

Abstract
The cholinergic system plays important roles in both learning and addiction. Medications that modify cholinergic tone can have pronounced effects on behaviors reinforced by natural and drug reinforcers. Importantly, enhancing the action of acetylcholine (ACh) in the nucleus accumbens and ventral tegmental area (VTA) dopamine system can either augment or diminish these behaviors. A threshold model is presented that can explain these seemingly contradictory results. Relatively low levels of ACh rise above a lower threshold, facilitating behaviors supported by drugs or natural reinforcers. Further increases in cholinergic tone that rise above a second upper threshold oppose the same behaviors. Accordingly, cholinesterase inhibitors, or agonists for nicotinic or muscarinic receptors, each have the potential to produce biphasic effects on reward behaviors. Pretreatment with either nicotinic or muscarinic antagonists can block drug- or food- reinforced behavior by maintaining cholinergic tone below its lower threshold. Potential threshold mediators include desensitization of nicotinic receptors and biphasic effects of ACh on the firing of medium spiny neurons. Nicotinic receptors with high- and low-affinity appear to play greater roles in reward enhancement and inhibition, respectively. Cholinergic inhibition of natural and drug rewards may serve as mediators of previously described opponent processes. Future studies should evaluate cholinergic agents across a broader range of doses, and include a variety of reinforced behaviors.
Keywords: acetylcholine, acetylcholinesterase inhibitors, cocaine, donepezil, galantamine, nicotinic receptor, muscarinic receptor, self-administration, reinforcement (Psychology), rivastigmine
Introduction
ACh is widely distributed in the central nervous system, where it functions as a signal for local circuits and projection neurons. Both types of cholinergic neuron are involved in brain learning and reward functions. Synaptic levels of ACh are regulated by choline acetyltransferase, the rate-limiting enzyme for formation of ACh, and cholinesterases that inactivate it. ACh activates two categories of receptor: nicotinic and muscarinic. Neuronal nicotinic ACh receptors (nAChRs) are a family of ligand-gated ion channels that are made of combinations of type 2 through 9 alpha subunits, and type 2 through 4 beta subunits, arranged to form a pentameric pattern. Different subunit combinations give rise to various types of nAChRs, which differ in sensitivity to nicotine, calcium conductance, and propensity to desensitize [1], discussed in greater detail below. In contrast, muscarinic receptors are members of the superfamily of G protein-coupled receptors. Five muscarinic subtypes have been cloned which function through either activation of phospholipase (types 1, 3, and 5) or inhibition of adenylate cyclase to decrease the concentration of intracellular cAMP (types 2 and 4) [2]. Dopamine neurons express multiple types of muscarinic and nicotinic ACh receptors, and a dense mingling of dopaminergic and cholinergic neurons in limbic areas of the brain allows coordinated functioning of these neurotransmitter systems [3,4].
The cholinergic system is well known for its role in learning, memory, and attention. In general, cholinergic activation modifies these functions with an inverted-U dose-effect relationship [5,6]. Accordingly, nicotinic or muscarinic cholinergic antagonists can disrupt learning and memory in human or animal experiments, with this effect reversed by restoring ACh function [7,8]. Either cholinesterase inhibitors or cholinergic agonists with nicotinic or muscarinic selectivity can enhance learning under conditions in which cholinergic function is diminished, but disrupt the same behaviors when administered at higher doses [9,10], which can be associated with signs of yawning, tremor, involuntary jaw movements, and diarrhea in animals [11]. Overall, these findings are consistent with an optimal level of central cholinergic activity for learning and memory, with deviations in either direction capable of impairing learning and memory. Parallel to this, interaction of the ACh and dopamine systems to modulate drug-reinforced and drug-seeking behaviors can also be interpreted using an inverted-U dose-effect relationship.
Behavioral Significance of Striatal Acetylcholine Elevations
Augmented release of ACh in the striatum and nucleus accumbens has been observed under a number of qualitatively different conditions [12]. Locomotor activity in rats is correlated with dialysate levels of ACh in the striatum, hippocampus, frontal cortex [13,14]. Handling of rats increases extracellular ACh in both the nucleus accumbens core and shell, with repeated exposure to an open field further increasing values in the shell but not the core region [15]. Importantly, disruption of an established contingency that requires learning of a new pattern of responding appears to increase extracellular ACh. In the dorsal striatum, reversal of maze requirements for food reward caused pronounced increases in ACh which resolve as rats learn to maximize correct responding [16].
Activation of cholinergic neurons has also been implicated in the rewarding effects of both natural and drug reinforcers [17]. Repeated exposure to different classes of abused substances can produce persistent increases in the activity of cholinergic neurons in the nucleus accumbens [18]. Psychostimulant-reinforced behavior can cause long-lasting decreases in levels of choline acetyltransferase in the nucleus accumbens [19]. During cocaine self-administration, greater increases in ACh occur in dialysate from the nucleus accumbens shell [20] or VTA [21], relative to neurotransmitter increases that occur in animals that receive drug noncontingently. This early-session accentuation also occurs in cocaine-trained animals evaluated during extinction (substitution of inert injections) [21]. As rats acquire reinforcement in a runway model, psychostimulant, opiate, or food induced elevations in ACh in the nucleus accumbens core increase over consecutive trials, while levels of dopamine do not change [22,23]. In these experiments, drug-induced increases in ACh also did not vary in magnitude for rats that received noncontingent injections.
ACh elevations have also been linked to satiety caused by feeding and aversive states [24]. In deprived rats, both ACh and dopamine in the nucleus accumbens increased in response to food or water [25]. For freely feeding rats, extracellular ACh in the nucleus accumbens increases and reaches a maximum as satiety occurs. In drug-dependent animals, withdrawal produced by blockade of opiate, nicotinic, or benzodiazepine receptors increases ACh concentration in the nucleus accumbens [26]. Exposure to a flavor that has been paired with lithium-induced illness increases ACh concentration in dialysate from the nucleus accumbens, and infusion of a cholinesterase inhibitor into the nucleus accumbens can produce conditioned taste aversions [27]. Aversive hypothalamic stimulation (AHS) releases ACh in the nucleus accumbens, and rats that lever press to terminate AHS decrease their concentration of ACh in accumbal dialysate [28]. Apparently, elevated levels of ACh in the nucleus accumbens can serve as a neural indicator of aversiveness, or as a signal that inhibits appetitive behaviors. In many instances, accumbal levels of dopamine change in an opposite direction to that of ACh in response to an aversive stimulus. For example, exposure to an aversively-conditioned flavor [27] or precipitated withdrawal [26] can cause decreases in dopamine that accompany increases in accumbal ACh. Taken together, these findings show that elevations of ACh occur in a variety of settings, with the common element being ecological significance.
A Threshold Model for Cholinergic Effects on Reinforced Behavior
Cholinergic influences can be broadly divided into treatments that either enhance or attenuate reinforced behavior. A two-phase model has been developed to explain seeming discrepancies between these competing actions. It is based on the following two opposing systems, described in Table 1 and shown schematically in Figure 1. Both thresholds are part of normal physiology, functioning in the absence of drug treatments. Short-term increases in VTA-accumbal cholinergic transmission that exceed a low-level threshold increase the probability of reward, but further increases above an upper threshold decrease its probability (reward enhancement and inhibition, respectively). Probability is the likelihood that an organism will choose a given behavior in the future. VTA-accumbal cholinergic transmission is defined as agonist activity at nicotinic and muscarinic receptors within these two brain regions.
Table 1.
Properties of the cholinergic systems underlying reward enhancement and inhibition.
| Threshold Level |
Timing | Reward [and Affective] Effects |
Description |
|---|---|---|---|
| Lower | Rapid, Short- Term |
Enhancement [positive] |
Under baseline, unstimulated conditions (Figure 1, State A), short-term, increases in VTA-accumbal cholinergic transmission that rise above a lower cholinergic threshold promote positive affective states, increasing the probability of reinforcement (Figure 1, State A is advanced to B). |
| Upper | Delayed, accumulates over Time |
Inhibition [negative] |
Larger and more prolonged increases in VTA-accumbal cholinergic transmission that exceed a second, higher value, have an opposite effect, facilitating negative affective states, and decreasing the likelihood of reinforcement (State B is advanced to C). |
Figure 1. Schematic Outline of Biphasic Cholinergic Effects on Reinforcing Events.
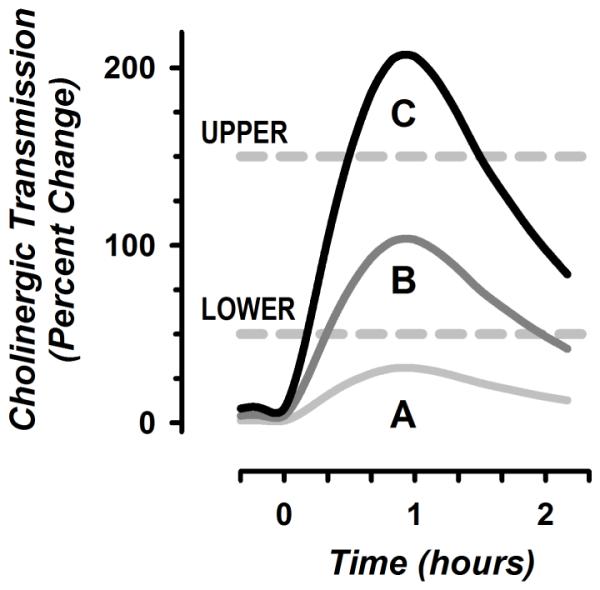
Hypothetical values for cholinergic transmission in the nucleus accumbens and VTA are shown on the vertical axis in arbitrary units, with time plotted horizontally. More precisely, the vertical axis corresponds to momentary agonist activity at cholinergic receptors. This can be provided by either endogenous ACh or medications with nicotinic or muscarinic agonist properties. Cholinergic transmission contributes to the rewarding properties of different events, if its level is increased to a magnitude between the lower and upper thresholds. Further increases above the upper threshold diminish the likelihood of reward. For example, pretreatment with the cholinesterase inhibitor donepezil prevents degradation of ACh in the synaptic cleft. Relatively low doses of donepezil typically used in humans can increase the subjective effects of low-dose cocaine as condition A is changed to condition B (reward enhancement) [186]. In contrast, if administered at relatively high doses in rodents, this agent can also change condition B to C, decreasing drug-reinforced behavior (reward inhibition) [172,178]. Because reward inhibition occurs at the upper limb of the dose-response for cholinergic agents, it is often associated with nonspecific disruption of behavior through adverse events.
The model predicts that behavior which influences cholinergic activation through non-pharmacologic mechanisms that are either psychological or physical can also modify the probability of reward through either threshold. Therefore, reward enhancement and inhibition encompass both psychological and pharmacological constructs. For example, behavior causing increases in endogenous cholinergic tone in the nucleus accumbens and VTA that are intermediate in magnitude, in the absence of drug treatments, are supported (positively reinforced). In contrast, alternative behavior causing larger and sustained increases of endogenous ACh in the same brain regions without drug exposure would be inhibited. Both scenarios can be mimicked by drug treatments leading to intermediate or large increases in cholinergic activation, respectively. As described by Solomon and Corbit [29,30], reward inhibition can be viewed as an opposing process that is a relatively delayed in onset and long-lasting. Reward enhancement and inhibition can occur through activation of nicotinic receptors, muscarinic receptors, or both classes of receptor, as is seen with administration of cholinesterase inhibitors. Desensitization of nicotinic receptors is one potential mediator of differential responding by cholinergic systems: phasic activation for brief periods increases the probability of reward, with an opposite effect after a period of prolonged receptor activation. This property allows the same molecule to produce different effects after dissimilar patterns of cholinergic activity.
In addition to ACh, dopamine and various other neurotransmitter systems work in concert as mediators of motivated behaviors. Because relatively few studies have attempted to decipher the influences of multiple neurotransmitters on reward behavior, the model focuses on the role of ACh. Positive and negative effects of dopaminergic tone may act additively with actions of ACh. More precise characterization of this interaction should be an important goal of future work.
In this review, relevant findings are interpreted in terms of the model and its potential mediators. For more comprehensive accounts of cholinergic influences on reward, the reader is referred to previous reviews by Williams and Adinoff [31], Sofuoglu and Mooney [32], and Mark et al. [33]. Key evidence for the threshold model is based on systemic treatments with cholinergic agonists and antagonists, which may involve more than one functional circuit. Therefore, the review is organized according to type of receptor (nicotinic or muscarinic) and or agent (agonist or antagonist).
Thresholds Mediated by Nicotinic Receptors
Background
As reviewed by Tuesta et al. [34], different subtypes of the nicotinic receptor underlie the reinforcing effects of nicotine, psychostimulants, and other drugs of abuse. Nicotine stimulates release of dopamine by direct activation of high-affinity nAChRs on both the cell bodies [35] and terminal fields [36,37] of midbrain dopaminergic neurons that project to the ventral striatum. VTA dopaminergic neurons express several types of nicotinic receptor subunit, including α3 to α7 and β2 to β4 [38-41]. The α4β2 subtype is the most abundant nicotinic receptor in mammalian brain [42], and variants of the CHRNA4 gene encoding the α4 subunit are associated with altered nicotine dependence in humans [43]. In the striatum, high affinity α4β2* and α6β2* nAChRs are expressed on dopaminergic and GABAergic neurons, where activation leads to calcium influx and greater excitability. This facilitates action potential firing and modulates dopamine release [44,45].
Homomeric α7 nAChRs are activated by both ACh and its ubiquitous breakdown product, choline [46]. In the VTA, receptors containing the α7 subunit are localized on glutamatergic axons that are distinct from cholinergic terminals, suggesting activation through diffusion of ACh or choline; a portion of these receptors are localized to cholinergic terminals in the nucleus accumbens [47]. Homomeric α7 nAChRs are typically activated only after relatively high agonist concentrations, in micromolar amounts [48]. In comparison, activity of α4β2* receptors is most often observed with only nanomolar concentrations of ACh or nicotine [49]. Although the α7-subunit may not form exclusively homomeric receptors in vivo, low agonist sensitivity also appears to be a property of native heteromeric receptors that include this subunit [50,51].
Nicotinic receptors exhibit a characteristic biphasic response to stimulation which is important for behavioral effects: brief, low levels of phasic stimulation can augment ion flux; but prolonged agonist exposure results in decreased responsiveness, termed desensitization [49]. Phasic events are those that are relatively brief, and resolve quickly. There is great variability in the degree to which the nicotinic receptors associated with individual neurons desensitize in response to agonist exposure. After more prolonged exposure to higher agonist concentration, recovery from desensitization typically requires longer periods. Certain subtypes of the nicotinic receptor are activated by phasic release of ACh but become desensitized after sustained ACh binding over seconds to minutes [52]. For VTA dopaminergic neurons, application of either micromolar concentrations of nicotine or rapid bursts of millimolar ACh desensitizes inward nicotinic currents [53]. At high firing rates under physiologic conditions, choline produced by ACh hydrolysis appears to be present at sufficient concentrations to cause homomeric α7 nAChRs to desensitize [54].
The firing of dopaminergic neurons in the VTA is also modulated by GABAergic and glutamatergic inputs that desensitize over different intervals after treatment with nicotine [55]. Because of enhanced glutamatergic transmission combined with inhibition of GABA release, there is a net facilitation of dopamine release that is thought to occur at micromolar concentrations of nicotine [56].
Nicotinic Agonists
Depending on the dose administered, systemic pretreatment with nicotine can have opposite effects on intravenous self-administration of cocaine in a rat model under a simple fixed-ratio-1 (FR-1) schedule in which each lever press resulted in a cocaine injection [57]. In this study, drug taking was enhanced by low-dose nicotine, but diminished after a higher dose. This biphasic effect of systemic nicotine on cocaine-reinforced behavior can be interpreted in as reward enhancement and inhibition mediated by the two thresholds shown in Figure 1. Low levels of nicotinic activation by ACh or other nicotinic agonists favor State A being advanced to State B as a lower threshold is exceeded, increasing the probability of reward. If nicotinic activation continues to increase, and reaches higher levels that exceed an upper threshold, the probability of reward is diminished, corresponding to State C.
Varenicline is a medication approved for use in smoking cessation that acts as a partial agonist at nicotinic α4β2* and α6β2* receptors, and a full agonist at α7 nAChRs [58]. In rhesus monkeys, varenicline can enhance cognitive function across a wide range of doses, 0.001 to 0.3 mg/kg [59]. In some instances, it has been associated with biphasic effects on cocaine-reinforced behavior which are consistent with reward enhancement and inhibition by low- and high-doses, respectively. For example, pretreatment with 0.01 to 0.56 orally in rhesus monkeys potentiated the reinforcing effects of cocaine [60], while 1.0 and 2.0 mg/kg administered subcutaneously to rats attenuated cocaine self-administration [61]. However, low (0.1 and 0.3 mg/kg) varenicline doses also attenuated cue- and cocaine- induced reinstatement of non-reinforced responding in the latter study. Low varenicline doses (0.004 and 0.04 mg/kg-hour) were reported to be ineffective in modifying cocaine reinforcement [62]. But, this protocol was not designed to detect increases in drug-reinforced responding, as monkeys self-administered at their limit of maximal available cocaine injections. Varenicline doses that attenuate cocaine-reinforced behavior in rats are well above those approved for clinical use. Consistent with this, varenicline treatment was found to be ineffective in facilitating abstinence from use of cocaine or opiates in treatment-seeking patients, but did decrease smoking in this population [63].
Knockouts and Antagonists
Nicotinic acetylcholine-receptor systems are important mediators of cognitive function. Pretreatment with the nonselective, noncompetitive nicotinic antagonist mecamylamine can impair learning and memory with threshold systemic doses of 1.0 to 5.0 mg/kg in rats [64]. For rats performing below criterion (less than 80% correct responses at baseline) on a food-reinforced, five-choice serial reaction time task [65], pretreatment with nicotine or an α4β2* agonist increased correct responding, and an α7-selective agonist was without effect. This may correspond to augmentation of nicotinic tone from a relatively low level to exceed a lower cholinergic threshold, causing reward enhancement. In rats that initially performed above criterion, mecamylamine pretreatment decreased accuracy. In addition to negative effects on learning and memory, nicotinic blockade may also decrease the value of food reinforcers, because cholinergic transmission falls short of its lower threshold. Systemic pretreatment with antagonists with α4β2* or α7 selectivity in this study did not alter responding [65], which may reflect involvement of other neurotransmitters.
After knock-out of the CHRNB2 gene which codes for the β2 nicotinic subunit, elderly mice exhibit impaired fear conditioning, diminished spatial learning, and elevations of basal plasma corticosterone [66,67]; implicating a role for endogenous ACh in learning and stress reactivity during aging. CHRNB2 deletion also disrupts nicotine-induced dopamine release by the ventral striatum [68], as well as nicotine self-administration [69]. In addition, intravenous self-administration of nicotine is prevented by deletion of either the CHRNA4 or CHRNA6 genes, which code for the α4 and α6 subunits respectively [70]. This same study showed that drug-reinforced behavior can be restored by re-expression of CHRNA4, CHRNA6, or CHRNB2 genes in the VTA. An additional experiment that used self-administration procedures based on chronic rather than acute drug taking found similar rates of nicotine-reinforced responding in CHRNA4 knockout and wild-type strains [71]. Diminished reward after genetic knockout of subunits that make up high-affinity nAChRs may be mediated by low levels of cholinergic transmission that fail to exceed a lower cholinergic threshold, preventing reward enhancement.
Acquisition of nicotine- or cocaine- induced conditioned-place preference is attenuated by a β2* antagonist delivered either intracerebroventricularly or to the nucleus accumbens, but not the septum [72]. Place preference conditioned by nicotine in this study was also disrupted by deletion of either the CHRNA4 or CHRNA6 gene. Although an earlier study found that that nicotine-induced conditioned-place preference was supported after CHRNA4 knockout, it used a different mouse strain, route of nicotine delivery, and conditioning procedure [71]. Effects of CHRNA6 deletion cited above could be overcome by high-dose nicotine, indicating a shift in its dose-response to the right [72]. In contrast, CHRNA4 knockout prevented place preferences for each of the nicotine doses evaluated. Place preference after CHRNA6 knockout produced by high-dose nicotine was blocked by pretreatment with an antagonist for β2* nAChRs [72]. Cocaine-induced conditioned-place preference was disrupted after knockout of the CHRNA6 but not the CHRNA4 genes in this study. Because the magnitude of nicotine-induced place preference can be increased by CHRNA6 knockout, a role for reward inhibition by α6β2* receptors is implicated after nicotine but not cocaine treatment. The broad role of the β2* subtype in preferences occurring under different conditions suggests an important contribution to reward enhancement (but see further discussion below, regarding reward inhibition after prolonged nicotine treatment).
The magnitude of conditioned-place preference for low-dose nicotine is increased after knockout of the CHRNA7 gene, which codes for the α7 nicotinic subunit; but decreased by knock-in of the same gene [73]. This is opposite to genetic effects of β2* expression, and implicates a role for low-affinity α7 homomeric nAChRs in reward inhibition after acute treatment with nicotine. Consistent with this, there is a graded increase in the strength of nicotine-induced place preference across genetically different mouse strains, which correlates with lower levels of α7 subunit expression in the nucleus accumbens [73]. Genetic variation in α7 subunit expression may explain why earlier studies failed to identify an effect of CHRNA7 knockouts on nicotine-induced conditioned-place preference or intravenous self-administration [70,74]. Pretreatment with a nicotinic agonist selective for α7 nAChRs prevented conditioning of place preference by nicotine, with this effect blocked by an antagonist for α7 receptors [73]. Cocaine-induced conditioned-place preference was unaffected by α7-selective agents in these studies, which could be explained by activation of other nicotinic subtypes or muscarinic receptors.
When each dose of cocaine self-administered under FR-3 in rats was combined with 70 μg of mecamylamine, escalation of drug taking with exposure to daily extended (6 hour) access was prevented, with no effect on self-administration during one-hour sessions [75]. Systemic pretreatment with 1.0, 2.0, or 4.0 mg/kg of mecamylamine also decreased self-administration of cocaine under FR-1 in rats, with only the highest dose decreasing food reinforcement [76]. This study is unusual in that mecamylamine pretreatment was administered daily over a seven-day period. Neither effects of single mecamylamine doses nor trends over daily sessions were reported, and it is unclear whether chronic exposure was used as a strategy to diminish variance or was required to enhance drug action. The requirement for a higher mecamylamine dose to decrease responding for food pellets was interpreted as a demonstration of the selective involvement of ACh for cocaine-reinforced behavior. However, food reinforcement supported a three-fold higher level of responding than cocaine in this study [76]. Palatable rewards can surpass cocaine as a motivator [77]. Use of a palatable non-drug reinforcer associated with greater responding may have caused drug reward to be more susceptible to blockade of nAChRs, with food reinforcement to appearing to be resistant. According to the threshold model, behavior motivated by either reinforcer is attenuated because cholinergic transmission is prevented from reaching its initial lower threshold for reward that is achieved in vehicle-treated animals, blocking reward enhancement (Figure 1, compare conditions A and B). Consistent with this, laboratory measures of cue-induced craving in human cocaine addicts can be enhanced or attenuated by acute pretreatment with nicotine [78] or mecamylamine [79], respectively.
Nicotinic Mechanisms
Activation of midbrain dopaminergic neurons in the VTA by either local drug treatments or optogenetic stimulation can enhance reward behavior (see review by Ikemoto et al. [80]). The threshold model predicts that delivery of agonists with selectivity for either nicotinic or muscarinic receptors to the VTA can exceed the lower cholinergic threshold. Delivery of 50 nM or more of nicotine after smoking appears to activate dopaminergic neurons [81], corresponding to in vitro estimates of its EC50 (50% effective concentration) [82]. By preventing breakdown of ACh, cholinesterase inhibitors can function in a similar manner. For example, local infusion of either the nonselective agonist carbachol or the cholinesterase inhibitor neostigmine into the VTA can produce conditioned place preferences, which are blocked by muscarinic or nicotinic antagonists [83]. Cocaine-seeking behavior can also be blocked by delivery of antagonists with either muscarinic or nicotinic selectivity to the VTA [84]. These results implicate midbrain dopaminergic neurons in the VTA as mediators of reward enhancement.
Desensitization of nAChRs can be viewed as a ‘nuisance’ that occurs only after drug treatments under experimental conditions [49]. Alternatively, it may contribute to physiological processing in the normal and diseased brain [85]. Consistent with this, desensitization could limit or reverse nicotinic effects after sustained increases in endogenous or drug-induced ACh, by acting as an upper, inhibitory cholinergic threshold. Although classically viewed as involving rapid oscillation between active and inactive conformational states across milliseconds, more detailed models encompass multiple states that include long-term loss of nicotinic receptor function that can persist over 24 hours, and resolve at least partially through synthesis of new receptors [86]. In vitro studies of muscle-type nAChRs have shown multiple desensitization states, with increased duration of agonist exposure associated with reductions in function over successively longer periods, described as ‘deeper’ desensitization states [87]. Deeper levels of desensitization may underlie reward inhibition that follows extended nAChR activation by drug treatments or sustained increases in endogenous ACh.
Delivery of mecamylamine to the VTA blocks increases in dopamine following systemic cocaine [33]. Cocaine-induced conditioned-place preference can be disrupted by either mecamylamine pretreatment, knockout of nAChRs containing the CHRNB2 subunit, or chronic intermittent nicotine treatment [88]. Disruption of cholinergic transmission by antagonist pretreatment or gene knockout is likely to block reward enhancement, as cholinergic transmission is prevented from rising above its lower threshold. For mice with a genetically deleted CHRNB2 nicotinic subunit, changes in dopamine metabolism and immediate-early gene-product expression were also deficient, consistent with a role of this receptor subtype in dopaminergic neurotransmission underlying reward processes [88]. Low, ineffective doses of cocaine and nicotine did cause a place preference if administered together, suggesting that greater nicotinic activation by combined treatment did allow the lower cholinergic threshold to be exceeded.
Disruption of cocaine-induced conditioned-place preference by chronic intermittent nicotine treatment [88] can be explained by reward inhibition, as prolonged exposure to agonist causes nicotinic receptors to become refractory. The pattern of intermittent nicotine dosing used in this study was aimed at causing tolerance to nicotine’s physiologic effects, including reductions in nicotine-induced dopamine release [89,90], perhaps through nAChR desensitization. Similarly, systemic dopamine blockade and microinjection of nicotine to the VTA can produce a place preference in vehicle-treated rats, but are aversive after chronic infusion of nicotine by osmotic pump [91]. This dichotomy may reflect acute β2* reward enhancement by activation beyond the lower threshold in nicotine-naïve animals which potentiates drug-motivated behavior, and reward inhibition after prolonged nicotine levels that raise cholinergic transmission above its upper threshold.
Through low sensitivity to agonist, α7-containing nAChRs may act as a mediator of the upper cholinergic threshold, favoring reward inhibition after acute exposure to relatively high agonist levels. As noted above, activation of the α7 nAChR decreases acute nicotine reward [73], with an opposite effect of high-affinity β2* receptors [70]. Knockout of the corresponding CHRNA7 gene in mice prolongs nicotine-induced increases in extracellular dopamine in the nucleus accumbens [92]. This suggests that α7 receptors inhibit dopamine release, which is again opposite to the effect of β2* receptors [68]. After relatively high levels of ACh, choline, or nicotinic agonist; α7-mediated reductions in dopamine release may underlie reward inhibition, serving an auto inhibitory role [93]. This may occur through phosphorylation of β2* receptors, associated with attenuated nicotine-induced increases in the firing of VTA dopaminergic neurons [94].
After nicotine dependence is established, systemic treatment with an antagonist for β2* nAChRs elevates anxiety-like behavior [95], and infusion of a β2* antagonist to the VTA increases the amount of current required to initiate intracranial self-stimulation [96]. Requirement for greater amounts of current to elicit intracranial self-stimulation to the medial forebrain bundle in the lateral hypothalamus is an animal model of human anhedonia and depressed mood [97]. For either spontaneous or precipitated withdrawal, knockout of the CHRNB2 gene prevents increases in anxiety measures [98] and elevations in reward threshold [99]. In contrast, an antagonist for α7 nAChRs can decrease locomotor activity and cause tremor and other somatic signs of nicotine withdrawal in dependent animals [100]. After genetic deletion of the CHRNA7 gene, withdrawal-induced increases in nociception are lost; with anxiety, reward-threshold, and somatic measures remaining intact [98]. In nicotine-dependent mice, genetic disruption of either the α7 or β4 nicotinic subunit delayed the onset of spontaneous withdrawal [101]. In the same study, genetic disruption of the α7 subunit caused biphasic effects on antagonist precipitated increases in reward threshold, which were potentiated and diminished by low- and high doses of mecamylamine, respectively.
Anxiety, elevated reward threshold, and somatic changes associated with withdrawal form an unconditioned contingency with changes in nicotinic activity. Prompt relief from the negative effects of withdrawal occurs after nicotine treatment [95], and supports further drug taking through negative reinforcement. A similar effect would also occur after actions that re-elevate endogenous ACh; reinforcing associated behavior. The differential sensitivity of low- and high- affinity nAChRs suggests a hierarchy of effects on reward that modify behavior through delayed consequences. Relatively modest, but sustained increases in either ACh or exogenous agonist that activate high-affinity receptors containing the β2 subunit are associated with delayed increases in anxiety [98] with an elevation of reward threshold [99]; larger agonist doses or increases in either endogenous ACh or choline which activate low-affinity homomeric receptors also produce heightened nociception, tremor, and other somatic effects [98,100]. In either case, behavior associated with nicotinic activation can either be punished by these effects, or allow negative reinforcement after relief of unpleasant consequences associated with reward inhibition. The key factor supporting motivation is short-term positive effects on mood, lowered reward threshold, and diminished somatic symptoms (reward enhancement); and delayed effects with an opposite (aversive) valence.
Thresholds Mediated by Muscarinic Receptors
Background, Muscarinic Systems
GABAergic medium spiny neurons make up more than 90% of neurons in the striatum, and serve as its major output [102]. They are named for their far-reaching dendritic trees that contain numerous small spines, allowing them to receive extensive synaptic input. Glutamatergic input from various cortical and thalamic structures provides the major excitatory input to medium spiny neurons, presumably allowing temporal and spatial direction [103]. Though activation of type 1 and 2 dopamine receptors, activities of medium spiny neurons are modulated in a biphasic manner, in part by facilitation or suppression of glutamatergic transmission [104,105]. Medium spiny neurons also express both M1 and M4 muscarinic receptors [106]. In the nucleus accumbens, glutamatergic input is generally potentiated or inhibited by nicotinic or muscarinic (M1 or M4) receptors, respectively [107].
Muscarinic M5 receptors on dopamine neuron terminals enhance striatal dopamine release, whereas M2 and M4 autoreceptors on cholinergic interneurons inhibit ACh release and subsequent nicotinic-receptor-dependent dopamine release [108,109]. Within the VTA and substantia nigra, RNA for the M5 receptor is the predominant muscarinic subtype expressed [110]. In these brain regions, most neurons express message for both the M5 muscarinic receptor and the dopamine D2 receptor [111].
ACh in the striatum and nucleus accumbens is released by cholinergic interneurons that express choline acetyltransferase. Striatal cholinergic interneurons are presumed to be the tonically active neurons of electrophysiologic studies, which serve as mediators of stimulus-reward associations [112]. They spontaneously produce wide action potentials at a frequency of 2 to 10 Hz in vivo, and comprise less than 2% of neurons in the nucleus accumbens [113]. Even so, cholinergic interneurons are the major source of ACh in the nucleus accumbens. The density of their local projections implies an important role of ACh in modulating medium spiny neuron function, which is supported by electrophysiologic and optogenetic studies. With exposure to behaviorally relevant stimuli or reward, most cholinergic interneurons respond with a brief pause in firing of approximately 200 to 300 milliseconds duration, which may be preceded or followed by increased firing rates [114]. Excitatory cortical and thalamic inputs appear to play a role in initiating pause-excitation events that follow reward-predicting stimuli [115]. Pauses are associated with increases in firing by midbrain dopaminergic neurons [116]. In addition, about 25% of GABA-releasing neurons in the VTA project to the nucleus accumbens [117]. Stimulation of GABA projection neurons in the VTA can produce inhibitory effects that facilitate pauses by accumbal cholinergic interneurons, which are associated with enhanced discrimination of motivationally important stimuli [118].
Optogenetic stimulation of burst firing by VTA dopaminergic neurons is followed by increased firing and a subsequent pause of cholinergic interneurons in the nucleus accumbens shell [119]. In contrast, optogenetic stimulation of cholinergic interneurons in the striatum augments dopamine release through β2* nAChRs [120,121]. In the nucleus accumbens, release by this mechanism relies at least partially on glutamatergic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors, appears to be opposed by muscarinic activation, and is independent of midbrain dopaminergic cell bodies [122]. Direct optogenetic stimulation of cholinergic interneurons in the nucleus accumbens suppresses most adjacent medium spiny neurons (81%), while exciting a minority of cells (19%); with an opposite effect of optogenetic inhibition [123]. In this study, systemic treatment with cocaine markedly increased the firing rate on cholinergic interneurons. Optogenetic suppression or facilitation of cholinergic interneuron firing has no obvious effect on spontaneous behavior, but suppression did attenuate cocaine-induced conditioned-place preference. Reward-associated pause-excitation events in cholinergic interneurons appear to inhibit neighboring medium spiny neurons by nicotinic excitation of GABAergic neurons [124]. Because of their specific cortical, hippocampal, and amygdalar afferents, individual medium spiny neurons in the nucleus accumbens are thought to encode different stimulus-action associations [125]. This allows selection of a certain action through inhibition of its corresponding medium spiny neurons.
Muscarinic Agonists
The threshold model predicts biphasic effects on reward behaviors, which are enhanced at low doses of muscarinic agonists, but suppressed by higher doses of the same agents. A recent study examined effects of the muscarinic agonist xanomeline, which is selective for both type M1 and M4 receptors, on cocaine- and food- reinforced behaviors [126]. Rats were allowed to choose reinforcement with either liquid food or different doses of cocaine. Daily treatment with xanomeline produced complex effects, generally shifting cocaine dose-effect curves to the right (suppressing responding for low doses of cocaine doses, while increasing drug taking for higher doses). For some cocaine-xanomeline dose combinations, behavior was reallocated from cocaine to food reinforcement. The net effect was that overall cocaine intake was augmented by lower xanomeline doses (1.8 and 3.2 mg/kg-day), with a trend for reduced intake in animals receiving a higher dose of xanomeline (5.6 mg/kg-day). This pattern likely reflects reward enhancement and inhibition by low- and high- xanomeline doses, respectively. It resembles biphasic effects of nicotine, which can also potentiate or diminish cocaine-reinforced behavior when administered at intermediate or high doses, respectively [57].
The study by Thomsen et al. described in the preceding paragraph [126] is unusual because of the broad range of muscarinic agonist doses evaluated during chronic dosing, with cocaine also tested across a broad range of doses that supported self-administration behavior. Furthermore, drug and food were both available under fixed-ratio-5 (FR-5), in which reinforcement was provided after 5 or more lever presses. This design is highly relevant for evaluation of potential human treatments for substance abuse disorders. Most other studies have evaluated a narrower dose range cholinergic agents, which may correspond to one limb of a biphasic dose-response. For example, pretreatment with single 1.0 mg/kg doses of xanomeline or two other muscarinic agonists attenuated self-administration under FR-1 across a broad range of cocaine doses in mice [127], implicating reward inhibition. Responding for the maximally reinforcing cocaine dose was attenuated by about two-thirds. Although food-reinforced behavior was not modified by pretreatment with M1-selective agonists, food pellets supported a higher response requirement than cocaine injections in this study as well as the earlier report by Thomsen et al. [126], again raising question as to whether this effect was selective for drug reward. Systemic pretreatment with nonselective muscarinic agonists or a partial agonist attenuated cocaine self-administration behavior in mice [128], again corresponding to reward inhibition. Lower agonist or cocaine doses which may have produced conditions leading to enhanced drug reward were not evaluated in this experiment.
Muscarinic Antagonists
If muscarinic receptors contribute to reward enhancement by augmenting cholinergic transmission above the lower threshold shown in Figure 1, preventing this action should disrupt behavior supported by natural or drug reinforcers. Pretreatment of rhesus monkeys during a single session with the muscarinic antagonist atropine increased self-administration of cocaine, with no effect observed after pretreatment with methylatropine which has limited brain penetration [129]. Enhanced responding under these conditions may correspond to a short-term increase in drug-seeking behavior that would have extinguished if it were evaluated over additional sessions or under a schedule with a greater response requirement. Consistent with this interpretation, cocaine combined with the nonselective muscarinic antagonist scopolamine, maintained lower rates of drug-reinforced responding in Rhesus monkeys evaluated under either a FR-25 or progressive-ratio schedule [130]. Systemic treatment with a relatively high 5.0 mg/kg dose of scopolamine attenuated cocaine-induced reinstatement by approximately one-half, with 0.5 mg/kg being ineffective [131]. Although not statistically significant, the 5.0 mg/kg dose of scopolamine also decreased responding during a sucrose-induced reinstatement procedure by more than one-third. It should be noted that a wide variety of memory-related tasks are impaired by scopolamine [132]. Scopolamine doses that exceed 0.1 mg/kg can nonselectively diminish performance by modifying attention, stimulus discrimination, and motor activity [133]. Anxiety, associated with peripheral effects of decreased salivation and pupillary dilatation, has been implicated.
In rats performing the runway task described above, microinjection of atropine or mecamylamine into the nucleus accumbens core blocked opiate reward (consistent with blockade of reward enhancement), without affecting performance maintained by sweetened, condensed milk [22,23]. Although not statistically significant, run times were more than 100% prolonged in atropine-treated food-reinforced rats in the study by Crespo et al. [23], relative to vehicle- or mecamylamine- treated animals. These longer values may reflect a role for accumbal muscarinic receptors in supporting food-reinforced behavior. Greater preferences for sucrose over cocaine reward in rodents [77] is an additional consideration that again makes it difficult to evaluate the relative selectivity for drug and food rewards. There is a need for further research that focuses on evaluating a broader range of alternative, non-drug reinforcers, which support different levels of responding.
Microinjection of scopolamine into the nucleus accumbens core attenuated non-reinforced responding for either cocaine or sucrose (again consistent with blockade of reward enhancement); but injection into the nucleus accumbens shell unambiguously attenuated responding for only cocaine [131]. An additional study reported that blockade of muscarinic but not nicotinic receptors in either the nucleus accumbens core or shell decreased sucrose-reinforced behavior [134]. Importantly, delivery of a muscarinic antagonist dose to the nucleus accumbens core that attenuated liquid food intake also produced flavor- and place- aversions [135]. Comparable effects were not observed after blockade of NMDA, dopamine D1, or opiate receptors. Rather than cognitive impairment, these results are more consistent with muscarinic blockade decreasing the reinforcing attributes of food or causing a “general hedonic suppression”.
Infusion of a muscarinic antagonist into the VTA of rats attenuated cocaine-induced increases in VTA dopamine by more than 50% [21]. With access to self-administration under FR-1, rats increased their rate of lever pressing and contingent injections, allowing them to maintain VTA elevations of dopamine that were comparable to animals not treated with an antagonist; use of a nicotinic antagonist was ineffective. After cocaine self-administration was completed, dopamine levels dropped more rapidly in rats pretreated with the muscarinic antagonist. The authors concluded that blocking muscarinic input to the VTA increased cocaine intake; this increase offset the reduction in cholinergic input, resulting in the same VTA dopamine levels as were seen in the absence of the ACh antagonists. Put another way, rats worked to maintain dopaminergic output from the VTA at an intermediate level by increasing lever pressing in the absence of muscarinic activation to this brain region that would normally enhance dopamine release [136] and drug reward. This appears similar to increases in cocaine-reinforced behavior under FR-1 after atropine treatment in monkeys [129]. An important message is that acute interventions under simple response requirements that decrease central mediators of reward via changes in cholinergic tone may actually increase drug motivated behavior. Presumably, evaluation under chronic conditions using a higher response requirement, such as multiple lever presses to obtain a drug injection, would lead to decreases in cocaine self-administration after cholinergic antagonist treatment that prevents reward enhancement.
Antagonists with selectivity for the M1 muscarinic receptor may exhibit greater selectivity for disrupting memory without modifying attention or food motivation [137]. In rats, infusion of a muscarinic agonist into the nucleus accumbens shell decreases cocaine self-administration behavior, with this effect blocked by pretreatment with an antagonist for the M1 muscarinic subtype [138]. This effect may be mediated by cholinergic transmission that exceeds an upper cholinergic threshold, causing locally- mediated reward inhibition in this brain region. Alternately, muscarinic agonists may also facilitate reward if administered at low enough doses, as cholinergic transmission rises above a lower cholinergic threshold.
Pretreatment with the M1 selective muscarinic antagonist biperiden alone does not cause either preference or aversion [139], but can attenuate the expression of cocaine-induced conditioned-place preference [140], consistent with blockade of reward enhancement. Conditioning of place preferences with cocaine is also diminished if biperiden is administered 5 minutes after exposure to cocaine pairings, implicating interference with the process of memory consolidation [141]. Both effects relied on a higher biperiden dose (10 mg/kg) associated with decreased response rate and diminished short-term memory, but without negative effects on attention or food-reinforced responding [137]. Lower doses of biperiden were ineffective in modifying expression of place preference [140]. In humans with cocaine-use disorder, biperiden administered over eight weeks (2 mg three-times-daily) improved treatment compliance and decreased craving intensity when combined with group-based counseling [142]. However, single 2 mg doses of biperiden cause broad declines on a subset of cognitive measures in humans, which include episodic memory, immediate recall, motor learning, and visual-spatial performance [143].
Morphine- and cocaine- induced conditioned-place preference are attenuated by either genetic disruption of the M1 muscarinic receptor or combining drug treatments with an antagonist for the M1 receptor [144]. In these studies, knock-out or antagonist treated animals correspond to State A in Figure 1, with wild-type and vehicle treated animals corresponding to State B in which activation of the M1 receptor allows ACh to facilitate food- or drug- reinforced behaviors.
Knock-out of the M5-muscarinic receptor decreases drug-induced place preference across a broad range of morphine doses, and also attenuates morphine-induced dopamine increases in the nucleus accumbens [145,146]. After knock-out of the M5-muscarinic receptor, mice self-administered some but not all cocaine doses at approximately one-half the rate of wild-type animals [147]. During conditioned- place preference testing, cocaine-induced increases in preference were also approximately one-half the magnitude of wild-type animals. A subsequent evaluation using more highly inbred M5-deficient mice observed lower rates of acquisition for cocaine self-administered under FR-1, but no differences in cocaine-experienced mice [148]. The same study found that lower break points were achieved by M5- deficient mice under a progressive-ratio schedule for 0.03 or 0.32 mg/kg per injection of cocaine (reductions of approximately 50 and 25%, respectively). Although no differences in food-reinforced behavior were observed for either fixed- or progressive- ratio schedules, food reinforcement supported a two-fold higher rate of responding. Accordingly, activation of M5 muscarinic receptors appears to preferentially contribute to reward enhancement, apparently through activation of VTA dopaminergic neurons. Nonetheless, M5-deficient mice did not differ in cocaine self-administration under many of the conditions evaluated (i.e., a full range of cocaine doses evaluated using FR-1, and the higher two of four cocaine doses tested under a progressive-ratio schedule) [148]. It may be that nicotinic signaling alone in the VTA can support drug-reinforced behavior in the absence of the M5 muscarinic receptor in these instances.
Electrical stimulation of laterodorsal tegmental (LDT) nucleus cholinergic neurons increases dopamine release through an initial rapid phase (over approximately 2 minutes) which is dependent on VTA nAChRs, and a late phase (8 to 50 minutes post stimulation) which is dependent on M5-muscarinic receptors [149]. Infusion of M5-antisense mRNA causes a reversible rightward shift in the stimulation frequency required to maintain electrical brain stimulation [150]. Intra-VTA infusion of muscarinic or nicotinic antagonists causes a similar shift in the frequency required to maintain electrical brain stimulation [151]. Based on these effects, activation of M5 muscarinic receptors on VTA dopaminergic neurons are believed to facilitate natural or drug reward, as cholinergic transmission exceeds its lower threshold. Because pharmacologic agents with M5-selectivity are not available, it is unclear whether reward inhibition can be produced by agonists with selectivity for this subtype.
Muscarinic Mechanisms
As cited above, low doses of the muscarinic agonist xanomeline augmented cocaine intake by up to 40.1% [126] In comparison, low doses of nicotine can enhance the amount of self-administered cocaine by approximately 120% [57]. Accordingly, nicotinic mechanisms appear to play a greater role in reward enhancement through the low-level threshold than that of the muscarinic system. This may be facilitated by more rapid nicotinic effects on ion flux, compared with slower muscarinic modulation of phospholipase and adenylate cyclase. Such a dichotomy echoes the time course of LDT activity cited above, with dopamine release stimulated over a several-minute rapid phase mediated by the nicotinic receptor, and a more prolonged phase produced by muscarinic receptors [149]. Nonetheless, both nicotinic and muscarinic agonists appeared to have the potential to produce either reward enhancement or inhibition in the studies cited above. Conclusions on the role of muscarinic receptors must again be qualified by the limitation that no direct agonists for the M5 receptor are currently available, with this subtype implicated in muscarinic reward-enhancing actions [148].
In striatal slice experiments performed in mice lacking different muscarinic receptors, potassium- evoked release of dopamine is modulated in a biphasic manner. The authors infer that dopamine release is stimulated by activation of either type M4 or M5 receptors, inhibited by M3 receptors, and unaffected by knock-out of M1 or M2 receptors [152,153]. Augmented release of dopamine is most consistent with reward enhancement, with reward inhibition expected after attenuation of neurotransmitter release. However, administration of an M4 positive allosteric modulator strongly attenuates cocaine-induced increases in dopamine measured in the striatum by microdialysis, as well as cocaine self-administration [154]. Similarly, knockout of type M4 muscarinic receptors appeared to facilitate dopamine efflux, augmenting cocaine- or amphetamine- induced locomotor activity [155]. Dissimilar actions of the muscarinic M4 subtype on dopamine release may reflect experimental conditions that evaluate either reward enhancement or inhibition, mediated by different cholinergic thresholds.
Tonic activity by a subset of nucleus accumbens neurons appears to inhibit appetitive responding, with several-second pauses in firing associated with initiation of food-reinforced behavior [156]. The subset of cells in the nucleus accumbens shell that exhibit a pause in firing during feeding behavior have been described as type 1 neurons. Feeding is disrupted by electrical stimulation of type 1 neurons [157]. Declines in the activity of nucleus accumbens medium spiny neurons are also implicated in drug- reinforced behavior [158]. For example, the most frequent pattern of neural activity in the nucleus accumbens that follows self-administration of an intermediate dose of cocaine is a decline in firing rate that begins within 0.2 minutes of lever pressing, and persists over approximately one minute [159]. Overall, these findings implicate interventions that quiet or excite accumbal type 1 neurons as underlying reward enhancement or inhibition, respectively.
In striatal slices, either cholinesterase inhibition or low, micromolar concentrations of a nonselective cholinergic agonist suppressed electrically-evoked activity, with periods of burst firing observed after higher agonist concentrations [160]. After iontophoretic application of ACh in awake rats, the activity of most spontaneously active or glutamate-excited neurons in the nucleus accumbens is suppressed [161]. As noted above, optogenetic stimulation of cholinergic interneurons plays a role in cocaine reward, and suppresses the activity of most adjacent medium spiny neurons [123]. A role for ACh-induced electrical pauses in reward enhancement is consistent with a literature linking decreases in the rate of tonically active cholinergic interneurons in the striatum with exposure to behaviorally relevant stimuli in non- human primates [162].
In brain slices from the nucleus accumbens shell, treatment with a low concentration of a nonselective cholinergic agonist (1 µM carbachol) suppresses repetitive firing in medium spiny neurons (consistent with reward enhancement), with this effect blocked by an M1 selective antagonist [163]. Carbachol applied at greater concentrations (50 µM) can enhance repetitive firing in medium spiny neurons (implicating reward inhibition) [107]. Short-term decreases in excitatory glutamatergic inputs mediated by muscarinic receptors [107,164] can also inhibit the activity of medium spiny neurons, possibly causing reward enhancement. In striatal brain slices, cholinergic agonists can depolarize GABAergic neurons through activation of nAChRs [165]. Inhibitory effects of enhanced GABA release may also increase the probability of pauses in firing by cholinergic interneurons and medium spiny neurons.
Biphasic effects of ACh on the firing rate of medium spiny neurons may underlie thresholds that modify reward behavior. Whether through ACh released by cholinergic interneurons, muscarinic agonists, or cholinesterase inhibition (see the following section), low-level binding at accumbal-VTA muscarinic receptors likely quiets the activity of type 1 accumbal medium spiny neurons [161,163]. This exceeds the lower cholinergic threshold, increasing the probability of reward behavior (reward enhancement). Further activation of the same muscarinic receptors rises above the upper cholinergic threshold, facilitating rhythmic activity by medium spiny neurons [107,160] and decreasing the probability of reward (reward inhibition).
Background, Cholinesterase Inhibition
Mammalian brain contains two forms of cholinesterase, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). The physiologic role of BuChE is unclear, but it can metabolize cocaine and other exogenous compounds and contributes to degradation of ACh. Cholinesterase inhibitors, which are widely used for treatment of Alzheimer’s disease, increase synaptic levels of ACh by preventing its inactivation by AChE or BuChE. In general, these agents attenuate declines in functional status associated with Alzheimer’s disease and other forms of dementia.
Clinically relevant doses of cholinesterase inhibitors can augment learning and memory in Alzheimer’s disease and other neuropsychiatric disorders [166,167]. Depending on the specific task being evaluated, performance in healthy individuals may be impaired, augmented, or unaffected [168,169]. Cholinesterase inhibition in some settings may disrupt performance by producing a hyper- vigilant state, enhancing sensitivity to irrelevant stimuli that serve as distractions [170]. In a subset of patients, cholinesterase inhibitors administered at relatively high doses cause symptoms of anorexia, nausea, diarrhea, and weight-loss [171].
Cholinesterase inhibition that attenuated morphine-induced conditioned-place preference in control animals was ineffective in after ablation of cholinergic neurons from the nucleus accumbens [172], implicating the involvement of this brain region in reward inhibition. Cholinergic lesions to the posterior nucleus accumbens and ventral pallidum caused a shift to the left in cocaine-reinforced behavior, with enhanced self-administration of relatively low doses of cocaine [173], again implicating removal of an inhibitory influence.
Thresholds and Cholinesterase Inhibition
By preventing degradation of ACh in the synaptic cleft, the threshold model predicts that cholinesterase inhibition will also produce biphasic effects on behavior that are mediated by a combination of the nicotinic and muscarinic mechanisms outlined in the preceding sections. When applied to striatal slices, different cholinesterase inhibitors share a common biphasic effect on dopamine efflux: low (nanomolar) concentrations enhance release that is either electrically evoked or spontaneous, with maximal effects of 12 to 24%, while higher (micromolar) concentrations strongly attenuate release [174]. These in vitro effects may correspond to in vivo enhancement and inhibition or reward, respectively.
An early study found that pretreatment with single doses of the cholinesterase inhibitor physostigmine (0.1 to 0.5 mg/kg) decreased cocaine self-administration in rhesus monkeys under FR-1 and caused vomiting and diarrhea [129], consistent with its administration at relatively high dose causing reward inhibition. When either cocaine or procaine were self-administered by rhesus monkeys under FR-10, 0.02 to 0.05 mg/kg doses of physostigmine increased responding in 3 of 10 sessions, with declines uniformly observed following greater physostigmine doses [175]. This inverted “U” dose-response relationship is again consistent with reward enhancement and inhibition, respectively. It is unclear why augmented reward behavior is not more consistently observed at lower cholinesterase inhibitor doses, but this may reflect differences in the time-action or dose-response functions for nicotinic and muscarinic receptors. Cocaine-reinforced behavior evaluated under FR-5 was also attenuated in rats pretreated with cholinesterase inhibitors delivered as single bolus doses [176] or by infusion [177,178]. Pretreatment with tacrine or other cholinesterase inhibitors can also produce a dose-related attenuation of food- or water-reinforced responding in rats [179,180].
Daily pretreatment with 0.1 mg/kg of physostigmine modestly impaired acquisition of heroin self- administration by rats, and also attenuated cue-induced reinstatement 14 days later, although no further physostigmine was administered during extinction and reinstatement session [181]. In the same study, single doses of 0.5 or 2.5 mg/kg of physostigmine also attenuated self-administration in heroin- experienced animals. Although lower physostigmine doses did not modify heroin reward, the number of daily self-administered heroin doses was limited by design, decreasing the potential to detect increases in self-administration. Reductions in self-administration after 0.5 mg/kg of physostigmine were prevented by scopolamine but not mecamylamine, implicating a role for muscarinic receptors underlying inhibition of opiate reward caused by treatment with cholinesterase inhibitors. Physostigmine doses of 0.1 or 0.5 mg/kg decreased drug-seeking behavior after cue exposure, with the higher dose also effective during extinction. Cue- and extinction- induced responding for heroin were increased after local infusion of physostigmine into the VTA; but cue- induced responding was decreased after delivery to the nucleus accumbens, with responses during extinction uneffected [181]. Under these conditions, increased synaptic ACh in the VTA appeared to exceed the lower cholinergic threshold, increasing the likelihood of behaviors associated with reward (enhancement). Reductions in heroin-seeking behavior (reward inhibition) after delivery of physostigmine to the nucleus accumbens [181] may be due to ACh levels that preferentially activate the upper cholinergic threshold.
Administration of cholinesterase inhibitors can prevent acquisition of cocaine- or morphine- induced conditioned-place preference in mice [172] or rats [182], which again reflects reward inhibition. For the latter study, cholinesterase inhibition also attenuated place preference expression. In contrast, methamphetamine-induced conditioned-place preference in mice was not attenuated by donepezil in one study [183]. Because it stimulates reverse transport by the dopamine transporter [184], amphetamines can augment dopamine release more potently the cocaine [185]. This difference may explain why donepezil modified cocaine- but not methamphetamine- induced place preference in the study by Takamatsu et al. [183].
Treatment of cocaine-dependent human volunteers with 5 mg daily of donepezil, the lowest dose approved for Alzheimer’s disease, increased ratings of ‘good’ drug or ‘any’ drug for low-dose of cocaine, without modifying the subjective effects intermediate-dose cocaine [186]. When donepezil was increased to a final dose of 10 mg daily in treatment-seeking patients receiving cognitive behavioral therapy, cocaine-positive urines were non-significantly increased, corresponding to values of 32 and 168% for placebo and active treatments respectively [187]. Apparently, donepezil administered at doses up to 10 mg daily interacts with the lower cholinergic threshold, augmenting drug-seeking behaviors through reward enhancement. Although a 23 mg daily dose of donepezil was approved which may offer greater improvements in some cognitive measures in Alzheimer’s disease [188], its effects on the course of cocaine-use disorder are unknown.
In another human laboratory study, an intermediate dose of the cholinesterase inhibitor rivastigmine attenuated methamphetamine-induced increases in blood pressure, anxiety, and “desire” to use drug, with a lower dose being ineffective [189]. Neither dose of rivastigmine modified the number of methamphetamine doses self-administered by dependent volunteers, but intermediate-dose rivastigmine was associated with a trend for attenuation of the positive subjective effects of self-administered methamphetamine [190]. Rivastigmine’s pharmacokinetics may favor reward inhibition, in parallel to actions of cholinesterase inhibitors in rodents [172,178,182].
Galantamine is an additional clinically-available, reversible cholinesterase inhibitor, which also acts to allosterically potentiate nicotinic function [191]. The latter mechanism appears to augment extracellular levels of dopamine in medial prefrontal cortex [192] and nicotine-induced norepinephrine release in hippocampus [193]. In recently detoxified alcoholics, treatment with galantamine treatment did not prolong abstinence, but decreased the number of self-reported drinks per alcohol-use episode [194]. Although it did not include an overt strategy for smoking cessation, this trial also reported decreased cigarette use after galantamine treatment [195]. For newly abstinent chronic cocaine users, daily treatment with galantamine improved reaction time and sustained attention [196]. In a relatively small preliminary trial (7 active and 7 placebo-treated cocaine-dependent patients receiving methadone maintenance), an escalating schedule of galantamine treatment (initiated at 8 mg daily over 4 weeks, subsequently advanced to 16 mg daily for an additional 4 weeks) was associated with trends for decreased drug use determined by either self-report or urine drug screen, relative to placebo treatment [197]. Four days of galantamine treatment in abstinent smokers decreased cigarette craving, improved response control and attention measures, and attenuated some subjective and physiologic effects of intravenous nicotine [198]. Galantamine’s unique dual mechanism of action of may allow it to preferentially cause reward inhibition by interacting with the upper inhibitory cholinergic threshold, without facilitating drug use through the lower threshold.
Alternative Interpretations
It has been proposed that reward processes are facilitated by VTA M5 muscarinic receptors, but attenuated by activation of M1 and M2 subtypes [199]. As noted above, the absence of muscarinic agonists with M5 selectivity prevents an evaluation of whether high agonist levels can prevent reward behavior. If the M1 muscarinic subtype only functioned to diminish the probability of reward behaviors, pretreatment with antagonists or genetic disruption would be expected to augment drug- or food- motivated responding. As outlined above, both of these strategies instead have an opposite effect, attenuating the magnitude of drug reward in rodents [140,141,144] and decreasing craving intensity in humans [142].
In response to various seemingly contradictory results of cholinergic medications on drug reward, Zernig and colleagues [200] hypothesize the existence of different ensembles of accumbal cholinergic interneurons that are dedicated to specific reinforcement scenarios. These include psychostimulant self- administration, social interaction, eating routine food, and consuming highly palatable treats. Variable effects of treatments that augment cholinergic transmission across different species and behaviors occur because of the specific ensemble type being activated. They posit considerable intra- and inter- individual variation in the spatial distribution of different ensembles. In support of this hypothesis, neuronal activation measured by expression of the early growth response 1 transcriptional regulator is shows distinct patterns in the nucleus accumbens across different individual rats. These can be described as diffuse, patchy, or mixed [200], and may allow information processing in the nucleus accumbens to provide specificity of responding based on individual differences or ecologically dissimilar circumstances. Even so, the existence a more generalized system which can facilitate or dampen reward through the thresholds described above allows a more parsimonious interpretation of existing data.
Dopaminergic systems in the nucleus accumbens also exert opposing influences on motivation, with the direct and indirect pathways facilitating and inhibiting behavior, respectively [201]. For example, stimulation of direct pathway medium spiny neurons expressing the D1 dopamine receptor can enhance cocaine-induced conditioned-place preference, with attenuation of place preference after activation of neurons expressing the dopamine D2 receptor that are part of the indirect pathway [202]. Self- administration of cocaine can also be inhibited by activation of indirect pathway D2 expressing medium spiny neurons in the nucleus accumbens core [203]. Interestingly, accumbal D1-expressing MSNs have an opposite effect on food-reinforced behavior, consistent with fundamental differences in the neural control of food- and drug- reinforcers [204]. As noted above, cholinergic interneurons can stimulate dopamine release through activation of nicotinic receptors on dopaminergic axons [120,121]. Therefore, biphasic effects of ACh on reward behaviors could be mediated through differential influences on dopaminergic direct and indirect pathways. Dopaminergic neurons projecting to the medial prefrontal cortex have also been implicated in aversive signaling [205].
A subset of neurons in the lateral habenula are activated by punishment and associated stimuli, but suppressed by rewarding events [206]. Intravenous cocaine can produce an initial inhibition of some neurons in the lateral habenula, followed by delayed excitation occurring 15 to 30 minutes later [207]. Local injection of an antagonist for α3β4 receptors into the habenular complex can have opposing effects on self-administration of intravenous nicotine: drug taking is decreased by injection into the medial habenula, the basolateral amygdala, or the dorsolateral tegmentum; but increased after delivery to the interpeduncular nucleus [208]. For primary cultures of neurons derived from the medial habenula and interpeduncular nucleus, repetitive activation of nAChRs can facilitate glutamatergic release for sustained periods of up to two hours; while prolonged exposure to nicotine can deactivate this response, requiring up to 24 hours for full recovery [86]. An important question is how function of the habenular complex interacts with thresholds mediated by the VTA-accumbal cholinergic transmission.
Clinical Implications of Cognitive Effects
Successful prevention of relapse in drug use disorders involves development of new coping strategies to situations that would otherwise stimulate drug-seeking behavior, and establishing new social contacts who do not use illicit substances. Depending upon the treatment program, relapse prevention may rely on attendance at clinical appointments, taking a medication to facilitate abstinence, and attempts at being employed. Cognitive-behavioral approaches can improve outcomes in cocaine use disorders, perhaps through enhancement of decision making. During recovery from each of the major forms of drug abuse, previous studies have demonstrated impairment in learning, memory, and attention [209-211]. Two outpatient-based studies have shown that greater delay after use of cocaine is associated with more severe impairment in cognitive performance [212,213]. Accordingly, protracted withdrawal from cocaine use leads to more pronounced cognitive deficits, which are less severe immediately following use of cocaine. If so, at least part of the motivation for relapse to drug use may be cocaine's ability to normalize mental function during recovery from cocaine dependence.
Because impaired learning, memory, and attention is one of the more well-validated actions of cholinergic antagonists, these agents have at least a relative contraindication for use in patients with substance abuse disorders due their potential for exacerbating cognitive deficits. Even so, recent findings of improved treatment compliance and decreased craving intensity after muscarinic blockade in treatment-seeking patients recovering from cocaine use [142] would support a lessor role of cognitive actions. It is also unclear whether use of cholinergic agonists or cholinesterase inhibition can ameliorate cognitive deficits in this setting. Treatment with galantamine appears to do so during either nicotine withdrawal [198] or recovery from cocaine dependence [196]. Further studies are needed which combine measures of cognitive function and drug-motivated behaviors.
Highlights.
Different classes of cholinergic agents produce biphasic effects on appetitive responding.
Phasic activation of cholinergic tone in the nucleus accumbens and VTA above a low-level threshold appears to increase the likelihood of rewarded behaviors.
Greater and more prolonged activation in the same brain regions above a second higher-level threshold is associated with a decreased likelihood of reward.
Cholinergic effects on the nicotinic receptor function, dopamine release, and the firing of medium spiny neurons are potential mediators of thresholds that shape behavior.
High-affinity β2* nicotinic receptors appear to play a greater role in reward enhancement, with low-affinity homomeric α7 receptors underlying reward inhibition.
Acknowledgments
This work was supported by grant R21-DA029787 issued to KG by the National Institutes of Health, Institute on Drug Abuse; and grant 589-KG-0012 issued to KG by the Medical Research Service, Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Stokes C, Treinin M, Papke RL. Looking below the surface of nicotinic acetylcholine receptors. Trends Pharmacol Sci. 2015 doi: 10.1016/j.tips.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117:232–43. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- [3].Lester DB, Rogers TD, Blaha CD. Acetylcholine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther. 2010;16:137–62. doi: 10.1111/j.1755-5949.2010.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhou FM, Wilson C, Dani JA. Muscarinic and nicotinic cholinergic mechanisms in the mesostriatal dopamine systems. Neuroscientist. 2003;9:23–36. doi: 10.1177/1073858402239588. [DOI] [PubMed] [Google Scholar]
- [5].Bentley P, Driver J, Dolan RJ. Cholinergic modulation of cognition: insights from human pharmacological functional neuroimaging. Prog Neurobiol. 2011;94:360–88. doi: 10.1016/j.pneurobio.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baldi E, Bucherelli C. The inverted "u-shaped" dose-effect relationships in learning and memory: modulation of arousal and consolidation. Nonlinearity Biol Toxicol Med. 2005;3:9–21. doi: 10.2201/nonlin.003.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Robinson L, Platt B, Riedel G. Involvement of the cholinergic system in conditioning and perceptual memory. Behav Brain Res. 2011;221:443–65. doi: 10.1016/j.bbr.2011.01.055. [DOI] [PubMed] [Google Scholar]
- [8].Deiana S, Platt B, Riedel G. The cholinergic system and spatial learning. Behav Brain Res. 2011;221:389–411. doi: 10.1016/j.bbr.2010.11.036. [DOI] [PubMed] [Google Scholar]
- [9].Smith RD, Kistler MK, Cohen-Williams M, Coffin VL. Cholinergic improvement of a naturally-occurring memory deficit in the young rat. Brain Res. 1996;707:13–21. doi: 10.1016/0006-8993(95)01207-9. [DOI] [PubMed] [Google Scholar]
- [10].Ogura H, Kosasa T, Kuriya Y, Yamanishi Y. Donepezil, a centrally acting acetylcholinesterase inhibitor, alleviates learning deficits in hypocholinergic models in rats. Methods Find Exp Clin Pharmacol. 2000;22:89–95. doi: 10.1358/mf.2000.22.2.796070. [DOI] [PubMed] [Google Scholar]
- [11].Ogura H, Kosasa T, Kuriya Y, Yamanishi Y. Central and peripheral activity of cholinesterase inhibitors as revealed by yawning and fasciculation in rats. Eur J Pharmacol. 2001;415:157–64. doi: 10.1016/s0014-2999(01)00824-x. [DOI] [PubMed] [Google Scholar]
- [12].Phillis JW. Acetylcholine release from the central nervous system: a 50-year retrospective. Crit Rev Neurobiol. 2005;17:161–217. doi: 10.1615/critrevneurobiol.v17.i3-4.30. [DOI] [PubMed] [Google Scholar]
- [13].Watanabe H, Shimizu H, Matsumoto K. Acetylcholine release detected by trans-striatal dialysis in freely moving rats correlates with spontaneous motor activity. Life Sci. 1990;47:829–32. doi: 10.1016/0024-3205(90)90556-7. [DOI] [PubMed] [Google Scholar]
- [14].Day J, Damsma G, Fibiger HC. Cholinergic activity in the rat hippocampus, cortex and striatum correlates with locomotor activity: an in vivo microdialysis study. Pharmacol Biochem Behav. 1991;38:723–9. doi: 10.1016/0091-3057(91)90233-r. [DOI] [PubMed] [Google Scholar]
- [15].Thiel CM, Huston JP, Schwarting RK. Cholinergic activation in frontal cortex and nucleus accumbens related to basic behavioral manipulations: handling, and the role of post-handling experience. Brain Res. 1998;812:121–32. doi: 10.1016/s0006-8993(98)00961-5. [DOI] [PubMed] [Google Scholar]
- [16].Ragozzino ME, Choi D. Dynamic changes in acetylcholine output in the medial striatum during place reversal learning. Learn Mem. 2004;11:70–7. doi: 10.1101/lm.65404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith JE, Vaughn TC, Co C. Acetylcholine turnover rates in rat brain regions during cocaine self-administration. J Neurochem. 2004;88:502–12. doi: 10.1046/j.1471-4159.2003.02222.x. [DOI] [PubMed] [Google Scholar]
- [18].Nestby P, Vanderschuren LJ, De Vries TJ, Hogenboom F, Wardeh G, Mulder, et al. Ethanol, like psychostimulants and morphine, causes long-lasting hyperreactivity of dopamine and acetylcholine neurons of rat nucleus accumbens: possible role in behavioural sensitization. Psychopharmacology. 1997;133:69–76. doi: 10.1007/s002130050373. [DOI] [PubMed] [Google Scholar]
- [19].Wilson JM, Carroll ME, Lac ST, DiStefano LM, Kish SJ. Choline acetyltransferase activity is reduced in rat nucleus accumbens after unlimited access to self-administration of cocaine. Neurosci Lett. 1994;180:29–32. doi: 10.1016/0304-3940(94)90906-7. [DOI] [PubMed] [Google Scholar]
- [20].Mark GP, Hajnal A, Kinney AE, Keys AS. Self-administration of cocaine increases the release of acetylcholine to a greater extent than response-independent cocaine in the nucleus accumbens of rats. Psychopharmacology. 1999;143:47–53. doi: 10.1007/s002130050918. [DOI] [PubMed] [Google Scholar]
- [21].You ZB, Wang B, Zitzman D, Wise RA. Acetylcholine release in the mesocorticolimbic dopamine system during cocaine seeking: conditioned and unconditioned contributions to reward and motivation. J Neurosci. 2008;28:9021–9. doi: 10.1523/JNEUROSCI.0694-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Crespo JA, Sturm K, Saria A, Zernig G. Activation of muscarinic and nicotinic acetylcholine receptors in the nucleus accumbens core is necessary for the acquisition of drug reinforcement. J Neurosci. 2006;26:6004–10. doi: 10.1523/JNEUROSCI.4494-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Crespo JA, Stockl P, Zorn K, Saria A, Zernig G. Nucleus accumbens core acetylcholine is preferentially activated during acquisition of drug- vs food-reinforced behavior. Neuropsychopharm. 2008;33:3213–20. doi: 10.1038/npp.2008.48. [DOI] [PubMed] [Google Scholar]
- [24].Avena NM, Rada PV. Cholinergic modulation of food and drug satiety and withdrawal. Physiol Behav. 2012;106:332–6. doi: 10.1016/j.physbeh.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mark GP, Rada P, Pothos E, Hoebel BG. Effects of feeding and drinking on acetylcholine release in the nucleus accumbens, striatum, and hippocampus of freely behaving rats. J Neurochem. 1992;58:2269–74. doi: 10.1111/j.1471-4159.1992.tb10973.x. [DOI] [PubMed] [Google Scholar]
- [26].Rada P, Hoebel BG. Acetylcholine in the accumbens is decreased by diazepam and increased by benzodiazepine withdrawal: a possible mechanism for dependency. Eur J Pharmacol. 2005;508:131–8. doi: 10.1016/j.ejphar.2004.12.016. [DOI] [PubMed] [Google Scholar]
- [27].Mark GP, Weinberg JB, Rada PV, Hoebel BG. Extracellular acetylcholine is increased in the nucleus accumbens following the presentation of an aversively conditioned taste stimulus. Brain Res. 1995;688:184–8. doi: 10.1016/0006-8993(95)00401-b. [DOI] [PubMed] [Google Scholar]
- [28].Rada PV, Hoebel BG. Aversive hypothalamic stimulation releases acetylcholine in the nucleus accumbens, and stimulation-escape decreases it. Brain Res. 2001;888:60–5. doi: 10.1016/s0006-8993(00)02865-1. [DOI] [PubMed] [Google Scholar]
- [29].Solomon RL, Corbit JD. An opponent-process theory of motivation: II. Cigarette addiction. J Abnorm Psych. 1973;81:158–71. doi: 10.1037/h0034534. [DOI] [PubMed] [Google Scholar]
- [30].Solomon RL, Corbit JD. An opponent-process theory of motivation: I. Temporal dynamics of affect. Psych Review. 1974;81:119–45. doi: 10.1037/h0036128. [DOI] [PubMed] [Google Scholar]
- [31].Williams MJ, Adinoff B. The role of acetylcholine in cocaine addiction. Neuropsychopharm. 2008;33:1779–97. doi: 10.1038/sj.npp.1301585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sofuoglu M, Mooney M. Cholinergic functioning in stimulant addiction: implications for medications development. CNS Drugs. 2009;23:939–52. doi: 10.2165/11310920-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mark GP, Shabani S, Dobbs LK, Hansen ST. Cholinergic modulation of mesolimbic dopamine function and reward. Physiol Behav. 2011;104:76–81. doi: 10.1016/j.physbeh.2011.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tuesta LM, Fowler CD, Kenny PJ. Recent advances in understanding nicotinic receptor signaling mechanisms that regulate drug self-administration behavior. Biochem Pharmacol. 2011;82:984–95. doi: 10.1016/j.bcp.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu L, Zhao-Shea R, McIntosh JM, Gardner PD, Tapper AR. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing alpha4 and alpha6 subunits. Mol Pharmacol. 2012;81:541–8. doi: 10.1124/mol.111.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jones IW, Bolam JP, Wonnacott S. Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol. 2001;439:235–47. doi: 10.1002/cne.1345. [DOI] [PubMed] [Google Scholar]
- [37].Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23:7820–9. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Livingstone PD, Wonnacott S. Nicotinic acetylcholine receptors and the ascending dopamine pathways. Biochem Pharmacol. 2009;78:744–55. doi: 10.1016/j.bcp.2009.06.004. [DOI] [PubMed] [Google Scholar]
- [39].Zhao-Shea R, Liu L, Soll LG, Improgo MR, Meyers EE, McIntosh JM, et al. Nicotine-mediated activation of dopaminergic neurons in distinct regions of the ventral tegmental area. Neuropsychopharm. 2011;36:1021–32. doi: 10.1038/npp.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–35. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- [41].Exley R, Maubourguet N, David V, Eddine R, Evrard A, Pons S, et al. Distinct contributions of nicotinic acetylcholine receptor subunit alpha4 and subunit alpha6 to the reinforcing effects of nicotine. Proc Natl Acad Sci U S A. 2011;108:7577–82. doi: 10.1073/pnas.1103000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Posadas I, Lopez-Hernandez B, Cena V. Nicotinic receptors in neurodegeneration. Curr Neuropharmacol. 2013;11:298–314. doi: 10.2174/1570159X11311030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hancock DB, Reginsson GW, Gaddis NC, Chen X, Saccone NL, Lutz SM, et al. Genome-wide meta-analysis reveals common splice site acceptor variant in CHRNA4 associated with nicotine dependence. Transl Psychiatry. 2015;5:e651. doi: 10.1038/tp.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- [45].Luetje CW. Getting past the asterisk: the subunit composition of presynaptic nicotinic receptors that modulate striatal dopamine release. Mol Pharmacol. 2004;65:1333–5. doi: 10.1124/mol.65.6.1333. [DOI] [PubMed] [Google Scholar]
- [46].Brunzell DH, McIntosh JM, Papke RL. Diverse strategies targeting alpha7 homomeric and alpha6beta2* heteromeric nicotinic acetylcholine receptors for smoking cessation. Ann N Y Acad Sci. 2014;1327:27–45. doi: 10.1111/nyas.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jones IW, Wonnacott S. Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci. 2004;24:11244–52. doi: 10.1523/JNEUROSCI.3009-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–59. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–78. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- [50].Anand R, Peng X, Lindstrom J. Homomeric and native alpha 7 acetylcholine receptors exhibit remarkably similar but non-identical pharmacological properties, suggesting that the native receptor is a heteromeric protein complex. FEBS Lett. 1993;327:241–6. doi: 10.1016/0014-5793(93)80177-v. [DOI] [PubMed] [Google Scholar]
- [51].Moretti M, Zoli M, George AA, Lukas RJ, Pistillo F, Maskos U, et al. The novel alpha7beta2-nicotinic acetylcholine receptor subtype is expressed in mouse and human basal forebrain: biochemical and pharmacological characterization. Mol Pharmacol. 2014;86:306–17. doi: 10.1124/mol.114.093377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–42. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- [53].Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–4. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- [54].Papke RL, Bencherif M, Lippiello P. An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the alpha 7 subtype. Neurosci Lett. 1996;213:201–4. doi: 10.1016/0304-3940(96)12889-5. [DOI] [PubMed] [Google Scholar]
- [55].Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–19. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- [56].Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–17. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- [57].Bechtholt AJ, Mark GP. Enhancement of cocaine-seeking behavior by repeated nicotine exposure in rats. Psychopharmacology. 2002;162:178–85. doi: 10.1007/s00213-002-1079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bordia T, Hrachova M, Chin M, McIntosh JM, Quik M. Varenicline is a potent partial agonist at alpha6beta2* nicotinic acetylcholine receptors in rat and monkey striatum. J Pharmacol Exp Ther. 2012;342:327–34. doi: 10.1124/jpet.112.194852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gould RW, Garg PK, Garg S, Nader MA. Effects of nicotinic acetylcholine receptor agonists on cognition in rhesus monkeys with a chronic cocaine self-administration history. Neuropharm. 2013;64:479–88. doi: 10.1016/j.neuropharm.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gould RW, Czoty PW, Nader SH, Nader MA. Effects of varenicline on the reinforcing and discriminative stimulus effects of cocaine in rhesus monkeys. J Pharmacol Exp Ther. 2011;339:678–86. doi: 10.1124/jpet.111.185538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Guillem K, Peoples LL. Varenicline effects on cocaine self administration and reinstatement behavior. Behav Pharmacol. 2010;21:96–103. doi: 10.1097/FBP.0b013e328336e9c5. [DOI] [PubMed] [Google Scholar]
- [62].Mello NK, Fivel PA, Kohut SJ, Carroll FI. Effects of chronic varenicline treatment on nicotine, cocaine, and concurrent nicotine+cocaine self-administration. Neuropsychopharm. 2014;39:1222–31. doi: 10.1038/npp.2013.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Poling J, Rounsaville B, Gonsai K, Severino K, Sofuoglu M. The safety and efficacy of varenicline in cocaine using smokers maintained on methadone: a pilot study. Am J Addict. 2010;19:401–8. doi: 10.1111/j.1521-0391.2010.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Roegge CS, Levin ED. Nicotinic receptor antagonists in rats. In: Levin ED, Buccafusco CJ, editors. Animal Models of Cognitive Impairment. CRC Press; Boca Raton, Florida: 2006. pp. 1–14. [Google Scholar]
- [65].Grottick AJ, Higgins GA. Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav Brain Res. 2000;117:197–208. doi: 10.1016/s0166-4328(00)00305-3. [DOI] [PubMed] [Google Scholar]
- [66].Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP. Increased neurodegeneration during ageing in mice lacking high-affinity nicotine receptors. EMBO J. 1999;18:1235–44. doi: 10.1093/emboj/18.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Caldarone BJ, Duman CH, Picciotto MR. Fear conditioning and latent inhibition in mice lacking the high affinity subclass of nicotinic acetylcholine receptors in the brain. Neuropharm. 2000;39:2779–84. doi: 10.1016/s0028-3908(00)00137-4. [DOI] [PubMed] [Google Scholar]
- [68].Grady SR, Meinerz NM, Cao J, Reynolds AM, Picciotto MR, Changeux JP, et al. Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: a function mediated by a different nAChR than dopamine release from striatum. J Neurochem. 2001;76:258–68. doi: 10.1046/j.1471-4159.2001.00019.x. [DOI] [PubMed] [Google Scholar]
- [69].Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–7. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- [70].Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–27. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cahir E, Pillidge K, Drago J, Lawrence AJ. The necessity of alpha4* nicotinic receptors in nicotine-driven behaviors: dissociation between reinforcing and motor effects of nicotine. Neuropsychopharm. 2011;36:1505–17. doi: 10.1038/npp.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sanjakdar SS, Maldoon PP, Marks MJ, Brunzell DH, Maskos U, McIntosh JM, et al. Differential roles of alpha6beta2* and alpha4beta2* neuronal nicotinic receptors in nicotine- and cocaine-conditioned reward in mice. Neuropsychopharm. 2015;40:350–60. doi: 10.1038/npp.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Harenza JL, Muldoon PP, De BM, Damaj MI, Miles MF. Genetic variation within the Chrna7 gene modulates nicotine reward-like phenotypes in mice. Genes Brain Behav. 2014;13:213–25. doi: 10.1111/gbb.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Walters CL, Brown S, Changeux JP, Martin B, Damaj MI. The beta2 but not alpha7 subunit of the nicotinic acetylcholine receptor is required for nicotine-conditioned place preference in mice. Psychopharmacology (Berl) 2006;184:339–44. doi: 10.1007/s00213-005-0295-x. [DOI] [PubMed] [Google Scholar]
- [75].Hansen ST, Mark GP. The nicotinic acetylcholine receptor antagonist mecamylamine prevents escalation of cocaine self-administration in rats with extended daily access. Psychopharmacology. 2007;194:53–61. doi: 10.1007/s00213-007-0822-z. [DOI] [PubMed] [Google Scholar]
- [76].Levin ED, Mead T, Rezvani AH, Rose JE, Gallivan C, Gross R. The nicotinic antagonist mecamylamine preferentially inhibits cocaine vs. food self-administration in rats. Physiol Behav. 2000;71:565–70. doi: 10.1016/s0031-9384(00)00382-6. [DOI] [PubMed] [Google Scholar]
- [77].Lenoir M, Serre F, Cantin L, Ahmed SH. Intense sweetness surpasses cocaine reward. PLoS ONE. 2007;2:e698. doi: 10.1371/journal.pone.0000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Reid MS, Mickalian JD, Delucchi KL, Hall SM, Berger SP. An acute dose of nicotine enhances cue-induced cocaine craving. Drug Alcohol Depend. 1998;49:95–104. doi: 10.1016/s0376-8716(97)00144-0. [DOI] [PubMed] [Google Scholar]
- [79].Reid MS, Mickalian JD, Delucchi KL, Berger SP. A nicotine antagonist, mecamylamine, reduces cue-induced cocaine craving in cocaine-dependent subjects. Neuropsychopharm. 1999;20:297–307. doi: 10.1016/S0893-133X(98)00076-1. [DOI] [PubMed] [Google Scholar]
- [80].Ikemoto S, Yang C, Tan A. Basal ganglia circuit loops, dopamine and motivation: A review and enquiry. Behav Brain Res. 2015;290:17–31. doi: 10.1016/j.bbr.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Dani JA, De BM. Cellular mechanisms of nicotine addiction. Pharmacol Biochem Behav. 2001;70:439–46. doi: 10.1016/s0091-3057(01)00652-9. [DOI] [PubMed] [Google Scholar]
- [82].Wonnacott S, Barik J. Nicotinic acetylcholine receptors. Tocris Reviews. 2007;28:1–19. [Google Scholar]
- [83].Ikemoto S, Wise RA. Rewarding effects of the cholinergic agents carbachol and neostigmine in the posterior ventral tegmental area. J Neurosci. 2002;22:9895–904. doi: 10.1523/JNEUROSCI.22-22-09895.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Schmidt HD, Famous KR, Pierce RC. The limbic circuitry underlying cocaine seeking encompasses the PPTg/LDT. Eur J Neurosci. 2009;30:1358–69. doi: 10.1111/j.1460-9568.2009.06904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang H, Sun X. Desensitized nicotinic receptors in brain. Brain Res Brain Res Rev. 2005;48:420–37. doi: 10.1016/j.brainresrev.2004.09.003. [DOI] [PubMed] [Google Scholar]
- [86].Girod R, Role LW. Long-lasting enhancement of glutamatergic synaptic transmission by acetylcholine contrasts with response adaptation after exposure to low-level nicotine. J Neurosci. 2001;21:5182–90. doi: 10.1523/JNEUROSCI.21-14-05182.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Reitstetter R, Lukas RJ, Gruener R. Dependence of nicotinic acetylcholine receptor recovery from desensitization on the duration of agonist exposure. J Pharmacol Exp Ther. 1999;289:656–60. [PubMed] [Google Scholar]
- [88].Zachariou V, Caldarone BJ, Weathers-Lowin A, George TP, Elsworth JD, Roth RH, et al. Nicotine receptor inactivation decreases sensitivity to cocaine. Neuropsychopharm. 2001;24:576–89. doi: 10.1016/S0893-133X(00)00224-4. [DOI] [PubMed] [Google Scholar]
- [89].Marks MJ, Campbell SM, Romm E, Collins AC. Genotype influences the development of tolerance to nicotine in the mouse. J Pharmacol Exp Ther. 1991;259:392–402. [PubMed] [Google Scholar]
- [90].Marks MJ, Grady SR, Collins AC. Downregulation of nicotinic receptor function after chronic nicotine infusion. J Pharmacol Exp Ther. 1993;266:1268–76. [PubMed] [Google Scholar]
- [91].Tan H, Bishop SF, Lauzon NM, Sun N, Laviolette SR. Chronic nicotine exposure switches the functional role of mesolimbic dopamine transmission in the processing of nicotine's rewarding and aversive effects. Neuropharm. 2009;56:741–51. doi: 10.1016/j.neuropharm.2008.12.008. [DOI] [PubMed] [Google Scholar]
- [92].Besson M, David V, Baudonnat M, Cazala P, Guilloux JP, Reperant C, et al. Alpha7-nicotinic receptors modulate nicotine-induced reinforcement and extracellular dopamine outflow in the mesolimbic system in mice. Psychopharmacology (Berl) 2012;220:1–14. doi: 10.1007/s00213-011-2422-1. [DOI] [PubMed] [Google Scholar]
- [93].Besson M, Granon S, Mameli-Engvall M, Cloez-Tayarani I, Maubourguet N, Cormier A, et al. Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A. 2007;104:8155–60. doi: 10.1073/pnas.0702698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Melis M, Scheggi S, Carta G, Madeddu C, Lecca S, Luchicchi A, et al. PPARalpha regulates cholinergic-driven activity of midbrain dopamine neurons via a novel mechanism involving alpha7 nicotinic acetylcholine receptors. J Neurosci. 2013;33:6203–11. doi: 10.1523/JNEUROSCI.4647-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Damaj MI, Kao W, Martin BR. Characterization of spontaneous and precipitated nicotine withdrawal in the mouse. J Pharmacol Exp Ther. 2003;307:526–34. doi: 10.1124/jpet.103.054908. [DOI] [PubMed] [Google Scholar]
- [96].Bruijnzeel AW, Markou A. Adaptations in cholinergic transmission in the ventral tegmental area associated with the affective signs of nicotine withdrawal in rats. Neuropharm. 2004;47:572–9. doi: 10.1016/j.neuropharm.2004.05.005. [DOI] [PubMed] [Google Scholar]
- [97].Barnes SA, Der-Avakian A, Markou A. Anhedonia, avolition, and anticipatory deficits: assessments in animals with relevance to the negative symptoms of schizophrenia. Eur Neuropsychopharmacol. 2014;24:744–58. doi: 10.1016/j.euroneuro.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jackson KJ, Martin BR, Changeux JP, Damaj MI. Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Ther. 2008;325:302–12. doi: 10.1124/jpet.107.132977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Stoker AK, Marks MJ, Markou A. Null mutation of the beta2 nicotinic acetylcholine receptor subunit attenuates nicotine withdrawal-induced anhedonia in mice. Eur J Pharmacol. 2015;753:146–50. doi: 10.1016/j.ejphar.2014.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Nomikos GG, Hildebrand BE, Panagis G, Svensson TH. Nicotine withdrawal in the rat: role of alpha7 nicotinic receptors in the ventral tegmental area. Neuroreport. 1999;10:697–702. doi: 10.1097/00001756-199903170-00007. [DOI] [PubMed] [Google Scholar]
- [101].Stoker AK, Olivier B, Markou A. Role of alpha7- and beta4-containing nicotinic acetylcholine receptors in the affective and somatic aspects of nicotine withdrawal: studies in knockout mice. Behav Genet. 2012;42:423–36. doi: 10.1007/s10519-011-9511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Girault JA. Integrating neurotransmission in striatal medium spiny neurons. Adv Exp Med Biol. 2012;970:407–29. doi: 10.1007/978-3-7091-0932-8_18. [DOI] [PubMed] [Google Scholar]
- [103].Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharm. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kiyatkin EA, Rebec GV. Striatal neuronal activity and responsiveness to dopamine and glutamate after selective blockade of D1 and D2 dopamine receptors in freely moving rats. J Neurosci. 1999;19:3594–609. doi: 10.1523/JNEUROSCI.19-09-03594.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].West AR, Grace AA. Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J Neurosci. 2002;22:294–304. doi: 10.1523/JNEUROSCI.22-01-00294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yan Z, Flores-Hernandez J, Surmeier DJ. Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience. 2001;103:1017–24. doi: 10.1016/s0306-4522(01)00039-2. [DOI] [PubMed] [Google Scholar]
- [107].Zhang L, Warren RA. Muscarinic and nicotinic presynaptic modulation of EPSCs in the nucleus accumbens during postnatal development. J Neurophysiol. 2002;88:3315–30. doi: 10.1152/jn.01025.2001. [DOI] [PubMed] [Google Scholar]
- [108].Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–63. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Shin JH, Adrover MF, Wess J, Alvarez VA. Muscarinic regulation of dopamine and glutamate transmission in the nucleus accumbens. Proc Natl Acad Sci U S A. 2015;112:8124–9. doi: 10.1073/pnas.1508846112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Raffa RB. The M5 muscarinic receptor as possible target for treatment of drug abuse. J Clin Pharm Ther. 2009;34:623–9. doi: 10.1111/j.1365-2710.2009.01059.x. [DOI] [PubMed] [Google Scholar]
- [111].Weiner DM, Levey AI, Brann MR. Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc Natl Acad Sci U S A. 1990;87:7050–4. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Aosaki T, Kimura M, Graybiel AM. Temporal and spatial characteristics of tonically active neurons of the primate's striatum. J Neurophysiol. 1995;73:1234–52. doi: 10.1152/jn.1995.73.3.1234. [DOI] [PubMed] [Google Scholar]
- [113].Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci. 2014;6:22. doi: 10.3389/fnsyn.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Apicella P, Deffains M, Ravel S, Legallet E. Tonically active neurons in the striatum differentiate between delivery and omission of expected reward in a probabilistic task context. Eur J Neurosci. 2009;30:515–26. doi: 10.1111/j.1460-9568.2009.06872.x. [DOI] [PubMed] [Google Scholar]
- [115].Doig NM, Magill PJ, Apicella P, Bolam JP, Sharott A. Cortical and thalamic excitation mediate the multiphasic responses of striatal cholinergic interneurons to motivationally salient stimuli. J Neurosci. 2014;34:3101–17. doi: 10.1523/JNEUROSCI.4627-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–43. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- [117].Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A. 2006;103:2938–42. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Brown MT, Tan KR, O'Connor EC, Nikonenko I, Muller D, Luscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492:452–6. doi: 10.1038/nature11657. [DOI] [PubMed] [Google Scholar]
- [119].Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron. 2014;81:901–12. doi: 10.1016/j.neuron.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- [121].Surmeier DJ, Graybiel AM. A feud that wasn't: acetylcholine evokes dopamine release in the striatum. Neuron. 2012;75:1–3. doi: 10.1016/j.neuron.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, et al. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–81. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsaki G, Deisseroth K, et al. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123–30. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Nicola SM. The nucleus accumbens as part of a basal ganglia action selection circuit. Psychopharmacology. 2007;191:521–50. doi: 10.1007/s00213-006-0510-4. [DOI] [PubMed] [Google Scholar]
- [126].Thomsen M, Fulton BS, Caine SB. Acute and chronic effects of the M1/M4-preferring muscarinic agonist xanomeline on cocaine vs. food choice in rats. Psychopharmacology. 2014;231:469–79. doi: 10.1007/s00213-013-3256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Thomsen M, Conn PJ, Lindsley C, Wess J, Boon JY, Fulton BS, et al. Attenuation of cocaine's reinforcing and discriminative stimulus effects via muscarinic M1 acetylcholine receptor stimulation. J Pharmacol Exp Ther. 2010;332:959–69. doi: 10.1124/jpet.109.162057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Rasmussen T, Sauerberg P, Nielsen EB, Swedberg MD, Thomsen C, Sheardown MJ, et al. Muscarinic receptor agonists decrease cocaine self-administration rates in drug-naive mice. Eur J Pharmacol. 2000;402:241–6. doi: 10.1016/s0014-2999(00)00442-8. [DOI] [PubMed] [Google Scholar]
- [129].Wilson MC, Schuster CR. Cholinergic influence on intravenous cocaine self-administration by rhesus monkeys. Pharmacol Biochem Behav. 1973;1:643–9. doi: 10.1016/0091-3057(73)90027-0. [DOI] [PubMed] [Google Scholar]
- [130].Ranaldi R, Woolverton WL. Self-administration of cocaine: scopolamine combinations by rhesus monkeys. Psychopharmacology. 2002;161:442–8. doi: 10.1007/s00213-002-1069-3. [DOI] [PubMed] [Google Scholar]
- [131].Yee J, Famous KR, Hopkins TJ, McMullen MC, Pierce RC, Schmidt HD. Muscarinic acetylcholine receptors in the nucleus accumbens core and shell contribute to cocaine priming-induced reinstatement of drug seeking. Eur J Pharmacol. 2011;650:596–604. doi: 10.1016/j.ejphar.2010.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Terry AV., Jr . Muscarinic receptor antagonists in rats. In: Levin ED, Buccafusco CJ, editors. Animal Models of Cognitive Impairment. CRC Press; Boca Raton, Florida: 2006. pp. 1–16. [Google Scholar]
- [133].Klinkenberg I, Blokland A. The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci Biobehav Rev. 2010;34:1307–50. doi: 10.1016/j.neubiorev.2010.04.001. [DOI] [PubMed] [Google Scholar]
- [134].Pratt WE, Kelley AE. Nucleus accumbens acetylcholine regulates appetitive learning and motivation for food via activation of muscarinic receptors. Behav Neurosci. 2004;118:730–9. doi: 10.1037/0735-7044.118.4.730. [DOI] [PubMed] [Google Scholar]
- [135].Pratt WE, Spencer RC, Kelley AE. Muscarinic receptor antagonism of the nucleus accumbens core causes avoidance to flavor and spatial cues. Behav Neurosci. 2007;121:1215–23. doi: 10.1037/0735-7044.121.6.1215. [DOI] [PubMed] [Google Scholar]
- [136].Gronier B, Perry KW, Rasmussen K. Activation of the mesocorticolimbic dopaminergic system by stimulation of muscarinic cholinergic receptors in the ventral tegmental area. Psychopharmacology. 2000;147:347–55. doi: 10.1007/s002130050002. [DOI] [PubMed] [Google Scholar]
- [137].Klinkenberg I, Blokland A. A comparison of scopolamine and biperiden as a rodent model for cholinergic cognitive impairment. Psychopharmacology. 2011;215:549–66. doi: 10.1007/s00213-011-2171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Mark GP, Kinney AE, Grubb MC, Zhu X, Finn DA, Mader SL, et al. Injection of oxotremorine in nucleus accumbens shell reduces cocaine but not food self-administration in rats. Brain Res. 2006;1123:51–9. doi: 10.1016/j.brainres.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Allahverdiyev O, Nurten A, Enginar N. Assessment of rewarding and reinforcing properties of biperiden in conditioned place preference in rats. Behav Brain Res. 2011;225:642–5. doi: 10.1016/j.bbr.2011.07.050. [DOI] [PubMed] [Google Scholar]
- [140].Ramos AC, Andersen ML, Oliveira MG, Soeiro AC, Galduroz JC. Biperiden (M(1) antagonist) impairs the expression of cocaine conditioned place preference but potentiates the expression of cocaine-induced behavioral sensitization. Behav Brain Res. 2012;231:213–6. doi: 10.1016/j.bbr.2012.03.030. [DOI] [PubMed] [Google Scholar]
- [141].Zacarias MS, Ramos AC, Alves DR, Galduroz JC. Biperiden (an M1 antagonist) reduces memory consolidation of cocaine-conditioned place preference. Neurosci Lett. 2012;513:129–31. doi: 10.1016/j.neulet.2012.01.073. [DOI] [PubMed] [Google Scholar]
- [142].Dieckmann LH, Ramos AC, Silva EA, Justo LP, Sabioni P, Frade IF, et al. Effects of biperiden on the treatment of cocaine/crack addiction: a randomised, double-blind, placebo-controlled trial. Eur Neuropsychopharmacol. 2014;24:1196–202. doi: 10.1016/j.euroneuro.2014.06.001. [DOI] [PubMed] [Google Scholar]
- [143].Wezenberg E, Verkes RJ, Sabbe BG, Ruigt GS, Hulstijn W. Modulation of memory and visuospatial processes by biperiden and rivastigmine in elderly healthy subjects. Psychopharmacology. 2005;181:582–94. doi: 10.1007/s00213-005-0083-7. [DOI] [PubMed] [Google Scholar]
- [144].Carrigan KA, Dykstra LA. Behavioral effects of morphine and cocaine in M1 muscarinic acetylcholine receptor-deficient mice. Psychopharmacology. 2007;191:985–93. doi: 10.1007/s00213-006-0671-1. [DOI] [PubMed] [Google Scholar]
- [145].Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, et al. Deletion of the M5 muscarinic acetylcholine receptor attenuates morphine reinforcement and withdrawal but not morphine analgesia. Proc Natl Acad Sci U S A. 2002;99:11452–7. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Steidl S, Miller AD, Blaha CD, Yeomans JS. M(5) muscarinic receptors mediate striatal dopamine activation by ventral tegmental morphine and pedunculopontine stimulation in mice. PLoS ONE. 2011;6:e27538. doi: 10.1371/journal.pone.0027538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Fink-Jensen A, Fedorova I, Wortwein G, Woldbye DP, Rasmussen T, Thomsen M, et al. Role for M5 muscarinic acetylcholine receptors in cocaine addiction. J Neurosci Res. 2003;74:91–6. doi: 10.1002/jnr.10728. [DOI] [PubMed] [Google Scholar]
- [148].Thomsen M, Woldbye DP, Wortwein G, Fink-Jensen A, Wess J, Caine SB. Reduced cocaine self-administration in muscarinic M5 acetylcholine receptor-deficient mice. J Neurosci. 2005;25:8141–9. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Forster GL, Yeomans JS, Takeuchi J, Blaha CD. M5 muscarinic receptors are required for prolonged accumbal dopamine release after electrical stimulation of the pons in mice. J Neurosci. 2002;22:RC190. doi: 10.1523/JNEUROSCI.22-01-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yeomans JS, Takeuchi J, Baptista M, Flynn DD, Lepik K, Nobrega J, et al. Brain-stimulation reward thresholds raised by an antisense oligonucleotide for the M5 muscarinic receptor infused near dopamine cells. J Neurosci. 2000;20:8861–7. doi: 10.1523/JNEUROSCI.20-23-08861.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Yeomans J, Baptista M. Both nicotinic and muscarinic receptors in ventral tegmental area contribute to brain-stimulation reward. Pharmacol Biochem Behav. 1997;57:915–21. doi: 10.1016/s0091-3057(96)00467-4. [DOI] [PubMed] [Google Scholar]
- [152].Yamada M, Lamping KG, Duttaroy A, Zhang W, Cui Y, Bymaster FP, et al. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:14096–101. doi: 10.1073/pnas.251542998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhang W, Yamada M, Gomeza J, Basile AS, Wess J. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice. J Neurosci. 2002;22:6347–52. doi: 10.1523/JNEUROSCI.22-15-06347.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Dencker D, Weikop P, Sorensen G, Woldbye DP, Wortwein G, Wess J, et al. An allosteric enhancer of M(4) muscarinic acetylcholine receptor function inhibits behavioral and neurochemical effects of cocaine. Psychopharmacology. 2012;224:277–87. doi: 10.1007/s00213-012-2751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Jeon J, Dencker D, Wortwein G, Woldbye DP, Cui Y, Davis AA, et al. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J Neurosci. 2010;30:2396–405. doi: 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Taha SA, Fields HL. Inhibitions of nucleus accumbens neurons encode a gating signal for reward-directed behavior. J Neurosci. 2006;26:217–22. doi: 10.1523/JNEUROSCI.3227-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Krause M, German PW, Taha SA, Fields HL. A pause in nucleus accumbens neuron firing is required to initiate and maintain feeding. J Neurosci. 2010;30:4746–56. doi: 10.1523/JNEUROSCI.0197-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Carlezon WA, Jr., Thomas MJ. Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharm. 2009;56(Suppl 1):122–32. doi: 10.1016/j.neuropharm.2008.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Peoples LL, West MO. Phasic firing of single neurons in the rat nucleus accumbens correlated with the timing of intravenous cocaine self-administration. J Neurosci. 1996;16:3459–73. doi: 10.1523/JNEUROSCI.16-10-03459.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Takagi M, Yamamoto C. Suppressing action of cholinergic agents on synaptic transmissions in the corpus striatum of rats. Exp Neurol. 1978;62:433–43. doi: 10.1016/0014-4886(78)90066-3. [DOI] [PubMed] [Google Scholar]
- [161].Windels F, Kiyatkin EA. Modulatory action of acetylcholine on striatal neurons: microiontophoretic study in awake, unrestrained rats. Eur J Neurosci. 2003;17:613–22. doi: 10.1046/j.1460-9568.2003.02492.x. [DOI] [PubMed] [Google Scholar]
- [162].Joshua M, Adler A, Mitelman R, Vaadia E, Bergman H. Midbrain dopaminergic neurons and striatal cholinergic interneurons encode the difference between reward and aversive events at different epochs of probabilistic classical conditioning trials. J Neurosci. 2008;28:11673–84. doi: 10.1523/JNEUROSCI.3839-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Ebihara K, Yamamoto K, Ueda K, Koshikawa N, Kobayashi M. Cholinergic interneurons suppress action potential initiation of medium spiny neurons in rat nucleus accumbens shell. Neuroscience. 2013;236:332–44. doi: 10.1016/j.neuroscience.2013.01.012. [DOI] [PubMed] [Google Scholar]
- [164].Pakhotin P, Bracci E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Koos T, Tepper JM. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci. 2002;22:529–35. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet. 2002;41:719–39. doi: 10.2165/00003088-200241100-00003. [DOI] [PubMed] [Google Scholar]
- [167].Aarsland D, Mosimann UP, McKeith IG. Role of cholinesterase inhibitors in Parkinson's disease and dementia with Lewy bodies. J Geriatr Psychiatry Neurol. 2004;17:164–71. doi: 10.1177/0891988704267463. [DOI] [PubMed] [Google Scholar]
- [168].Balsters JH, O'Connell RG, Martin MP, Galli A, Cassidy SM, Kilcullen SM, et al. Donepezil impairs memory in healthy older subjects: behavioural, EEG and simultaneous EEG/fMRI biomarkers. PLoS ONE. 2011;6:e24126. doi: 10.1371/journal.pone.0024126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Pepeu G, Giovannini MG. Cholinesterase inhibitors and memory. Chem Biol Interact. 2010;187:403–8. doi: 10.1016/j.cbi.2009.11.018. [DOI] [PubMed] [Google Scholar]
- [170].Bentley P, Driver J, Dolan RJ. Cholinesterase inhibition modulates visual and attentional brain responses in Alzheimer's disease and health. Brain. 2008;131:409–24. doi: 10.1093/brain/awm299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Imbimbo BP. Pharmacodynamic-tolerability relationships of cholinesterase inhibitors for Alzheimer's disease. CNS Drugs. 2001;15:375–90. doi: 10.2165/00023210-200115050-00004. [DOI] [PubMed] [Google Scholar]
- [172].Hikida T, Kitabatake Y, Pastan I, Nakanishi S. Acetylcholine enhancement in the nucleus accumbens prevents addictive behaviors of cocaine and morphine. Proc Natl Acad Sci U S A. 2003;100:6169–73. doi: 10.1073/pnas.0631749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Smith JE, Co C, Yin X, Sizemore GM, Liguori A, Johnson WE, III, et al. Involvement of cholinergic neuronal systems in intravenous cocaine self-administration. Neurosci Biobehav Rev. 2004;27:841–50. doi: 10.1016/j.neubiorev.2003.11.002. [DOI] [PubMed] [Google Scholar]
- [174].Zhang L, Zhou FM, Dani JA. Cholinergic drugs for Alzheimer's disease enhance in vitro dopamine release. Mol Pharmacol. 2004;66:538–44. doi: 10.1124/mol.104.000299. [DOI] [PubMed] [Google Scholar]
- [175].De La Garza R, Johanson CE. Effects of haloperidol and physostigmine on self-administration of local anesthetics. Pharmacol Biochem Behav. 1982;17:1295–9. doi: 10.1016/0091-3057(82)90138-1. [DOI] [PubMed] [Google Scholar]
- [176].Grasing K, He S, Yang Y. Dose-related effects of the acetylcholinesterase inhibitor tacrine on cocaine and food self-administration in rats. Psychopharmacology. 2008;196:133–42. doi: 10.1007/s00213-007-0944-3. [DOI] [PubMed] [Google Scholar]
- [177].Grasing K, He S, Yang Y. Long-lasting decreases in cocaine-reinforced behavior following treatment with the cholinesterase inhibitor tacrine in rats selectively bred for drug self-administration. Pharmacol Biochem Behav. 2009;94:169–78. doi: 10.1016/j.pbb.2009.08.004. [DOI] [PubMed] [Google Scholar]
- [178].Grasing K, Yang Y, He S. Reversible and persistent decreases in cocaine self-administration after cholinesterase inhibition: different effects of donepezil and rivastigmine. Behav Pharmacol. 2011;22:58–70. doi: 10.1097/FBP.0b013e3283428cd8. [DOI] [PubMed] [Google Scholar]
- [179].van Haaren F, Haworth SC, Bennett SM, Cody BA. The effects of pyridostigmine bromide on progressive ratio performance in male and female rats. Pharmacol Biochem Behav. 2001;68:81–5. doi: 10.1016/s0091-3057(00)00438-x. [DOI] [PubMed] [Google Scholar]
- [180].Liu WF. Effects of cholinesterase inhibitors on a two-component chained schedule performance in rats. Neurotoxicol Teratol. 2000;22:389–96. doi: 10.1016/s0892-0362(99)00086-0. [DOI] [PubMed] [Google Scholar]
- [181].Zhou W, Liu H, Zhang F, Tang S, Zhu H, Lai M, et al. Role of acetylcholine transmission in nucleus accumbens and ventral tegmental area in heroin-seeking induced by conditioned cues. Neuroscience. 2007;144:1209–18. doi: 10.1016/j.neuroscience.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Gawel K, Labuz K, Jenda M, Silberring J, Kotlinska JH. Influence of cholinesterase inhibitors, donepezil and rivastigmine on the acquisition, expression, and reinstatement of morphine-induced conditioned place preference in rats. Behav Brain Res. 2014;268:169–76. doi: 10.1016/j.bbr.2014.04.019. [DOI] [PubMed] [Google Scholar]
- [183].Takamatsu Y, Yamanishi Y, Hagino Y, Yamamoto H, Ikeda K. Differential Effects of Donepezil on Methamphetamine and Cocaine Dependencies. Ann N Y Acad Sci. 2006;1074:418–26. doi: 10.1196/annals.1369.042. [DOI] [PubMed] [Google Scholar]
- [184].Riddle EL, Fleckenstein AE, Hanson GR. Role of monoamine transporters in mediating psychostimulant effects. AAPS J. 2005;7:E847–E851. doi: 10.1208/aapsj070481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Grasing K, Ghosh S. Selegiline prevents long-term changes in dopamine efflux and stress immobility during the second and third weeks of abstinence following opiate withdrawal. Neuropharm. 1998;37:1007–17. doi: 10.1016/s0028-3908(98)00093-8. [DOI] [PubMed] [Google Scholar]
- [186].Grasing K, Mathur D, Newton TF, DeSouza C. Donepezil treatment and the subjective effects of intravenous cocaine in dependent individuals. Drug Alcohol Depend. 2010;107:69–75. doi: 10.1016/j.drugalcdep.2009.09.010. [DOI] [PubMed] [Google Scholar]
- [187].Winhusen TM, Somoza EC, Harrer JM, Mezinskis JP, Montgomery MA, Goldsmith RJ, et al. A placebo-controlled screening trial of tiagabine, sertraline and donepezil as cocaine dependence treatments. Addiction. 2005;100(Suppl 1):68–77. doi: 10.1111/j.1360-0443.2005.00992.x. [DOI] [PubMed] [Google Scholar]
- [188].Farlow MR, Salloway S, Tariot PN, Yardley J, Moline ML, Wang Q, et al. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer's disease: A 24-week, randomized, double-blind study. Clin Ther. 2010;32:1234–51. doi: 10.1016/j.clinthera.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].De La Garza R, Shoptaw S, Newton TF. Evaluation of the cardiovascular and subjective effects of rivastigmine in combination with methamphetamine in methamphetamine-dependent human volunteers. Int J Neuropsychopharmacol. 2008:1–13. doi: 10.1017/S1461145708008456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].De La Garza R, Mahoney JJ, III, Culbertson C, Shoptaw S, Newton TF. The acetylcholinesterase inhibitor rivastigmine does not alter total choices for methamphetamine, but may reduce positive subjective effects, in a laboratory model of intravenous self-administration in human volunteers. Pharmacol Biochem Behav. 2008;89:200–8. doi: 10.1016/j.pbb.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Coyle JT, Geerts H, Sorra K, Amatniek J. Beyond in vitro data: a review of in vivo evidence regarding the allosteric potentiating effect of galantamine on nicotinic acetylcholine receptors in Alzheimer's neuropathology. J Alzheimers Dis. 2007;11:491–507. doi: 10.3233/jad-2007-11411. [DOI] [PubMed] [Google Scholar]
- [192].Schilstrom B, Ivanov VB, Wiker C, Svensson TH. Galantamine enhances dopaminergic neurotransmission in vivo via allosteric potentiation of nicotinic acetylcholine receptors. Neuropsychopharm. 2007;32:43–53. doi: 10.1038/sj.npp.1301087. [DOI] [PubMed] [Google Scholar]
- [193].Sharp BM, Yatsula M, Fu Y. Effects of galantamine, a nicotinic allosteric potentiating ligand, on nicotine-induced catecholamine release in hippocampus and nucleus accumbens of rats. J Pharmacol Exp Ther. 2004;309:1116–23. doi: 10.1124/jpet.103.063586. [DOI] [PubMed] [Google Scholar]
- [194].Mann K, Ackermann K, Diehl A, Ebert D, Mundle G, Nakovics H, et al. Galantamine: a cholinergic patch in the treatment of alcoholism: a randomized, placebo-controlled trial. Psychopharmacology. 2006;184:115–21. doi: 10.1007/s00213-005-0243-9. [DOI] [PubMed] [Google Scholar]
- [195].Diehl A, Nakovics H, Croissant B, Smolka MN, Batra A, Mann K. Galantamine reduces smoking in alcohol-dependent patients: a randomized, placebo-controlled trial. Int J Clin Pharmacol Ther. 2006;44:614–22. doi: 10.5414/cpp44614. [DOI] [PubMed] [Google Scholar]
- [196].Sofuoglu M, Waters AJ, Poling J, Carroll KM. Galantamine improves sustained attention in chronic cocaine users. Exp Clin Psychopharmacol. 2011;19:11–9. doi: 10.1037/a0022213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Sofuoglu M, Carroll KM. Effects of galantamine on cocaine use in chronic cocaine users. Am J Addict. 2011;20:302–3. doi: 10.1111/j.1521-0391.2011.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Sofuoglu M, Herman AI, Li Y, Waters AJ. Galantamine attenuates some of the subjective effects of intravenous nicotine and improves performance on a Go No-Go task in abstinent cigarette smokers: a preliminary report. Psychopharmacology. 2012;224:413–20. doi: 10.1007/s00213-012-2763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Thomsen M, Lindsley CW, Conn PJ, Wessell JE, Fulton BS, Wess J, et al. Contribution of both M1 and M4 receptors to muscarinic agonist-mediated attenuation of the cocaine discriminative stimulus in mice. Psychopharmacology. 2012;220:673–85. doi: 10.1007/s00213-011-2516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Prast JM, Kummer KK, Barwitz CM, Humpel C, Dechant G, Zernig G. Acetylcholine, drug reward and substance use disorder treatment: intra- and interindividual striatal and accumbal neuron ensemble heterogeneity may explain apparent discrepant findings. Pharmacology. 2012;90:264–73. doi: 10.1159/000342636. [DOI] [PubMed] [Google Scholar]
- [201].Britt JP, Bonci A. Optogenetic interrogations of the neural circuits underlying addiction. Curr Opin Neurobiol. 2013;23:539–45. doi: 10.1016/j.conb.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Lobo MK, Covington HE, III, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–90. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, et al. Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci. 2013;16:632–8. doi: 10.1038/nn.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].O'Connor EC, Kremer Y, Lefort S, Harada M, Pascoli V, Rohner C, et al. Accumbal D1R neurons projecting to lateral hypothalamus authorize feeding. Neuron. 2015;88:553–64. doi: 10.1016/j.neuron.2015.09.038. [DOI] [PubMed] [Google Scholar]
- [205].Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491:212–7. doi: 10.1038/nature11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Matsumoto M, Hikosaka O. Representation of negative motivational value in the primate lateral habenula. Nat Neurosci. 2009;12:77–84. doi: 10.1038/nn.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Jhou TC, Good CH, Rowley CS, Xu SP, Wang H, Burnham NW, et al. Cocaine drives aversive conditioning via delayed activation of dopamine-responsive habenular and midbrain pathways. J Neurosci. 2013;33:7501–12. doi: 10.1523/JNEUROSCI.3634-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Glick SD, Sell EM, McCallum SE, Maisonneuve IM. Brain regions mediating alpha3beta4 nicotinic antagonist effects of 18-MC on nicotine self-administration. Eur J Pharmacol. 2011;669:71–5. doi: 10.1016/j.ejphar.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Baldacchino A, Balfour DJ, Passetti F, Humphris G, Matthews K. Neuropsychological consequences of chronic opioid use: a quantitative review and meta-analysis. Neurosci Biobehav Rev. 2012;36:2056–68. doi: 10.1016/j.neubiorev.2012.06.006. [DOI] [PubMed] [Google Scholar]
- [210].Jovanovski D, Erb S, Zakzanis KK. Neurocognitive deficits in cocaine users: a quantitative review of the evidence. J Clin Exp Neuropsychol. 2005;27:189–204. doi: 10.1080/13803390490515694. [DOI] [PubMed] [Google Scholar]
- [211].Schrimsher GW, Parker JD, Burke RS. Relation between cognitive testing performance and pattern of substance use in males at treatment entry. Clin Neuropsychol. 2007;21:498–510. doi: 10.1080/13803390600674441. [DOI] [PubMed] [Google Scholar]
- [212].Woicik PA, Moeller SJ, ia-Klein N, Maloney T, Lukasik TM, Yeliosof O, et al. The neuropsychology of cocaine addiction: recent cocaine use masks impairment. Neuropsychopharm. 2009;34:1112–22. doi: 10.1038/npp.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Pace-Schott EF, Morgan PT, Malison RT, Hart CL, Edgar C, Walker M, et al. Cocaine users differ from normals on cognitive tasks which show poorer performance during drug abstinence. Am J Drug Alcohol Abuse. 2008;34:109–21. doi: 10.1080/00952990701764821. [DOI] [PubMed] [Google Scholar]