

Abstract
Protein dynamics on the microsecond (μs) time scale were investigated by temperature-jump fluorescence spectroscopy as a function of temperature in two variants of a thermophilic alcohol dehydrogenase: W87F and W87F:H43A. Both mutants exhibit a fast, temperature-independent μs decrease in fluorescence followed by a slower full recovery of the initial fluorescence. The results, which rule out an ionizing histidine as the origin of the fluorescence quenching, are discussed in the context of a Trp49-containing dimer interface that acts as a conduit for thermally activated structural change within the protein interior.
Protein dynamics on the microsecond (μs) time scale have been shown to be important in processes involving concerted fluctuations or protein networks that affect catalysis, protein folding, and allostery.1−5 Moreover, many efforts are aimed at extending MD simulations out to this time scale, as they are thought to be most informative for long-range motions of biological interest.6−8
A thermophilic alcohol dehydrogenase isolated from Bacillus stearothermophilus (ht-ADH) has been shown kinetically to sample two distinct conformational ensembles controlling hydride transfer above and below 30 °C.9,10 Previous H/D exchange-mass spectrometry (HDX) studies on ht-ADH revealed that nearly half of the total peptides analyzed undergo a discontinuous increase in amide-backbone deuteron incorporation from the solvent occurring between 20 and 55 °C.11 While HDX studies provided the first structural evidence for separate equilibrium processes in the apo-state of protein at low and high temperatures, the time scale of these measurements (seconds to hours) represents equilibrium local unfolding processes that are too long to enable conformational interconversions to be monitored in real time. Therefore, subsequent attempts were made to interrogate the conformational phase transition on the picosecond (ps) to nanosecond (ns) time scale using time-resolved fluorescence relaxation and Stokes shift studies.12 Of particular interest is V260A, a cofactor binding site mutant that drastically enhances the difference in activation enthalpies at low and high temperature for hydride transfer. When introduced to the single active site tryptophan variant (W87in), two conformational ensembles with distinctly different fluorescence properties were revealed above and below 30 °C.13
It was concluded that the protein motions causing the temperature-dependent transition in ht-ADH activity are essentially frozen on the fast time scale of fluorescence measurements, arguing for methods that can detect motions on time scales that are longer than ns and shorter than the ms time scale of catalysis. In the present study we have implemented a series of temperature-jump fluorescence (TJF) studies capable of examining μs relaxations in response to very rapid ns jumps in temperature of approximately 7 °C (Figure S1). A striking transient fluorescence quenching and recovery is observed, attributed to a thermally activated uptake and subsequent release of solvent water(s) that is accompanied by structural perturbation within the protein interior.
Initial TJF studies of wild-type ht-ADH (WT) were found to be unreliable due to persistent cavitation effects (Figure S2). Subsequent screening of several modified Trp variants of ht-ADH led to a series of cavitation-free TJF experiments for W87F over a range of temperatures; W87F retains Trp at both position 49 within a dimer interface that is close to the active site and at position 167, a control position near the protein surface (Figure S3). Kinetic characterization of W87F indicated that W87F retains the signature break at 30 °C and a difference in EA of approximately 6.0 ± 0.7 kcal/mol, within error of that observed in WT (Table S1).
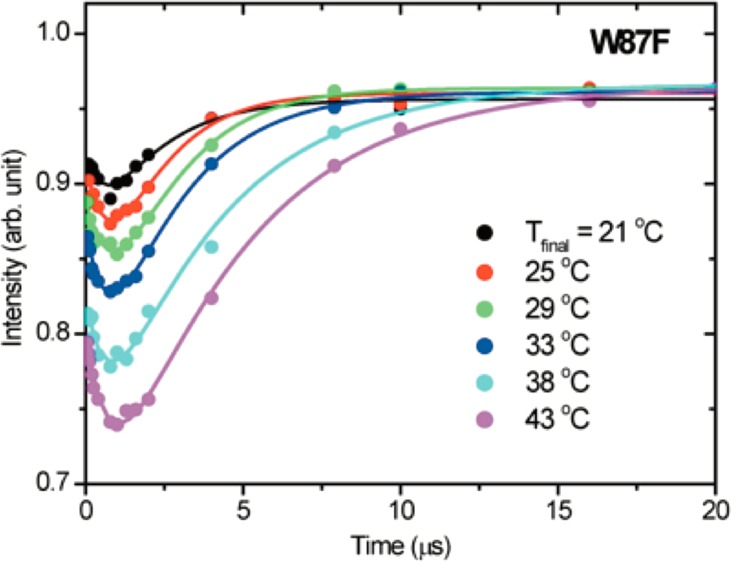
The temperature dependence of the TJF relaxation kinetic traces for W87F is shown in Figure 1. Of immediate note is a novel biphasic behavior in which the fluorescence intensity drops and recovers to almost its initial value. To our knowledge, such behavior is rare in this time regime, with only a single earlier example from TJF studies of apo-Myoglobin unfolding.14 In that case, the fluorescence evolved in an opposite fashion from the present study, leading to a transient increase in fluorescence intensity that subsequently decayed to the initial quenched state. T-jump fluorescence studies on lactate dehydrogenase and triosephosphate isomerase revealed similar kinetic traces on the ns time scale.15,16 However, neither system exhibited a full recovery to the initial fluorescence intensity and the associated μs dynamics evolved in a unidirectional manner.
Figure 1.
T-jump (=7 °C) induced fluorescence (λex = 295 nm and λem = 355 nm) time courses as a function of final temperature for the W87F form of ht-ADH. The plotted intensities were normalized to “probe-only” emission spectra obtained at −0.6 μs, and the experimental data were fit to a double exponential (Table 1). The final level of protein fluorescence is within 4% of the initial fluorescence in each case.
A biexponential fitting of the data in Figure 1 yields two kinetically important processes: an initial, temperature independent loss of fluorescence (τ1) with an average relaxation lifetime of 0.77 ± 0.06 μs, followed by a slower microsecond relaxation that returns the system to its initial intensity (τ2). In contrast to the temperature independence seen for τ1, the recovery phase describing τ2 shows a trend in which the rate is reduced with increasing temperature (Table 1). This slowing of the recovery phase is associated with increased accumulation of the quenched state at elevated temperatures.
Table 1. Temperature Dependence of TJF Relaxation Times.
| W87F |
W87F:H43A |
|||||
|---|---|---|---|---|---|---|
| Tfinal (°C)a | τ1 (μs)b | τ2 (μs)b | intensity loss %c,d | τ1 (μs)b | τ2 (μs)b | intensity loss %c,d |
| 21e | 0.6 ± 0.3 | 1.5 ± 0.1 | 10 | 0.6 ± 0.3 | 0.8 ± 0.2 | 17 |
| 25 | 0.8 ± 0.5 | 2.1 ± 0.4 | 12 | N.D.f | N.D.f | N.D.f |
| 29e | 1.0 ± 0.5 | 2.8 ± 0.3 | 14 | 0.5 ± 0.3 | 0.9 ± 0.6 | 13 |
| 33g | 0.7 ± 0.1 | 3.1 ± 0.4 | 17 | N.D.f | 2.1 ± 1.0 | 19 |
| 38 | 0.7 ± 0.2 | 4.6 ± 0.7 | 22 | 0.7 ± 0.2 | 1.9 ± 0.2 | 24 |
| 43 | 0.8 ± 0.1 | 5.6 ± 0.8 | 26 | 0.4 ± 0.1 | 5.6 ± 1.6 | 25 |
Final temperature following the T-jump of 7 °C.
Error reported is the standard error.
Determined as the difference between the fluorescence intensity at time zero and the fluorescence intensity at the inflection point for each respective temperature.
Absolute error <1% for all measurements.
Temperatures 22 and 28 °C are reported here for W87F:H43A instead of 21 and 29 °C.
N.D. denotes not determined.
For W87F:H43A at 33 °C, the fast transient could not be resolved and only the recovery was fit to a monoexponential.
The observed fluorescence is associated with the two tryptophans in our variant of ht-ADH, Trp49 and Trp167. However, Trp167 at the protein surface is unlikely to be subjected to the quenching seen in TJF for two reasons. First, the nanosecond collisional quenching data at Trp167 revealed a ΔH‡ in line with that observed for diffusion between water and free Trp (3.7 kcal/mol),17 in stark contrast with the observed temperature independence assigned to the τ1 quenching phenomenon (Table 1). Second, Trp167 exhibits a very small Stokes Shift with rates unaffected by increasing temperature, indicating that there is little interaction between this locale and bulk solvent or neighboring side chains.13,18,19 Moreover, neither of these fluorescence properties at Trp167 is altered in the presence of mutants at either the Tyr-Tyr dimer interface or cofactor binding site.13
On this basis, we eliminate a contribution from Trp167 to the TJF quenching and ascribe the observed behavior to Trp49. One mechanism for fluorescence quenching could be attributed to a T-jump-induced change in protonation of a nearby His43 (Figure 2) as a result of a temperature-dependent perturbation of its side chain pKa. Precedence for temperature-dependent pKa shifts of histidine residues in enzymes has been well documented by NMR studies.20,21
Figure 2.
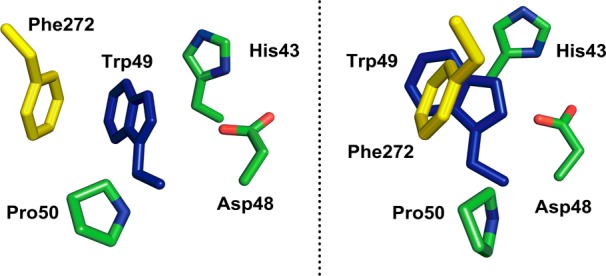
Close-up of the residues surrounding Trp49 in ht-ADH (left). On the right is a rotated view that shows the extent of Trp49 shielding (blue subunit) by an intersubunit π-stack with Phe272 (yellow subunit), see Figure 4 for color code. The remaining residues (green) all reside within the same subunit as Trp49.
To test this hypothesis, an additional mutant of ht-ADH was prepared, W87F:H43A. While its turnover rate is reduced relative to W87F, it exhibits the signature break in behavior at 30 °C, coupled with activation parameters and kinetic isotope effects that are within error of that found for W87F (Table S1). However, W87F:H43A exhibits TJF behavior very similar to that of W87F (Figure S4). The quenching phase is slightly faster on average for W87F:H43A (τ1 = 0.55 ± 0.06 μs) and remains temperature independent. The recovery phase, albeit slightly faster than W87F, also slows with increasing temperature in a manner similar to that observed for W87F (Table 1). Because the Arrhenius plots for τ2 are parallel (Figure 3), any local perturbation of the protonation state at His43 can be eliminated as the mechanism for Trp49 quenching.
Figure 3.
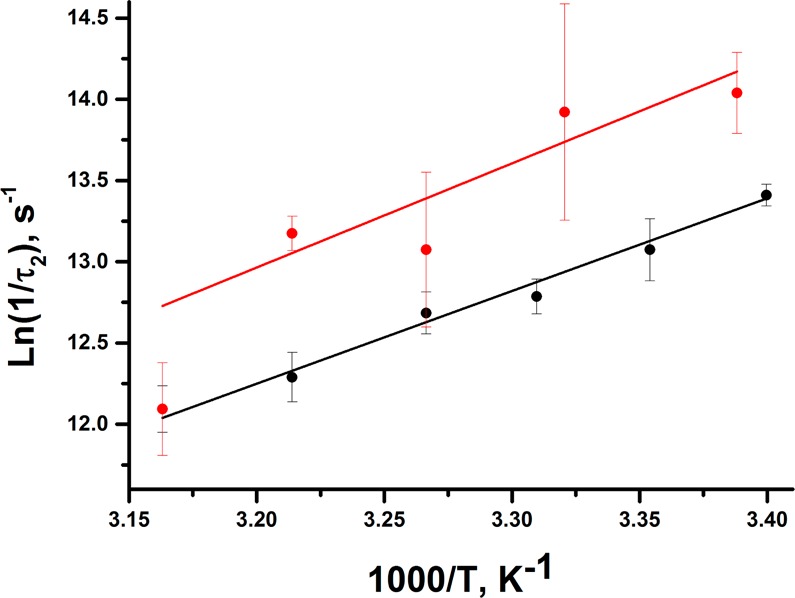
Arrhenius behavior associated with the slow phase (τ2) for W87F (black) and W87F:H43A (red).
Hence, we propose a mechanism that takes into account a more global view of the protein structure to explain the origin of the ht-ADH TJF behavior; this involves two detectable intermediates (E(II) and E(III), Scheme 1). While the final fluorescence intensities evolve to an almost fully recovered intensity after 20 μs in both W87F and W87F:H43A (Figures 1 and S4), the temperature dependence of the static fluorescence intensities (Figure S5) implicates a third kinetic process extending beyond the μs regime; eventually, protein reverts to its initial structure upon cooling.
Scheme 1. Upon ps Excitation, Native Enzyme, E(I), Undergoes a Sub-μs Transition to an Intermediate, E(II), That Is Attributed to an Ingress of Solvent Water(s).

This intermediate can either partition back to E(I) or evolve in a few μs to a new, structurally distinct intermediate, E(III); both processes are accompanied by an expulsion of water from the dimer interface.
Previous studies of ht-ADH have identified a symmetrical Tyr-Tyr π-stack at a subunit interface that plays an important role in the rigidification of ht-ADH that occurs below 30 °C and appears to be critical in the adaptation of prokaryotic ADHs to function within different temperature niches.22 To explain the TJF quenching and recovery, we propose that the newly implicated Phe272-Trp49 pair provides a local pathway for the transmission of thermally induced structural changes (Figure 4). While Trp49 is close to solvent, it is shielded by the Phe272 side chain from the adjacent subunit, increasing the hydrophobicity around Trp49 and yielding a relatively high initial fluorescence intensity (Figure 2, right). We suggest that the subsequent decrease in fluorescence intensity (τ1) is due to an influx of bulk solvent around Trp49 triggered by a thermally induced rearrangement of Phe272, thereby widening the diffusive channel at the intersubunit interface. Examination of the TJ fluorescence spectra reveals a red shift in the quenched state, E(II), consistent with Trp49 exposure to water; however, the extent of the shift is difficult to ascertain because of concomitant changes in intensity and spectral width. Water penetration is often interpreted as a fast initial phase in thermally induced protein unfolding mechanisms in a variety of systems.14,23−25 It has been well documented that π-stacks in proteins can be greatly weakened when displaced from optimal orientations by as little as 1 Å.26,27 Because finite rates are measured for τ1 without a significant ΔH‡, the rearrangement that allows water penetration is almost exclusively entropically driven.
Figure 4.
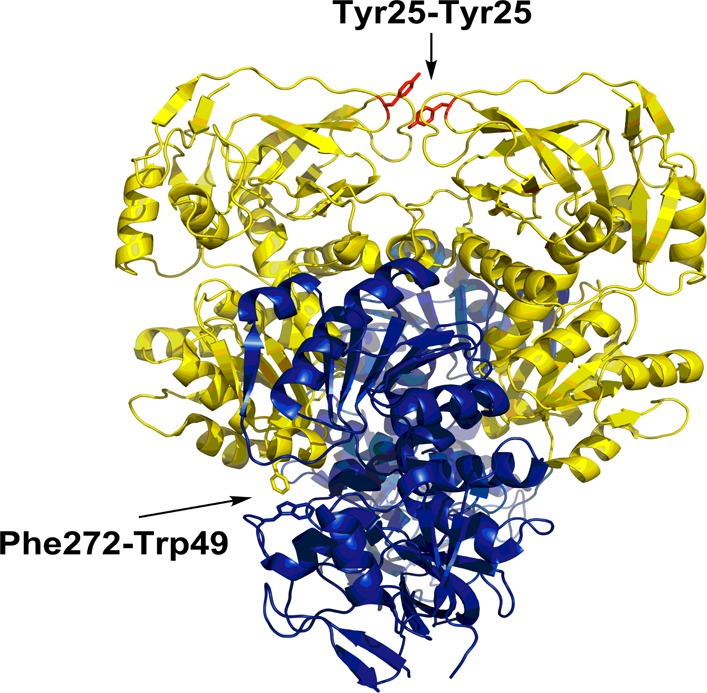
Tetrameric view of ht-ADH showing the relationship of Trp49 (blue subunit) to Phe272 (yellow subunit). The proximity of these two side chains is consistent with an asymmetric π-stack that contributes to oligomeric stability. Disruption at this site via T-jump results in quenched fluorescence from solvent penetration. This interaction contrasts with the symmetrical Tyr25-Tyr25 interaction (red sticks) found at a different subunit interface that has been correlated with the adaptation of ADH to different temperature regimes.
The recovery of the initial fluorescence (τ2) results from restoration of the protecting Phe272 contact by reclosing at this subunit interface and ultimate formation of E(III). The negative ΔH‡ associated with this process is reminiscent of protein folding studies, where negative enthalpies of activation have been attributed to a large differential in heat capacity between the hydrated, unfolded protein and a more compact transition state leading to the folded product.28 For the TJF studies described herein, an increase in the number of bound water molecules at elevated temperature is also a possible factor in decreasing the rate of the recovery phase. This thermally activated structural changes within ht-ADH adjacent to the active site may be related to a reported anisotropic conversion of heat released during single molecule enzyme catalytic turnover into vectorial changes in protein diffusion.29
The present TJF results on ht-ADH have uncovered μs motions that are anticipated to be critical for global conformational sampling in proteins. They also suggest that solvent accessible regions between subunits in protein oligomers may play a role in facilitating heat transfer from a protein surface to catalytically relevant protein residues. It will be of considerable interest to see if a similar role for an asymmetric dimer interface can be identified in other ADHs functioning optimally over various temperature regimes.
Acknowledgments
Support from NIGMS (025765 to J.P.K. and 25158 to T.G.S.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04413.
Methods, static fluorescence spectra, monomeric protein structure, enzyme kinetics, and WT and W87F:H43A TJF traces (PDF)
Author Present Address
# Fuels Synthesis Division, Joint BioEnergy Institute, 5885 Hollis St 4th Floor, Emeryville, CA 94608, United States.
Author Present Address
∇ Mass Spectrometry and Biophysics Center of Excellence, Bristol Myers Squibb, 311 Pennington Rocky Hill Road, Pennington, New Jersey 08354, United States.
Author Contributions
⊥ C.W.M. and G.B. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Lange O. F.; et al. Science 2008, 320, 1471. 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Volkman B. F.; Lipson D.; Wemmer D. E.; Kern D. Science 2001, 291, 2429. 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- Liu F.; Gruebele M. Chem. Phys. Lett. 2008, 461, 1. 10.1016/j.cplett.2008.04.075. [DOI] [Google Scholar]
- Yang H.; et al. Science 2003, 302, 262. 10.1126/science.1086911. [DOI] [PubMed] [Google Scholar]
- Chakrapani S.; Auerbach A. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 87. 10.1073/pnas.0406777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino P. L.; Liu F.; Gruebele M.; Schulten K. Biophys. J. 2008, 94, L75. 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayachandran G.; Vishal V.; Pande V. S. J. Chem. Phys. 2006, 124, 164902. 10.1063/1.2186317. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; et al. Nature 2007, 450, 838. 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- Kohen A.; Cannio R.; Bartolucci S.; Klinman J. P. Nature 1999, 399, 496. 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Dong M.; Bahnson B. J.; Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10520. 10.1073/pnas.1104989108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z.-X.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9556. 10.1073/pnas.0403337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows C. W.; Ou R.; Klinman J. P. J. Phys. Chem. B 2014, 118, 6049. 10.1021/jp500825x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows C. W.; Tsang J. E.; Klinman J. P. J. Am. Chem. Soc. 2014, 136, 14821. 10.1021/ja506667k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruebele M.; Sabelko J.; Ballew R.; Ervin J. Acc. Chem. Res. 1998, 31, 699. 10.1021/ar970083x. [DOI] [Google Scholar]
- Gulotta M.; Deng H.; Deng H.; Dyer R. B.; Callender R. H. Biochemistry 2002, 41, 3353. 10.1021/bi016009a. [DOI] [PubMed] [Google Scholar]
- Desamero R.; Rozovsky S.; Zhadin N.; McDermott A.; Callender R. Biochemistry 2003, 42, 2941. 10.1021/bi026994i. [DOI] [PubMed] [Google Scholar]
- Eftink M. R.; Ghiron C. A. Biochemistry 1977, 16, 5546. 10.1021/bi00644a024. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- Badea M. G.; Brand L. Meth. Enz. 1979, 61, 378. 10.1016/0076-6879(79)61019-4. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Lecomte J. T. J. Biophys. J. 1997, 73, 3241. 10.1016/S0006-3495(97)78349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrescu A. T.; Mills D. A.; Ulrich E. L.; Chinami M.; Markley J. L. Biochemistry 1988, 27, 2158. 10.1021/bi00406a051. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Cun S.; Klinman J. P. J. Biol. Chem. 2013, 288, 14087. 10.1074/jbc.M113.453951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioni P.; Gabellieri E.; Marchal S.; Lange R. Proteins: Struct., Funct., Genet. 2014, 82, 1787. 10.1002/prot.24532. [DOI] [PubMed] [Google Scholar]
- Caflisch A.; Karplus M. J. Mol. Biol. 1995, 252, 672. 10.1006/jmbi.1995.0528. [DOI] [PubMed] [Google Scholar]
- Yoda T.; et al. Proteins: Struct., Funct., Genet. 2001, 42, 49.. [DOI] [PubMed] [Google Scholar]
- McGaughey G. B.; Gagne M.; Rappe A. K. J. Biol. Chem. 1998, 273, 15458. 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- Chipot C.; Jaffe R.; Maigret B.; Pearlman D. A.; Kollman P. A. J. Am. Chem. Soc. 1996, 118, 11217. 10.1021/ja961379l. [DOI] [Google Scholar]
- Oliveberg M.; Tan Y. J.; Fersht A. R. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 8926. 10.1073/pnas.92.19.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C.; et al. Nature 2015, 517, 227. 10.1038/nature14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.