effect of gemcitabine (GEM) on the expression of CD46, CD55 and CD59 mRNA and protein. TK (a, b) and KML‐1 (c, d) cells were incubated with GEM (1000 ng/mL). After 0, 6, 12, 24 and 48 h incubation, the cells were analyzed for CD46, CD55 and CD59 expression by flow cytometry. The results were presented as ∆MFI (mean fluorescence intensity [MFI] of subtracting MFI of isotype control) (a, c). CD46, CD55 and CD59 mRNA expression was analyzed by qRT‐PCR. The results of qRT‐PCR were quantified by the delta‐Ct method using 18S as a reference (b, d). P‐values were calculated by Student's t‐test (*P < 0.01). Error bars represent SD.
Gemcitabine‐activated NF‐kB signaling is associated with CD20 upregulation
We investigated the activation status of different signaling pathways in untreated and agent‐treated TK cells. GEM treatment for 6 h activated the NF‐kB signaling pathway, but this activation was attenuated following 12 h of GEM treatment (Fig. 6a). Treatment with LEN did not activate the NF‐kB signaling pathway (Fig. 6b). Activation of AKT, ERK and STAT3 was not observed in TK cells treated with GEM (Fig. 6a).
Figure 6.
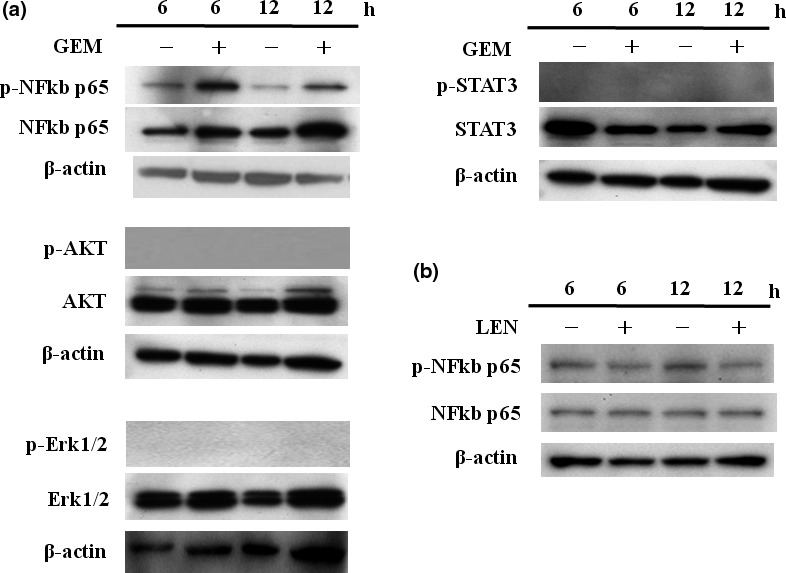
Gemcitabine (GEM)‐activated NF‐kB signaling. TK cells were cultured with GEM (1000 ng/mL) for 6 and 12 h, and the phosphorylation status of NF‐kB, AKT, ERK and STAT3 was examined by western blotting. P‐AKT, p‐ERK and p‐STAT3 expression in other cell types is shown. The same experiments were repeated three times, and identical results were obtained.
The association between NF‐kB activation and GEM‐induced CD20 upregulation was examined using an NF‐kB inhibitor. GEM‐induced CD20 upregulation was significantly suppressed by treatment with the NF‐kB inhibitor BAY 11‐7082 at the mRNA and protein levels (Fig. 7a,b).
Figure 7.
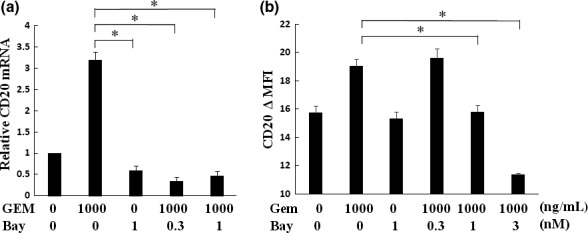
Gemcitabine (GEM)‐induced CD20 upregulation was inhibited by NF‐kB inhibitor treatment. TK cells were treated with BAY 11‐7082 (0.3, 1 μM) for 1 h prior to treatment with GEM (1000 ng/mL). After a 6‐h incubation with both GEM and BAY 11‐7082, (a) CD20 mRNA expression was analyzed by qRT‐PCR. The results of qRT‐PCR were quantified by the delta‐Ct method using 18S as a reference. (b) Surface CD20 expression was analyzed by flow cytometry (n = 3). P‐values were calculated by Student's t‐test (*P < 0.01). Error bars represent SD.
Discussion
CD20 is predominantly expressed as a tetramer on the cell surface, and it functions in the regulation of the store‐operated calcium entry channel.26 CD20 is not internalized upon antibody binding nor is it shed from the cell surface, properties that are considered desirable for a target molecule of antibody therapy.27
Gemcitabine has played an important role in DLBCL treatment, and it was reported that a complete response was achieved in 34% of DLBCL patients in 12 months. The overall survival rate and progression‐free survival rate were 41% and 29% at 12 months, respectively.15 In the present study, our results indicate that surface CD20 protein expression on DLBCL cells was upregulated by GEM treatment. As GEM‐induced upregulation of CD20 increases rituximab binding to CD20 and enhances rituximab‐mediated CDC activity in vivo, it is likely that combination treatment with GEM and rituximab might provide a beneficial therapeutic effect in DLBCL patients. Rituximab treatment is used not only in the treatment of DLBCL but also in relapsed or refractory low‐grade or follicular lymphomas. Mantle cell lymphoma and follicular lymphoma cells also express CD20 on the cell surface. Therefore, combining GEM with rituximab might enhance the efficacy of treatment for these lymphomas by upregulating surface CD20.
Although CD20 upregulation and enhanced rituximab binding on CD20 were observed in both GEM‐treated TK and KML‐1 cells, KML‐1 cells were more resistant to rituximab‐mediated CDC activity than TK cells. It was found that expression of complement regulatory proteins, CD46 and CD59, was also upregulated in GEM‐treated KML‐1 cells but not in GEM‐treated TK cells. Because CD46 and CD59 act as a complement inhibitor,19 GEM‐induced upregulation of these molecules might make KML‐1 cells more resistant to rituximab‐mediated CDC activity.
CD20 protein expression in DLBCL cells was upregulated by GEM‐induced NF‐kB activation. Previous reports demonstrated that GEM also activated NF‐kB in pancreatic cancer cells.28, 29 The CD20 gene promoter contains binding sites to Sp1, NF‐kB, Oct and PU.1.20, 22 Based on these findings, we presume that GEM‐activated NF‐kB binds to the CD20 promoter to induce CD20 transcriptional activation. We evaluated signaling pathways mediated by AKT, ERK and STAT3 that also play important roles in the survival and proliferation of DLBCL cells.30, 31, 32 We found that GEM did not promote the phosphorylation of AKT, ERK or STAT3. Furthermore, the NF‐kB inhibitor BAY 11‐7082 significantly suppressed GEM‐induced CD20 upregulation. Collectively, our results indicate that the activation of NF‐kB is closely associated with GEM‐induced CD20 upregulation.
NF‐kB activation and elevation of CD20mRNA expression were induced 6 h after the initiation of GEM treatment. Intracellular CD20 expression was upregulated in 36–48 h, and surface CD20 expression increased in 48 h following GEM treatment. Our western blot results (Fig. 2a) demonstrated that CD20 expression was enhanced in 36 h and declined in 48 h. Flow cytometry analyses revealed that GEM‐induced CD20 upregulation occurred in 48 h. Viable cells at 48 h were selectively analyzed by gating using flow cytometry for CD20 expression. Both viable cells and the many dead cells resulting from GEM treatment were analyzed together at 48 h by western blotting, which revealed a decrease in CD20 expression.
One important question is whether GEM treatment increases CD20 expression in normal B cells. Given that surface CD20 expression in normal B cells is increased by GEM treatment, normal B cells might also be killed by enhanced rituximab‐mediated CDC activity. As it was reported that CD20 expression in normal B cells was increased by bortezomib treatment,19 it is feasible that surface CD20 in normal cells was similarly upregulated by GEM treatment. However, rituximab does not target hematopoietic stem cells or pro‐B cells due to the absence of surface CD20 expression in these cells. Accordingly, normal B cells can successfully differentiate and proliferate from hematopoietic stem cells and pro‐B cells, and, therefore, a permanent depletion of B‐cells would not result from the combined treatment of GEM and rituximab.
Development of other anti‐CD20 monoclonal antibodies for clinical or pre‐clinical studies is currently in progress.33 The radio‐labeled antibody 90yttrium ibritumomab tiuxetan is a CD20‐directed radio‐therapeutic antibody approved for the treatment of relapsed or refractory low‐grade or follicular B cell non‐Hodgkin lymphoma,34 and several trials evaluating its therapeutic activity in DLBCL are in progress.35 It was reported that patients with relapsed or primary refractory DLBCL received 90yttrium ibritumomab tiuxetan and that the overall response rate was over 50%.36 GEM treatment might enhance 90yttrium ibritumomab tiuxetan binding to CD20 and, thus, elicit a stronger direct cytotoxic effect on lymphoma cells. Ofatumumab is a newly approved CD20 monoclonal antibody used for the treatment of chronic lymphocytic leukemia. Ofatumumab binds an epitope of CD20 distinct from that of rituximab.37 The overall response rate with ofatumumab was 61%, and the complete response rate was 37%.38 The mechanisms of the anti‐tumor effect of ofatumumab include CDC and ADCC.39 Combination treatment with GEM and ofatumumab in patients with DLBCL might similarly demonstrate improved efficacy being that GEM pre‐treatment enhances the expression of CD20, the molecule targeted by ofatumumab.
We found that GEM induced the upregulation of CD20 expression, but the effect of GEM on the expression of other B cell surface markers such as CD19, CD22 and CD23 is unknown. The CD19 gene is known to be expressed under the control of the transcription factor B‐cell‐specific activator protein (BSAP),40 but the interaction between BSAP and NF‐kB is unclear. In contrast, potential AP‐1 and NF‐kB binding sites in the CD22 and CD23 promotor region have been identified.41, 42 Therefore, it is possible that GEM might upregulate CD22 and CD23 expression, and, in doing so, GEM treatment might also contribute to the efficacy of monoclonal antibody therapies targeting these antigens.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgements
This research was supported by The Jikei University Research Fund for Graduate Students. Rituximab was kindly provided by Zenyaku Kogyo (Tokyo, Japan).
Cancer Sci 107 (2016) 682–689
Funding Information
The Jikei University Research Fund for Graduate Students.
References
- 1. Armitage JO, Weisenburger DD. New approach to classifying non‐Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non‐Hodgkin's Lymphoma Classification Project. J Clin Oncol 1998; 16: 2780–95. [DOI] [PubMed] [Google Scholar]
- 2. Pettengell R, Linch D; Haemato‐Oncology Task Force of the British Committee for Standards in Haematology . Position paper on the therapeutic use of rituximab in CD20‐positive diffuse large B‐cell non‐Hodgkin's lymphoma. Br J Haematol 2003; 121: 44–8. [DOI] [PubMed] [Google Scholar]
- 3. Sehn LH, Donaldson J, Chhanabhai M et al Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B‐cell lymphoma in British Columbia. J Clin Oncol 2005; 23: 5027–33. [DOI] [PubMed] [Google Scholar]
- 4. Ardeshna KM, Qian W, Smith P et al Rituximab versus a watch‐and‐wait approach in patients with advanced‐stage, asymptomatic, non‐bulky follicular lymphoma: an open‐label randomised phase 3 trial. Lancet Oncol 2014; 15: 424–35. [DOI] [PubMed] [Google Scholar]
- 5. Hallek M, Fischer K, Fingerle‐Rowson G et al Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open‐label, phase 3 trial. Lancet 2010; 376: 1164–74. [DOI] [PubMed] [Google Scholar]
- 6. Edwards JC, Szczepanski L, Szechinski J et al Efficacy of B‐cell‐targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 2004; 350: 2572–81. [DOI] [PubMed] [Google Scholar]
- 7. Beck L, Bomback AS, Choi MJ et al KDOQI US commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis. Am J Kidney Dis 2013; 62: 403–41. [DOI] [PubMed] [Google Scholar]
- 8. Stone JH, Merkel PA, Spiera R et al Rituximab versus cyclophosphamide for ANCA‐associated vasculitis. N Engl J Med 2010; 363: 221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manches O, Lui G, Chaperot L et al In vitro mechanisms of action of rituximab on primary non‐Hodgkin lymphomas. Blood 2003; 101: 949–54. [DOI] [PubMed] [Google Scholar]
- 10. Cardarelli PM, Quinn M, Buckman D et al Binding to CD20 by anti‐B1 antibody or F(ab’)(2) is sufficient for induction of apoptosis in B‐cell lines. Cancer Immunol Immunother 2002; 51: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akhtar S, Maghfoor I. Rituximab plus CHOP for diffuse large‐B‐cell lymphoma. N Engl J Med 2002; 346: 1830–1; author reply 1830–31. [DOI] [PubMed] [Google Scholar]
- 12. Nishimori H, Matsuo K, Maeda Y et al The effect of adding rituximab to CHOP‐based therapy on clinical outcomes for Japanese patients with diffuse large B‐cell lymphoma: a propensity score matching analysis. Int J Hematol 2009; 89: 326–31. [DOI] [PubMed] [Google Scholar]
- 13. Purroy N, Bergua J, Gallur L et al Long‐term follow‐up of dose‐adjusted EPOCH plus rituximab (DA‐EPOCH‐R) in untreated patients with poor prognosis large B‐cell lymphoma. A phase II study conducted by the Spanish PETHEMA Group. Br J Haematol 2015; 169: 188–98. [DOI] [PubMed] [Google Scholar]
- 14. Aviles A, Neri N, Huerta‐Guzman J, de Jesus Nambo M. ESHAP versus rituximab‐ESHAP in frail patients with refractory diffuse large B‐cell lymphoma. Clin Lymphoma Myeloma Leuk 2010; 10: 125–8. [DOI] [PubMed] [Google Scholar]
- 15. Lopez A, Gutierrez A, Palacios A et al GEMOX‐R regimen is a highly effective salvage regimen in patients with refractory/relapsing diffuse large‐cell lymphoma: a phase II study. Eur J Haematol 2008; 80: 127–32. [DOI] [PubMed] [Google Scholar]
- 16. Larouche JF, Berger F, Chassagne‐Clement C et al Lymphoma recurrence 5 years or later following diffuse large B‐cell lymphoma: clinical characteristics and outcome. J Clin Oncol 2010; 28: 2094–100. [DOI] [PubMed] [Google Scholar]
- 17. Taylor RP, Lindorfer MA. Antigenic modulation and rituximab resistance. Semin Hematol 2010; 47: 124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith MR. Rituximab (monoclonal anti‐CD20 antibody): mechanisms of action and resistance. Oncogene 2003; 22: 7359–68. [DOI] [PubMed] [Google Scholar]
- 19. Bil J, Winiarska M, Nowis D et al Bortezomib modulates surface CD20 in B‐cell malignancies and affects rituximab‐mediated complement‐dependent cytotoxicity. Blood 2010; 115: 3745–55. [DOI] [PubMed] [Google Scholar]
- 20. Winiarska M, Nowis D, Bil J et al Prenyltransferases regulate CD20 protein levels and influence anti‐CD20 monoclonal antibody‐mediated activation of complement‐dependent cytotoxicity. J Biol Chem 2012; 287: 31983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wojciechowski W, Li H, Marshall S, Dell'Agnola C, Espinoza‐Delgado I. Enhanced expression of CD20 in human tumor B cells is controlled through ERK‐dependent mechanisms. J Immunol 2005; 174: 7859–68. [DOI] [PubMed] [Google Scholar]
- 22. Shimizu R, Kikuchi J, Wada T, Ozawa K, Kano Y, Furukawa Y. HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells. Leukemia 2010; 24: 1760–8. [DOI] [PubMed] [Google Scholar]
- 23. Sivaraman S, Venugopal P, Ranganathan R et al Effect of interferon‐alpha on CD20 antigen expression of B‐cell chronic lymphocytic leukemia. Cytokines Cell Mol Ther 2000; 6: 81–7. [DOI] [PubMed] [Google Scholar]
- 24. Crump M, Baetz T, Couban S et al Gemcitabine, dexamethasone, and cisplatin in patients with recurrent or refractory aggressive histology B‐cell non‐Hodgkin lymphoma: a phase II study by the National Cancer Institute of Canada Clinical Trials Group (NCIC‐CTG). Cancer 2004; 101: 1835–42. [DOI] [PubMed] [Google Scholar]
- 25. Crump M, Kuruvilla J, Couban S et al Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem‐cell transplantation for relapsed and refractory aggressive lymphomas: NCIC‐CTG LY.12. J Clin Oncol 2014; 32: 3490–6. [DOI] [PubMed] [Google Scholar]
- 26. Rose AL, Smith BE, Maloney DG. Glucocorticoids and rituximab in vitro: synergistic direct antiproliferative and apoptotic effects. Blood 2002; 100: 1765–73. [PubMed] [Google Scholar]
- 27. Li H, Ayer LM, Polyak MJ et al The CD20 calcium channel is localized to microvilli and constitutively associated with membrane rafts: antibody binding increases the affinity of the association through an epitope‐dependent cross‐linking‐independent mechanism. J Biol Chem 2004; 279: 19893–901. [DOI] [PubMed] [Google Scholar]
- 28. Chen D, Niu M, Jiao X, Zhang K, Liang J, Zhang D. Inhibition of AKT2 enhances sensitivity to gemcitabine via regulating PUMA and NF‐kappaB signaling pathway in human pancreatic ductal adenocarcinoma. Int J Mol Sci 2012; 13: 1186–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arlt A, Gehrz A, Muerkoster S et al Role of NF‐kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine‐induced cell death. Oncogene 2003; 22: 3243–51. [DOI] [PubMed] [Google Scholar]
- 30. Uddin S, Hussain AR, Siraj AK et al Role of phosphatidylinositol 3′‐kinase/AKT pathway in diffuse large B‐cell lymphoma survival. Blood 2006; 108: 4178–86. [DOI] [PubMed] [Google Scholar]
- 31. Dai B, Zhao XF, Mazan‐Mamczarz K et al Functional and molecular interactions between ERK and CHK2 in diffuse large B‐cell lymphoma. Nat Commun 2011; 2: 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang X, Meng B, Iqbal J et al Activation of the STAT3 signaling pathway is associated with poor survival in diffuse large B‐cell lymphoma treated with R‐CHOP. J Clin Oncol 2013; 31: 4520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lim SH, Beers SA, French RR, Johnson PW, Glennie MJ, Cragg MS. Anti‐CD20 monoclonal antibodies: historical and future perspectives. Haematologica 2010; 95: 135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uike N, Choi I, Tsuda M et al Factors associated with effects of 90Y‐ibritumomab tiuxetan in patients with relapsed or refractory low‐grade B cell non‐Hodgkin lymphoma: single‐institution experience with 94 Japanese patients in rituximab era. Int J Hematol 2014; 100: 386–92. [DOI] [PubMed] [Google Scholar]
- 35. Auger‐Quittet S, Duny Y, Daures JP, Quittet P. Outcomes after (90) Yttrium‐ibritumomab tiuxetan‐BEAM in diffuse large B‐cell lymphoma: a meta‐analysis. Cancer Med 2014; 3: 927–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morschhauser F, Illidge T, Huglo D et al Efficacy and safety of yttrium‐90 ibritumomab tiuxetan in patients with relapsed or refractory diffuse large B‐cell lymphoma not appropriate for autologous stem‐cell transplantation. Blood 2007; 110: 54–8. [DOI] [PubMed] [Google Scholar]
- 37. Teeling JL, Mackus WJ, Wiegman LJ et al The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol 2006; 177: 362–71. [DOI] [PubMed] [Google Scholar]
- 38. Matasar MJ, Czuczman MS, Rodriguez MA et al Ofatumumab in combination with ICE or DHAP chemotherapy in relapsed or refractory intermediate grade B‐cell lymphoma. Blood 2013; 122: 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin TS. Ofatumumab: a novel monoclonal anti‐CD20 antibody. Pharmgenomics Pers Med 2010; 3: 51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mei HE, Schmidt S, Dorner T. Rationale of anti‐CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther 2012; 14(Suppl 5): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilson GL, Najfeld V, Kozlow E, Menniger J, Ward D, Kehrl JH. Genomic structure and chromosomal mapping of the human CD22 gene. J Immunol 1993; 150: 5013–24. [PubMed] [Google Scholar]
- 42. Li H, Chehade M, Liu W, Xiong H, Mayer L, Berin MC. Allergen‐IgE complexes trigger CD23‐dependent CCL20 release from human intestinal epithelial cells. Gastroenterology 2007; 133: 1905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]