

Abstract
The fungal kingdom is a hyperdiverse group of multicellular eukaryotes with profound impacts on human society and ecosystem function. The challenge of documenting and describing fungal diversity is exacerbated by their typically cryptic nature, their ability to produce seemingly unrelated morphologies from a single individual and their similarity in appearance to distantly related taxa. This multiplicity of hurdles resulted in the early adoption of DNA-based comparisons to study fungal diversity, including linking curated DNA sequence data to expertly identified voucher specimens. DNA-barcoding approaches in fungi were first applied in specimen-based studies for identification and discovery of taxonomic diversity, but are now widely deployed for community characterization based on sequencing of environmental samples. Collectively, fungal barcoding approaches have yielded important advances across biological scales and research applications, from taxonomic, ecological, industrial and health perspectives. A major outstanding issue is the growing problem of ‘sequences without names’ that are somewhat uncoupled from the traditional framework of fungal classification based on morphology and preserved specimens. This review summarizes some of the most significant impacts of fungal barcoding, its limitations, and progress towards the challenge of effective utilization of the exponentially growing volume of data gathered from high-throughput sequencing technologies.
This article is part of the themed issue ‘From DNA barcodes to biomes’.
Keywords: internal transcribed spacer, fungi, barcoding, diversity, metabarcoding, taxonomy
1. Introduction
Diversity in the fungal kingdom is estimated to range from 1.5 to more than 5 million species [1–3], but only a small fraction of these species (approx. 100 000) have so far been described [4], despite their essential roles in ecological systems in terms of global chemical cycling, decomposition, nutrient acquisition in symbiosis and pathogenicity [5–8]. Because these eukaryotic organisms have microscopic life-history stages with simple and often convergent morphological features, genetic data are essential for quantifying the extent and distribution of their diversity. Early molecular studies focused on fungi relevant to medical and industrial applications, but within little more than a decade, surveys of the natural environment were being used to uncover hidden fungal diversity, all based on universal nuclear ribosomal primers developed by White et al. [9]. The development of these primers was perhaps the most important advance in establishing a barcoding approach—using standard, short sequences to identify taxa, facilitating comparative research across diverse fungal groups and ultimately becoming the standard practice.
The formal acceptance of the internal transcribed spacer (ITS) region in the nuclear ribosomal cistron as the standard fungal barcode was based on a phylogenetically wide-ranging test showing reasonable discriminatory power at the species level in many groups [10]. This built on an extensive body of literature showing that discontinuities in sequence variation often correspond to data from morphology, chemistry, biogeography and ecology [11]. Comparisons with mitochondrial cytochrome oxidase 1 (CO1), the standard barcode marker for animals, showed that in many fungi CO1 is prone to having multiple introns and is difficult to amplify with universal primers [12–14]. There is now an extensive set of resources for fungal barcoding, including sampling protocols and laboratory techniques, summarized in the electronic supplementary material.
The aim of this paper is to review how DNA barcoding has been deployed to enhance understanding of global fungal diversity, including both scientific advances and societal applications, focusing on ITS barcoding and extending to genome-wide sequencing. We build on more than 20 years of data collection using the originally de facto and now formal ITS barcode marker, and we reiterate the challenge to integrate DNA sequence data into the wider historical classification framework for fungi [15–17]. Given the scale of this challenge, and the increasingly urgent need to rationalize these two approaches, it is clear that one important role for DNA barcoding will be to generate novel species hypotheses as well as evaluating existing taxon concepts. Although unrelated lineages such as oomycetes share fungus-like lifestyles, research challenges and even barcoding target loci [18], we restrict this review to true fungi.
2. Fungal barcoding databases
Effective DNA barcoding requires comparing newly generated sequences to a well-established reference database. This voucher-based approach enabling reproducibility and re-examination was advocated from the early stages of fungal barcoding, with sequence data routinely accompanied by curated and annotated specimens or strains [19]. However, two important challenges have persisted. First, despite concerted efforts to fill the gap, only a small proportion of fungal species have ITS data in the public databases, such as GenBank, which form part of the International Nucleotide Sequence Database Collaboration (INSDC) [20–23]. Second is the accumulation of misidentified and unspecified sequences in public sequence databases [24,25], which make identifications using these sequences problematic.
GenBank and UNITE are the main repositories of fungal sequence data. These data are augmented by various specialized sequence databases for barcoding and barcoding-related work, which collate and curate databases of reference material (voucher specimens or cultures) linked to sequence data (table 1). In addition, several specialized bioinformatics pipelines for high-throughput fungal analyses have been devised [36–38]. The UNITE database and PlutoF [39] workbench include modules for ITS extraction, chimera checking, and identification, including matching query sequences with species hypotheses (including varying similarity cut-offs) and reference sequences determined by expert users [36]. The Ribosomal Database Project (RDP) employs a naive Bayesian approach to classify unknown sequences, relying on initially selected training sets [40], including ITS [41]. Another important initiative at the US National Center for Biotechnology Information (NCBI) is focused on curating and re-annotating ITS sequences from type material that is already publically available at the INSDC, i.e. the RefSeq Targeted Loci ITS project [32,42]. Curated databases with a guarantee of long-term support are critically important, because the community-led specialist fungal databases often lack such funding commitments.
Table 1.
A selection of actively curated specialist databases containing ITS reference sequences for fungi, including single and multilocus sequence typing (MLST) resources.
| name | scope | contents | web address, NCBI nucleotide Entrez search term(s) |
|---|---|---|---|
| CBS-KNAW: Centraalbureau voor Schimmelcultures BioloMICS Databases [26] |
Aspergillus and Penicillium Dermatophytes Fusarium indoor fungi medical fungi Phaeoacremonium Pseudallescheria/Scedosporium Resupinate Russulales Russula yeasts |
ITS and MLST | www.cbs.knaw.nl |
| Fusarium-ID [27,28] | Fusarium | multiple markers, ITS | http://isolate.fusariumdb.org |
| International Society of Human and Animal Mycology (ISHAM) ITS database [29] | human and animal pathogens | ITS |
http://its.mycologylab.org loprovishamits[filter] |
| MaarjAM [30] | Glomeromycota | multiple markers, ITS | http://maarjam.botany.ut.ee |
| Q-Bank [31] | quarantine organisms | ITS and MLST | www.q-bank.eu/fungi |
| RefSeq Targeted Loci [32] | mainly sequences from type material, re-annotated from INSDC | ITS, LSU |
http://www.ncbi.nlm.nih.gov/refseq/targetedloci/ 177353[bioproject] 51803[bioproject] |
| RDP Classifier [33] | wide taxonomic range, training set selected from INSDC | ITS, LSU | https://rdp.cme.msu.edu/classifier/classifier.jsp |
| Targeted host-associated Fungi ITS Database (THF) [34] | wide taxonomic range, re-annotated from INSDC | ITS | https://risccweb.csmc.edu/microbiome/thf/ |
| TrichoBLAST [35] | Trichoderma and its Hypocrea synonyms | TEF1, RPB2, ITS | www.isth.info/tools/blast/index.php |
| UNITE [36] | wide taxonomic range, re-annotated from INSDC | ITS |
http://Unite.ut.ee loprovunite[filter] |
3. Linking names and sequences
Ultimately, linking scientific names to molecular data requires reference sequences generated from type material. Recent efforts to add barcodes for cultures and specimens are beginning to make inroads into this problem [32,43]. However, the problem of ‘dark taxa’ represented by sequences lacking formal binomials is steadily growing [44,45]. The latest comparisons of the names in the NCBI Taxonomy Database [46] indicate a shift in the early 2000s where more sequences were released without, rather than with, species-level identification; this trend was recognized by Hibbett et al. [17] in 2011 and has not diminished (figure 1). Compounding the ‘sequences without names’ issue is the ‘names without sequences’ problem (figure 2). From a 10 year period up to 2009, more than 70% of new fungal species described had no ITS sequence deposited [17]. This in part is driven by some researchers having limited access or resources for DNA sequencing, and in part by researchers not choosing to generate the sequence data for new species. After the requirement for online deposition of new fungal names was proposed in 2011, the percentage of new species with sequences has increased to 55%. Another important improvement to NCBI, implemented in 2013, is allowing material to be retrospectively designated by curators as from a type in the taxonomy database; links can also be made directly to outside biorepositories [47]. As of 2016, 23% (7308) of current fungal species with binomials (32 431) in the INSDC databases (November 2015) can be tied to sequences from type material with 14% (4759) having quality verified ITS sequences in the UNITE database.
Figure 1.
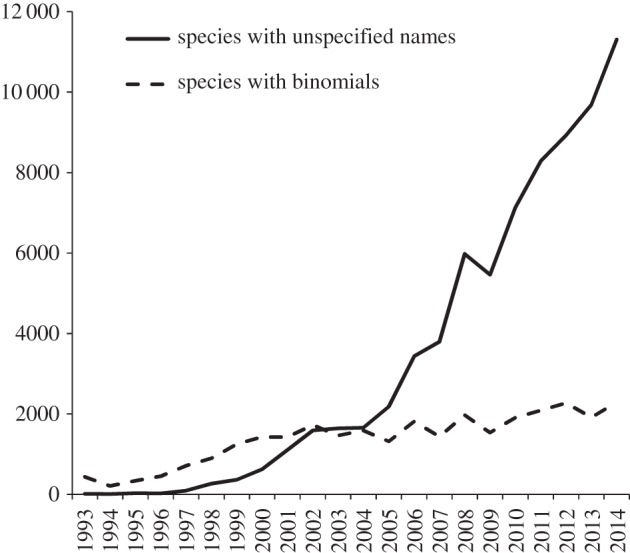
Number of fungal binomial species names and unspecified binomials added each year—‘dark taxa’ in GenBank.
Figure 2.
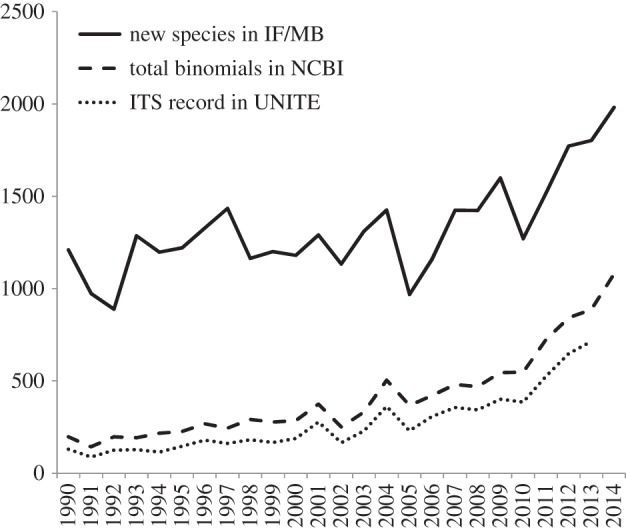
Number of new species listed in Index Fungorum by year, and their representation in the NCBI Taxonomy Database. New species names in Index Fungorum per year (solid line), their presence in NCBI Taxonomy (dashed line) and which of them have ITS sequences in the UNITE database (version 7; dotted line).
Massively parallel metabarcoding, the use of high-throughput barcoding to analyse community composition, is also resulting in ever-increasing numbers of unidentified fungal ITS sequences [48], a challenge clearly articulated in the recent review by Hibbett et al. [17]. The main public repository for these sequences is the Sequence Read Archive (SRA) of NCBI. Current bioinformatic tools and techniques diagnose molecular operational taxonomic units (MOTUs) or species hypotheses from these sequences using similarity thresholds (e.g. 0.03 for ectomycorrhizal (EM) fungi or 0.05 for endophytes). This standardization of unit diagnosis using sequence data allows fungal ecologists to compare across studies and geographical areas. Although sequence clusters are potentially uncoupled from other biologically meaningful information and may not always correspond to recognized species [49–51], it is clear that closely related fungi can be detected repeatedly with this approach, enabling diversity comparisons to be made.
The outputs of conventional specimen-based barcoding and community sequencing thus share the common problem of linking sequences (and sequence clusters) to names. One approach to tackle this is akin to that of the barcode identification number system used in animal barcoding [52]. This involves the establishment of a separate nomenclature based around sequence clusters; this approach is used in the UNITE database [36]. This sequence cluster framework can then be mapped to existing taxonomic infrastructure where sequence clusters/MOTUs overlap with named specimens. The alternative approach, advocated by Hibbett and co-workers [17,53], is for mycologists to collectively work to alter common practice in applying the International Code of Nomenclature, allowing species to be formally named with binomials based on sequence types alone or based on environmental samples.
Regardless of the mechanism, the importance of using well-curated fungal collections as a source of expert taxonomic opinion and authoritative-type material cannot be overstated, and can serve to integrate new sequence data with taxonomy and its important legacy of associated biological and evolutionary knowledge [54,55]. Although ITS has been successfully sequenced from fungus-type material over 200 years old [56,57], this is the exception. Shotgun sequencing of historic material [58] may represent a new opportunity to access genetic information in historical specimens, potentially revolutionizing our ability to stabilize nomenclature and improve connections between sequences, specimens and names. It is clear that both integrating retrospective data from existing collections and routinely sequencing new collections (including generating sequences from all new species) are needed.
4. Barcoding successes
An important success of fungal ITS barcoding and the tools devoted to its use (e.g. table 1) is the increased ability to include fungi in studies of biological diversity. Although few fungal researchers use the term barcoding, ITS sequencing is now often a routine part of diversity assessment, particularly for unexplored habitats and regions. Because most of the world's fungi have not been detected by traditional surveys, basic distributional data about the species diversity for most geographical regions and fungal groups are often lacking [59].
(a). Location-based insights into diversity
At the level of a local assemblage, barcoding approaches have relatively rarely been carried out on individual specimens, because most fungi are usually microscopic. However, lichens and EM fungi produce perennial structures and can provide tests of the method. In lichens, a floristic specimen-based barcoding approach identified a larger proportion of specimens than taxonomists owing to its greater ability to identify scanty, immature and poorly developed material [60]. This does depend on having a reference database available, and in the less well-known lower latitudes, the lack of suitable sequenced reference material for comparison still acts as a constraint [61]. For EM fungi, barcoding of root tips shows greater diversity at sites than above-ground identification of fruiting bodies, even with up to 50 years of fruit–body surveys [11,51,62,63].
Metabarcoding studies have been on scales ranging from the tiny (e.g. the size of insect guts [64] and leaves [65]) to whole-forest soils [48]. In most such studies, the two-stage process of species discrimination and species identification entirely relies on bioinformatics pipelines to streamline analysis of many thousands of newly generated sequences (see section Fungal barcoding databases). Not surprisingly, using this approach, the number of new species clusters discovered by ITS has been growing relative to specimen-based sequences, with little overlap between taxa found in specimen-based compared with environmental samples [17], and total estimates of diversity limited by the inability of studies to reach saturation in rarefaction curves [1,48,66]. For instance, endophyte diversity using ITS sequence data typically far exceeds that found using morphotypes (even with a conservative sequence similarity of 95%; [67]).
(b). Taxon-based studies
Because of the cryptic nature of the fungal lifecycle, a large degree of unseen diversity is expected. This exists across taxonomic ranks, with new class and even phylum-level divergences being documented, with a particularly rapid expansion in known fungal diversity stemming from sequencing of environmental samples [68,69].
Although multilocus sequencing is likely to remain the gold standard for the unambiguous definition of new species [70–72], data from ITS have been a steady component of fungal diversity description since the early 2000s. Numerous examples of cryptic species have been described, with unrecognized genetic diversity hidden in what was assumed to be a single lineage, e.g. [73], even from quite small sampling areas (e.g. 400 m2, [74]). Biologists have long been aware of cryptic species [75], with perhaps the most extreme example from a single basidiolichen now known to represent at least 126 species based on ITS divergence, each with a recognizable combination of traits, including morphology, habitat and distribution. Hundreds more species belonging to this morphology were predicted from unsampled geographical areas [76]. Other lichens and form genera in asexual fungi offer similar cases of extreme polyphyly hidden by seemingly similar morphologies [77,78].
At larger spatial scales, one repeated finding is that fungal taxa with wide distributions are likely to comprise different and isolated genetic lineages sharing exceedingly similar morphology [79–81]. Often, names based on first-described types have been applied to similar morphologies as the nearest approximation of a species hypothesis in another geographical area [82,83], and fungal species with broad geographical distributions are likely to represent fertile areas for discovery of cryptic species [84,85]. The one caveat to this is the trend of widespread high-latitude distributions for many fungal taxa across the arctic [86–88]. A key practical issue in unravelling the complexity of widespread named taxa is effective sampling. Low-intensity sampling from a restricted part of the distribution may generate apparently distinct sequence clusters that then merge as further sampling across the range is undertaken [89].
(c). Ecology and biogeography
Although a succession of individual species-based studies have shown that fungi are distributed in biogeographically distinct patterns [90], the availability of large datasets from high-throughput studies means that global trends can begin to be examined for fungi in a meaningful way. Ongoing debates about primer choice notwithstanding (electronic supplementary material), the rapidly accumulating findings are at last opening a window into biogeography and diversity for unseen, uncultured and uncollected fungi [59]. For both endophytes and soil fungi, the general trend of species diversity increasing with decreasing latitude has been supported by ITS data [65,91,92]. In contrast, EM fungi appear to be more diverse in the temperate zone [92–95], corresponding to general trends of high Basidiomycota diversity in temperate Fagus [96] and pine [1] forests. Similarly, in a global sample of indoor air, latitude was the best predictor of fungal diversity rather than the details of the buildings sampled, and temperate diversity was higher than tropical [97]. Analyses across the arctic have shown no decline in EM fungal diversity from two host plants with increasing latitude [98], and increasing dominance of Ascomycota, including the majority of lichens [99].
Metabarcoding studies of fungi have shown similar biogeographic patterns to other organisms [100], but have also revealed a surprising level of local distributions, potentially sensitive enough for determination of geographical origins of dust for forensic or archaeological application [101]. Supporting the idea that fungal endemism is widespread, metadata mined from unidentified fungal ITS sequences in the INSDC databases allowed a comparison across EM genera, showing that a small handful of poorly known genera such as Inocybe, Tomentella, Cortinarius and Russula are often encountered, with high numbers of unidentified sequences (e.g. widespread genera, but not widespread species) [102]. In one example, 0–40% of sequences of Inocybe were identified to the species level, depending on their continent of origin, with lower numbers of identified sequences come from Asia and Australia, where reference material is poorly represented in databases [102] and where tropical regions have higher degrees of endemism [92]. However, there are exceptions, and some species were apparently widespread with over 35% of species found on more than one continent [102]. A meta-analysis of published ITS sequences from the truffle genus Tuber documented 126 ITS phylotypes, with none sharing intercontinental distributions [103].
One of the best-studied groups of fungi, the EM plant associates, has been used to address the long-standing question about how the diversity of fungi is associated with the diversity of plants. Although there is a general geographical bias in studies of EM fungi favouring Western Europe and North America, in a review of 100 studies, EM fungal diversity was shown to be better explained by host-plant genera than by plant species or family-level diversity [104]. Even some of the best examples of highly specific plant–fungus symbioses have associations that link fungal species groups to host-plant genera [105,106]. Similarly, in fungal–algal associations in lichens, extreme host specificity at the species or strain level tends to be the exception rather than the rule [107]. Typical patterns demonstrate the specificity of fungi for their algal hosts above the species level [108–111]. Likewise, in a global meta-analysis of arbuscular mycorrhizal fungi in the Glomeromycota, fungal community differences are related to geographical distance, climate and plant community [112].
(d). Conservation applications
Although fungi are often poorly represented in conservation plans compared with plants and animals, they are of considerable conservation relevance. Fungal species can act as bioindicators of habitat status and type, and indicate sites with long ecological continuity [113]. Fungi are also involved in a myriad of complex, often unseen, interactions that are crucial in the functioning of many ecosystems. Individual species can also provide societal benefits in terms of nutrition, medicine, aesthetics and/or cultural values, and hence warrant conservation in their own right. Given that DNA barcoding can improve species discovery and an understanding of fungal distributions [114,115], it can, by extension, improve conservation decision-making.
Metabarcoding datasets have been compared with specimen-based inventory data for invertebrates and birds, and have shown general comparability in relative assessments of alpha and beta diversity, in addition to having the advantages of being much more efficient in terms of person-hours, and amenable to audit by third parties [116]. Such approaches have great potential to understand diversity, distributions and trends in fungi to inform conservation policy and practice. However, one challenge is the potential for disengagement of conservation agencies (e.g. conservation non-governmental organizations) and natural history societies whose working ethos is based around named species and whose efforts have been critical to establishing data on fungal diversity and distribution studies to date. Thus, effective systems to connect sequence data to the existing taxonomic framework are important from a conservation perspective [117] for maintaining cultural connections to fungal diversity. Convenient but non-Linnean names (e.g. soil clone group 1, [69]) can represent a barrier to uptake by land managers, local agencies and decision-makers in many countries.
(e). Wider societal applications
The practical applications of insights from barcoding may be profound for natural and human systems. Detection of plant pathogens has huge economic implications for both forestry [118] and crop plant systems, where a single pathogen can potentially impact a crop worth billions [119]. At tactical timescales, the detection of cryptic species is crucial to understand major ecological change in European woodlands: divergent ITS types distinguished Hymenoscyphus fraxineus, the novel disease agent causing ash dieback [120], with the increasingly apparent impact in the UK [121]. From a human-health perspective, barcoding can extend to indoor mycology [97,122,123] and the importance of fungi contributing to both health and illness in the human microbiome [124], in addition to the obvious application to identification of human and animal pathogens [6], for which diagnostic inaccuracy represents a serious shortcoming [125]. Barcoding approaches are also applicable to industry, in food traceability and understanding industrial composting processes [126–128].
5. Where ITS barcoding fails
It is estimated that barcoding using the ITS amplicon is effective for species discrimination across more than 70% of fungi tested [10]. For a barcoding approach to be successful, the variation between species should exceed that within species, with barcodes from a given species best matching conspecifics. The use of ITS sequences for species diagnosis was questioned early on when divergent ITS2 sequences were detected in Fusarium [129], and although uncommonly reported, they appear to be taxonomically widespread [130–132] and sometimes linked to hybridization [133]. Intragenomic heterogeneity in ITS may be more prevalent than is currently appreciated, found in several unrelated ascomycete and basidiomycete genera [134,135]. In Glomeromycota, species are multinucleate with extreme intraspecies divergence in nuclear ribosomal sequences, which creates additional challenges for the use of ITS for species discrimination [136].
On the other hand, the lack of sufficient ITS variability has also been a problem, especially in Ascomycota. In some species-rich genera, ITS amplicons that are shorter than the 500 bp recommended for an effective barcode marker are typical [137], resulting in many species having insufficient variation to discriminate important biologically significant groups or closely related species [23]. Although the ITS cistron can correctly identify fungi to the genus level, species discrimination is poor for many plant pathogenic fungi in economically important genera such as Alternaria, Diaporthe, Fusarium, Teratosphaeria and others [23].
6. Secondary barcode markers
In some lineages, protein-coding genes may have equal or better resolving power than ITS, although these suffer from the lack of universal primers and unreliable amplification [10,138]. In some economically important fungi, genus-specific techniques have been developed which sometimes incorporate ITS along with other markers in multilocus sequence typing [139,140]. This can be used for sequence matching and strain identification [31]. Efforts are underway to propose protein-coding secondary barcodes for specific groups of fungi [141–143]. Additionally, the broad application of using protein coding markers to directly sample environments has been demonstrated recently [144]. This suggests that improved amplification methods may allow for protein-coding genes to act as near-universal DNA barcodes and it will be worthwhile to consider an expansion beyond ITS alone as the barcode marker.
7. Integrating DNA barcoding with genomic studies
Fungal genomes provide robust scaffolds that can improve phylogenetic resolution [145], and phylogenomic analyses have proved key for understanding evolutionary relationships in some fungal groups, such as yeasts [146,147]. Sequencing costs continue to drop, new technologies promise rapid and portable platforms that increase the accessibility of genomic sequencing (e.g. Oxford Nanopore's MinION), and ambitious efforts to compile large-scale genome-level data have been proceeding, such as the 1000 fungal genomes [148] and the Plant and Fungal Tree of Life [149]. Already, over 2000 fungal genome projects are underway or complete [150]. However, these initiatives still require considerably more research investment in data gathering and data analysis. For example, multispecies coalescent approaches sacrifice scalability and efficiency in addition to computational time for multilocus versus single-locus approaches [151], and phylogenomic analysis is complicated by gene tree incongruence and the increased sensitivity to long branch attraction from concatenated alignments [146,152]. However, these problems are surmountable, and many nuclear phylogenomic datasets confirm current phylogenetic hypotheses [153].
Although it seems unlikely that whole genome comparisons will displace ITS-based barcoding for fungi in the near future owing to consumables costs and especially the degree of bioinformatics expertise required, there are already several approaches to compare whole genomes that would mirror a DNA-barcoding approach without the need for full-scale phylogenomics. A sizable percentage of known bacterial species have multiple genomes deposited at GenBank. This includes genome data obtained from type cultures for close to 30% of all bacterial species. The use of average nucleotide identity (ANI) and kmer score comparisons are feasible for fast identification of misidentified bacterial genomes [47,154]. Although eukaryotic genomes certainly pose more complex challenges, some of the bacterial approaches could be scalable to fungi [155]. It seems likely that the yeasts will be the first lineages of fungi where this will become a reality in the near future [156]. An important step in this process is linking standard ITS barcoding with genome sequencing projects. Public genome assemblies frequently do not include sequences from nuclear ribosomal RNA cistron, and when they are included, it is often as incorrect or low-quality assemblies. A simple practical step to promote future comparability of fungal datasets is to increase efforts in providing reliable ribosomal data for samples that have their genomes sequenced.
8. Conclusion
Fungal research has benefited tremendously from DNA-barcoding approaches and the growing collection of sequences in public, curated databases. Applications range from critical identifications of pathogens to global-scale investigations of fungal diversity. However, the scale of the challenge posed by the sheer diversity of fungi is enormous. Pooling resources to identify and tackle knowledge gaps is therefore essential, and the mycological community has already actively promoted several large-scale collaborations [23,157,158].
The world's preserved fungal collections in herbaria represent an underused resource for building up voucher-based reference datasets [20]: collections-based sequencing is an important priority for the coming decades. Likewise, another step of key importance is to increase the proportion of newly described species that have barcode sequences from the type material. Nevertheless, it is also clear that most fungal diversity will remain uncollected and uncultured, and for the foreseeable future will be known only from environmental samples and sequences. There is thus an urgent need for the fungal research community to unite behind a common approach linking sequences to an effective, scalable method of naming. This approach needs to maximize linkages between ITS barcode sequences and the existing taxonomic framework encompassing specimens, morphological taxonomic descriptions and species concepts. It also needs to encompass the growing depth of sequence coverage given the inevitable increase in genome-level sequencing and the need for multilocus data to provide species-level resolution in many fungal groups.
Supplementary Material
Acknowledgements
Katy Hayden, Pete Hollingsworth and two reviewers improved earlier drafts of this manuscript. Paul Kirk, Nathalie van de Wiele and Barbara Robbertse are thanked for assistance with data for figures 1 and 2.
Authors' contributions
R.Y., C.L.S. and B.T.M.D. drafted the manuscript. C.L.S. and B.T.M.D. provided data for the figures. All gave final approval for publication.
Competing interests
We have no competing interests.
Funding
C.L.S. acknowledges support from the Intramural Research Program of the US National Institutes of Health, National Library of Medicine. R.Y. was supported by the Scottish Government's Rural and Environment Science and Analytical Services.
References
- 1.O'Brien HE, Parrent JL, Jackson JA, Moncalvo J-M, Vilgalys R. 2005. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microbiol. 71, 5544–5550. ( 10.1128/AEM.71.9.5544-5550.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass D, Richards TA. 2011. Three reasons to re-evaluate fungal diversity ‘on Earth and in the ocean’. Fungal Biol. Rev. 25, 159–164. ( 10.1016/j.fbr.2011.10.003) [DOI] [Google Scholar]
- 3.Hawksworth DL. 1991. The fungal dimension of biodiversity - magnitude, significance, and conservation. Mycol. Res. 95, 641–655. ( 10.1016/S0953-7562(09)80810-1) [DOI] [Google Scholar]
- 4.Kirk P, Cannon P, Minter D, Stalpers J. 2008. Dictionary of the fungi. Wallingford, UK: CABI. [Google Scholar]
- 5.Martin F, Cullen D, Hibbett D, Pisabarro A, Spatafora JW, Baker SE, Grigoriev IV. 2011. Sequencing the fungal tree of life. New Phytol. 190, 818–821. ( 10.1111/j.1469-8137.2011.03688.x) [DOI] [PubMed] [Google Scholar]
- 6.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484, 186–194. ( 10.1038/nature10947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Read DJ, Leake JR, Perez-Moreno J. 2004. Mycorrhizal fungi as drivers of ecosystem processes in heathland and boreal forest biomes. Can. J. Bot.-Revue Canadienne De Botanique 82, 1243–1263. [Google Scholar]
- 8.Taylor LL, Banwart SA, Valdes PJ, Leake JR, Beerling DJ. 2012. Evaluating the effects of terrestrial ecosystems, climate and carbon dioxide on weathering over geological time: a global-scale process-based approach. Phil. Trans. R. Soc. B 367, 565–582. ( 10.1098/rstb.2011.0251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White TJ, Bruns T, Lee S, Taylor JW. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR protocols: a guide to methods and applications (eds Innis MA, Gelfand DH, Sninsky JJ, White TJ), pp. 315–322. New York, NY: Academic Press. [Google Scholar]
- 10.Schoch CL, Seifert KA, Huhndorf C, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl Acad. Sci. USA 109, 6241–6246. ( 10.1073/pnas.1117018109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton TR, Bruns TD. 2001. The molecular revolution in ectomycorrhizal ecology: peeking into the black-box. Mol. Ecol. 10, 1855–1871. ( 10.1046/j.0962-1083.2001.01333.x) [DOI] [PubMed] [Google Scholar]
- 12.Begerow D, Nilsson H, Unterseher M, Maier W. 2010. Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 87, 99–108. ( 10.1007/s00253-010-2585-4) [DOI] [PubMed] [Google Scholar]
- 13.Dentinger BT, Didukh MY, Moncalvo J-M. 2011. Comparing COI and ITS as DNA barcode markers for mushrooms and allies (Agaricomycotina). PLoS ONE 6, e25081 ( 10.1371/journal.pone.0025081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seifert KA, Samson RA, Houbraken J, Lévesque CA, Moncalvo J-M, Louis-Seize G, Hebert PD. 2007. Prospects for fungus identification using CO1 DNA barcodes, with Penicillium as a test case. Proc. Natl Acad. Sci. USA 104, 3901–3906. ( 10.1073/pnas.0611691104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor JW, Hibbett DS. 2013. Toward sequence-based classification of fungal species. IMA Fungus 4, 33–34. [Google Scholar]
- 16.Herr JR, Öpik M, Hibbett DS. 2015. Towards the unification of sequence-based classification and sequence-based identification of host-associated microorganisms. New Phytol. 205, 27–31. ( 10.1111/nph.13180) [DOI] [PubMed] [Google Scholar]
- 17.Hibbett DS, Ohman A, Glotzer D, Nuhn M, Kirk P, Nilsson RH. 2011. Progress in molecular and morphological taxon discovery in Fungi and options for formal classification of environmental sequences. Fungal Biol. Rev. 25, 38–47. ( 10.1016/j.fbr.2011.01.001) [DOI] [Google Scholar]
- 18.Robideau GP, et al. 2011. DNA barcoding of oomycetes with cytochrome c oxidase subunit I and internal transcribed spacer. Mol. Ecol. Res. 11, 1002–1011. ( 10.1111/j.1755-0998.2011.03041.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratnasingham S, Hebert PD. 2007. bold: the barcode of life data system (www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364. ( 10.1111/j.1471-8286.2007.01678.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brock PM, Döring H, Bidartondo MI. 2009. How to know unknown fungi: the role of a herbarium. New Phytol. 181, 719–724. ( 10.1111/j.1469-8137.2008.02703.x) [DOI] [PubMed] [Google Scholar]
- 21.Osmundson TW, Robert VA, Schoch CL, Baker LJ, Smith A, Robich G, Mizzan L, Garbelotto MM. 2013. Filling gaps in biodiversity knowledge for macrofungi: contributions and assessment of an herbarium collection DNA barcode sequencing project. PLoS ONE 8, e62419 ( 10.1371/journal.pone.0062419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreakis N, Høj L, Kearns P, Hall MR, Ericson G, Cobb RE, Gordon BR, Evans-Illidge E. 2015. Diversity of marine-derived fungal cultures exposed by DNA barcodes: the algorithm matters. PLoS ONE 10, e0136130 ( 10.1371/journal.pone.0136130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crous PW, Hawksworth DL, Wingfield MJ. 2015. Identifying and naming plant-pathogenic fungi: past, present, and future. Annu. Rev. Phytopathol. 53, 247–267. ( 10.1146/annurev-phyto-080614-120245) [DOI] [PubMed] [Google Scholar]
- 24.Bridge PD, Roberts PJ, Spooner BM, Panchal G. 2003. On the unreliability of published DNA sequences. New Phytol. 160, 43–48. ( 10.1046/j.1469-8137.2003.00861.x) [DOI] [PubMed] [Google Scholar]
- 25.Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson K-H, Kõljalg U. 2006. Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS ONE 1, e59 ( 10.1371/journal.pone.0000059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robert V, Szoke S, Jabas B, Vu D, Chouchen O, Blom E, Cardinali G. 2011. BioloMICS software: biological data management, identification, classification and statistics. Open Appl. Informatics J. 5, 87–98. ( 10.2174/1874136301005010087) [DOI] [Google Scholar]
- 27.Geiser DM, Jiménez-Gasco MD, Kang SC, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, O'Donnell K. 2004. FUSARIUM-ID v. 1.0: A DNA sequence database for identifying Fusarium. Eur. J. Plant Pathol. 110, 473–479. ( 10.1023/B:Ejpp.0000032386.75915.A0) [DOI] [Google Scholar]
- 28.Park B, et al. 2011. Cyber infrastructure for Fusarium: three integrated platforms supporting strain identification, phylogenetics, comparative genomics and knowledge sharing. Nucleic Acids Res. 39, D640–D646. ( 10.1093/nar/gkq1166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irinyi L, et al. 2015. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—the quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med. Mycol. 53, 313–337. ( 10.1093/mmy/myv008) [DOI] [PubMed] [Google Scholar]
- 30.Öpik M, Vanatoa A, Vanatoa E, Moora M, Davison J, Kalwij J, Reier Ü, Zobel M. 2010. The online database MaarjAM reveals global and ecosystemic distribution patterns in arbuscular mycorrhizal fungi (Glomeromycota). New Phytol. 188, 223–241. ( 10.1111/j.1469-8137.2010.03334.x) [DOI] [PubMed] [Google Scholar]
- 31.Bonants P, Edema M, Robert V. 2013. Q-bank, a database with information for identification of plant quarantine plant pest and diseases. EPPO Bull. 43, 211–215. ( 10.1111/epp.12030) [DOI] [Google Scholar]
- 32.Schoch CL, et al. 2014. Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database-J. Biol. Databases Curation 2014, bau061. ( 10.1093/database/bau061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. ( 10.1128/AEM.00062-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang J, Iliev ID, Brown J, Underhill DM, Funari VA. 2015. Mycobiome: approaches to analysis of intestinal fungi. J. Immunol. Methods 421, 112–121. ( 10.1016/j.jim.2015.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopchinskiy A, Komón M, Kubicek CP, Druzhinina IS. 2005. TrichoBLAST: a multilocus database for Trichoderma and Hypocrea identifications. Mycol. Res. 109, 658–660. ( 10.1017/S0953756205233397) [DOI] [PubMed] [Google Scholar]
- 36.Kõljalg U, et al. 2013. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. ( 10.1111/mec.12481) [DOI] [PubMed] [Google Scholar]
- 37.Gweon HS, Oliver A, Taylor J, Booth T, Gibbs M, Read DS, Griffiths RI, Schonrogge K. 2015. PIPITS: an automated pipeline for analyses of fungal internal transcribed spacer sequences from the Illumina sequencing platform. Methods Ecol. Evol. 6, 973–980. ( 10.1111/2041-210X.12399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bálint M, Schmidt P-A, Sharma R, Thines M, Schmitt I. 2014. An Illumina metabarcoding pipeline for fungi. Ecol. Evol. 4, 2642–2653. ( 10.1002/ece3.1107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abarenkov K, et al. 2010. PlutoF—a web based workbench for ecological and taxonomic research, with an online implementation for fungal ITS sequences. Evol. Bioinform. Online 6, 189. [Google Scholar]
- 40.Porras-Alfaro A, Liu K-L, Kuske CR, Xie G. 2014. From genus to phylum: large-subunit and internal transcribed spacer rRNA operon regions show similar classification accuracies influenced by database composition. Appl. Environ. Microbiol. 80, 829–840. ( 10.1128/AEM.02894-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deshpande V, Wang Q, Greenfield P, Charleston M, Porras-Alfaro A, Kuske CR, Cole JR, Midgley DJ, Tran-Dinh N. 2016. Fungal identification using a Bayesian classifier and the Warcup training set of internal transcribed spacer sequences. Mycologia 1, 1–5. ( 10.3852/14-293) [DOI] [PubMed] [Google Scholar]
- 42.O'Leary NA, et al. 2015. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. ( 10.1093/nar/gkv1189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crous PW, et al. 2014. The Genera of Fungi: fixing the application of type species of generic names. IMA Fungus 5, 141–160. ( 10.5598/imafungus.2014.05.01.14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Page RD. 2013. BioNames: linking taxonomy, texts, and trees. PeerJ 1, e190 ( 10.7717/peerj.190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Page RDM. 2016. DNA barcoding and taxonomy: dark taxa and dark texts. Phil. Trans. R. Soc. B 371, 20150334 ( 10.1098/rstb.2015.0334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Federhen S. 2012. The NCBI taxonomy database. Nucleic Acids Res. 40, D136–D143. ( 10.1093/nar/gkr1178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Federhen S. 2015. Type material in the NCBI taxonomy database. Nucleic Acids Res. 43, D1086–D1098. ( 10.1093/nar/gku1127) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F. 2009. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol. 184, 449–456. ( 10.1111/j.1469-8137.2009.03003.x) [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto N, Bibby K. 2014. Clustering of fungal community internal transcribed spacer sequence data obscures taxonomic diversity. Environ. Microbiol. 16, 2491–2500. ( 10.1111/1462-2920.12390) [DOI] [PubMed] [Google Scholar]
- 50.Ryberg M. 2015. Molecular operational taxonomic units as approximations of species in the light of evolutionary models and empirical data from Fungi. Mol. Ecol. 24, 5770–5777. ( 10.1111/mec.13444) [DOI] [PubMed] [Google Scholar]
- 51.Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H. 2008. Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. Online 4, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ratnasingham S, Hebert PD. 2013. A DNA-based registry for all animal species: the barcode index number (BIN) system. PLoS ONE 8, e66213 ( 10.1371/journal.pone.0066213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hibbett DS, Taylor JW. 2013. Fungal systematics: is a new age of enlightenment at hand? Nat. Rev. Microbiol. 11, 129–133. ( 10.1038/nrmicro2963) [DOI] [PubMed] [Google Scholar]
- 54.Parr CS, Guralnick R, Cellinese N, Page RD. 2012. Evolutionary informatics: unifying knowledge about the diversity of life. Trends Ecol. Evol. 27, 94–103. ( 10.1016/j.tree.2011.11.001) [DOI] [PubMed] [Google Scholar]
- 55.Korf RP. 2005. Reinventing taxonomy: a curmudgeon's view of 250 years of fungal taxonomy, the crisis in biodiversity, and the pitfalls of the phylogenetic age. Mycotaxon 93, 407–416. [Google Scholar]
- 56.Larsson E, Jacobsson S. 2004. Controversy over Hygrophorus cossus settled using ITS sequence data from 200 year-old type material. Mycol. Res. 108, 781–786. ( 10.1017/S0953756204000310) [DOI] [PubMed] [Google Scholar]
- 57.Hawksworth DL. 2013. The oldest sequenced fungal specimen. The Lichenologist 45, 131–132. ( 10.1017/S0024282912000710) [DOI] [Google Scholar]
- 58.Staats M, Erkens RHJ, van de Vossenberg B, Wieringa JJ, Kraaijeveld K, Stielow B, Geml J, Richardson JE, Bakker FT. 2013. Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS ONE 8, e69189 ( 10.1371/journal.pone.0069189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peay KG, Bidartondo MI, Arnold AE. 2010. Not every fungus is everywhere: scaling to the biogeography of fungal–plant interactions across roots, shoots and ecosystems. New Phytol. 185, 878–882. ( 10.1111/j.1469-8137.2009.03158.x) [DOI] [PubMed] [Google Scholar]
- 60.Kelly LJ, Hollingsworth PM, Coppins BJ, Ellis CJ, Harrold P, Tosh J, Yahr R. 2011. DNA barcoding of lichenized fungi demonstrates high identification success in a floristic context. New Phytol. 191, 288–300. ( 10.1111/j.1469-8137.2011.03677.x) [DOI] [PubMed] [Google Scholar]
- 61.Orock EA, Leavitt SD, Fonge BA, St Clair LL, Lumbsch HT. 2012. DNA-based identification of lichen-forming fungi: can publicly available sequence databases aid in lichen diversity inventories of Mount Cameroon (West Africa)? Lichenologist 44, 833–839. ( 10.1017/s0024282912000424) [DOI] [Google Scholar]
- 62.Porter TM, Skillman JE, Moncalvo JM. 2008. Fruiting body and soil rDNA sampling detects complementary assemblage of Agaricomycotina (Basidiomycota, Fungi) in a hemlock-dominated forest plot in southern Ontario. Mol. Ecol. 17, 3037–3050. ( 10.1111/j.1365-294X.2008.03813.x) [DOI] [PubMed] [Google Scholar]
- 63.Johnson J, Evans C, Brown N, Skeates S, Watkinson S, Bass D. 2014. Molecular analysis shows that soil fungi from ancient semi-natural woodland exist in sites converted to non-native conifer plantations. Forestry 87, 705–717. ( 10.1093/forestry/cpu031) [DOI] [Google Scholar]
- 64.Suh S-O, McHugh JV, Pollock DD, Blackwell M. 2005. The beetle gut: a hyperdiverse source of novel yeasts. Mycol. Res. 109, 261–265. ( 10.1017/S0953756205002388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arnold AE, Lutzoni F. 2007. Diversity and host range of foliar fungal endophytes: are tropical leaves biodiversity hotspots? Ecology 88, 541–549. ( 10.1890/05-1459) [DOI] [PubMed] [Google Scholar]
- 66.Jumpponen A. 2007. Soil fungal communities underneath willow canopies on a primary successional glacier forefront: rDNA sequence results can be affected by primer selection and chimeric data. Microb. Ecol. 53, 233–246. ( 10.1007/s00248-004-0006-x) [DOI] [PubMed] [Google Scholar]
- 67.Arnold AE, Henk DA, Eells RL, Lutzoni F, Vilgalys R. 2007. Diversity and phylogenetic affinities of foliar fungal endophytes in loblolly pine inferred by culturing and environmental PCR. Mycologia 99, 185–206. ( 10.3852/mycologia.99.2.185) [DOI] [PubMed] [Google Scholar]
- 68.Grossart H-P, Wurzbacher C, James TY, Kagami M. 2015. Discovery of dark matter fungi in aquatic ecosystems demands a reappraisal of the phylogeny and ecology of zoosporic fungi. Fungal Ecol. 19, 28–38. ( 10.1016/j.funeco.2015.06.004) [DOI] [Google Scholar]
- 69.Rosling A, Cox F, Cruz-Martinez K, Ihrmark K, Grelet G-A, Lindahl BD, Menkis A, James TY. 2011. Archaeorhizomycetes: unearthing an ancient class of ubiquitous soil fungi. Science 333, 876–879. ( 10.1126/science.1206958) [DOI] [PubMed] [Google Scholar]
- 70.Taylor JW, Fisher MC. 2003. Fungal multilocus sequence typing—it's not just for bacteria. Curr. Opin. Microbiol. 6, 351–356. ( 10.1016/S1369-5274(03)00088-2) [DOI] [PubMed] [Google Scholar]
- 71.Roe AD, Rice AV, Bromilow SE, Cooke JEK, Sperling FAH. 2010. Multilocus species identification and fungal DNA barcoding: insights from blue stain fungal symbionts of the mountain pine beetle. Mol. Ecol. Res. 10, 946–959. ( 10.1111/j.1755-0998.2010.02844.x) [DOI] [PubMed] [Google Scholar]
- 72.Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC. 2000. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 31, 21–32. ( 10.1006/fgbi.2000.1228) [DOI] [PubMed] [Google Scholar]
- 73.O'Donnell K, et al. 2004. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J. Clin. Microbiol. 42, 5109–5120. ( 10.1128/JCM.42.11.5109-5120.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith ME, Douhan GW, Rizzo DM. 2007. Intra-specific and intra-sporocarp ITS variation of ectomycorrhizal fungi as assessed by rDNA sequencing of sporocarps and pooled ectomycorrhizal roots from a Quercus woodland. Mycorrhiza 18, 15–22. ( 10.1007/s00572-007-0148-z) [DOI] [PubMed] [Google Scholar]
- 75.Bickford D, Lohman DJ, Sodhi NS, Ng PK, Meier R, Winker K, Ingram KK, Das I. 2007. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 22, 148–155. ( 10.1016/j.tree.2006.11.004) [DOI] [PubMed] [Google Scholar]
- 76.Lücking R, et al. 2014. A single macrolichen constitutes hundreds of unrecognized species. Proc. Natl Acad. Sci. USA 111, 11 091–11 096. ( 10.1073/pnas.1403517111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shenoy BD, Jeewon R, Hyde KD. 2007. Impact of DNA sequence-data on the taxonomy of anamorphic fungi. Fungal Divers. 26, 1–54. [Google Scholar]
- 78.Lumbsch HT, Leavitt SD. 2011. Goodbye morphology? A paradigm shift in the delimitation of species in lichenized fungi. Fungal Divers. 50, 59–72. ( 10.1007/s13225-011-0123-z) [DOI] [Google Scholar]
- 79.Nguyen HDT, Jančič S, Meijer M, Tanney JB, Zalar P, Gunde-Cimerman N, Seifert KA. 2015. Application of the phylogenetic species concept to Wallemia sebi from house dust and indoor air revealed by multi-locus genealogical concordance. PLoS ONE 10, e0120894 ( 10.1371/journal.pone.0120894) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dettman JR, Jacobson DJ, Taylor JW. 2003. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 57, 2703–2720. ( 10.1111/j.0014-3820.2003.tb01514.x) [DOI] [PubMed] [Google Scholar]
- 81.Geml J, Tulloss RE, Laursen GA, Sazanova NA, Taylor D. 2008. Evidence for strong inter-and intracontinental phylogeographic structure in Amanita muscaria, a wind-dispersed ectomycorrhizal basidiomycete. Mol. Phylogent. Evol. 48, 694–701. ( 10.1016/j.ympev.2008.04.029) [DOI] [PubMed] [Google Scholar]
- 82.Kinoshita A, Sasaki H, Nara K. 2011. Phylogeny and diversity of Japanese truffles (Tuber spp.) inferred from sequences of four nuclear loci. Mycologia 103, 779–794. ( 10.3852/10-138) [DOI] [PubMed] [Google Scholar]
- 83.Amo de Paz G, Cubas P, Crespo A, Elix JA, Lumbsch HT. 2012. Transoceanic dispersal and subsequent diversification on separate continents shaped diversity of the Xanthoparmelia pulla group (Ascomycota). PLoS ONE 7, e39683 ( 10.1371/journal.pone.0039683) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Molina MD, Crespo A, Blanco O, Lumbsch HT, Hawksworth DL. 2004. Phylogenetic relationships and species concepts in Parmelia s.str. (Parmeliaceae) inferred from nuclear ITS rDNA and beta-tubulin sequences. Lichenologist 36, 37–54. ( 10.1017/S0024282904013933) [DOI] [Google Scholar]
- 85.Leavitt SD, Esslinger TL, Divakar PK, Lumbsch HT. 2012. Miocene divergence, phenotypically cryptic lineages, and contrasting distribution patterns in common lichen-forming fungi (Ascomycota: Parmeliaceae). Biol. J. Linn. Soc. 107, 920–937. ( 10.1111/j.1095-8312.2012.01978.x) [DOI] [Google Scholar]
- 86.Buschbom J. 2007. Migration between continents: geographical structure and long-distance gene flow in Porpidia flavicunda (lichen-forming Ascomycota). Mol. Ecol. 16, 1835–1846. ( 10.1111/j.1365-294X.2007.03258.x) [DOI] [PubMed] [Google Scholar]
- 87.Geml J, Kauff F, Brochmann C, Taylor DL. 2010. Surviving climate changes: high genetic diversity and transoceanic gene flow in two arctic-alpine lichens, Flavocetraria cucullata and F. nivalis (Parmeliaceae, Ascomycota). J. Biogeogr. 37, 1529–1542. ( 10.1111/j.1365-2699.2010.02287.x) [DOI] [Google Scholar]
- 88.Geml J, Timling I, Robinson CH, Lennon N, Nusbaum HC, Brochmann C, Noordeloos ME, Taylor DL. 2012. An arctic community of symbiotic fungi assembled by long-distance dispersers: phylogenetic diversity of ectomycorrhizal basidiomycetes in Svalbard based on soil and sporocarp DNA. J. Biogeogr. 39, 74–88. ( 10.1111/j.1365-2699.2011.02588.x) [DOI] [Google Scholar]
- 89.Bergsten J, et al. 2012. The effect of geographical scale of sampling on DNA barcoding. Syst. Biol. 61, 851–869. ( 10.1093/sysbio/sys037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taylor JW, Turner E, Townsend JP, Dettman JR, Jacobson D. 2006. Eukaryotic microbes, species recognition and the geographic limits of species: examples from the kingdom Fungi. Phil. Trans. R. Soc. B 361, 1947–1963. ( 10.1098/rstb.2006.1923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arnold AE, Miadlikowska J, Higgins KL, Sarvate SD, Gugger P, Way A, Hofstetter V, Kauff F, Lutzoni F. 2009. A phylogenetic estimation of trophic transition networks for Ascomycetous fungi: are lichens cradles of symbiotrophic fungal diversification? Syst. Biol. 58, 283–297. ( 10.1093/sysbio/syp001) [DOI] [PubMed] [Google Scholar]
- 92.Tedersoo L, et al. 2014. Global diversity and geography of soil fungi. Science 346, 1078 ( 10.1126/science.1256688) [DOI] [PubMed] [Google Scholar]
- 93.Sánchez-Ramírez S, Etienne RS, Moncalvo JM. 2015. High speciation rate at temperate latitudes explains unusual diversity gradients in a clade of ectomycorrhizal fungi. Evolution 69, 2196–2209. ( 10.1111/evo.12722) [DOI] [PubMed] [Google Scholar]
- 94.Looney BP, Ryberg M, Hampe F, Sánchez-García M, Matheny PB. 2016. Into and out of the tropics: global diversification patterns in a hyperdiverse clade of ectomycorrhizal fungi. Mol. Ecol. 25, 630–647. ( 10.1111/mec.13506) [DOI] [PubMed] [Google Scholar]
- 95.Tedersoo L, Nara K. 2010. General latitudinal gradient of biodiversity is reversed in ectomycorrhizal fungi. New Phytol. 185, 351–354. ( 10.1111/j.1469-8137.2009.03134.x) [DOI] [PubMed] [Google Scholar]
- 96.Wubet T, Christ S, Schöning I, Boch S, Gawlich M, Schnabel B, Fischer M, Buscot F. 2012. Differences in soil fungal communities between European beech (Fagus sylvatica L.) dominated forests are related to soil and understory vegetation. PLoS ONE 7, e47500 ( 10.1371/journal.pone.0047500) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amend AS, Seifert KA, Samson R, Bruns TD. 2010. Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics. Proc. Natl Acad. Sci. USA 107, 13 748–13 753. ( 10.1073/pnas.1000454107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Timling I, Dahlberg A, Walker DA, Gardes M, Charcosset J-Y, Welker JM, Taylor DL. 2012. Distribution and drivers of ectomycorrhizal fungal communities across the North American Arctic. Ecosphere 3, 1–25. ( 10.1890/ES12-00217.1) [DOI] [Google Scholar]
- 99.Timling I, Walker DA, Nusbaum C, Lennon NJ, Taylor D. 2014. Rich and cold: diversity, distribution and drivers of fungal communities in patterned-ground ecosystems of the North American Arctic. Mol. Ecol. 23, 3258–3272. ( 10.1111/mec.12743) [DOI] [PubMed] [Google Scholar]
- 100.Talbot JM, et al. 2014. Endemism and functional convergence across the North American soil mycobiome. Proc. Natl Acad. Sci. USA 111, 6341–6346. ( 10.1073/pnas.1402584111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grantham NS, et al. 2015. Fungi identify the geographic origin of dust samples. PLoS ONE 10, e0122605 ( 10.1371/journal.pone.0122605) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ryberg M, Nilsson RH, Kristiansson E, Töpel M, Jacobsson S, Larsson E. 2008. Mining metadata from unidentified ITS sequences in GenBank: a case study in Inocybe (Basidiomycota). BMC Evol. Biol. 8, 50 ( 10.1186/1471-2148-8-50) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bonito GM, Gryganskyi AP, Trappe JM, Vilgalys R. 2010. A global meta-analysis of Tuber ITS rDNA sequences: species diversity, host associations and long-distance dispersal. Mol. Ecol. 19, 4994–5008. ( 10.1111/j.1365-294X.2010.04855.x) [DOI] [PubMed] [Google Scholar]
- 104.Gao C, et al. 2013. Host plant genus-level diversity is the best predictor of ectomycorrhizal fungal diversity in a Chinese subtropical forest. Mol. Ecol. 22, 3403–3414. ( 10.1111/mec.12297) [DOI] [PubMed] [Google Scholar]
- 105.Bidartondo MI, Bruns TD. 2001. Extreme specificity in epiparasitic Monotropoideae (Ericaceae): widespread phylogenetic and geographical structure. Mol. Ecol. 10, 2285–2295. ( 10.1046/j.1365-294X.2001.01358.x) [DOI] [PubMed] [Google Scholar]
- 106.Bidartondo MI, Bruns TD. 2002. Fine-level mycorrhizal specificity in the Monotropoideae (Ericaceae): specificity for fungal species groups. Mol. Ecol. 11, 557–569. ( 10.1046/j.0962-1083.2001.01443.x) [DOI] [PubMed] [Google Scholar]
- 107.Otálora MAG, Martínez I, O'Brien H, Molina MC, Aragón G, Lutzoni F. 2010. Multiple origins of high reciprocal symbiotic specificity at an intercontinental spatial scale among gelatinous lichens (Collemataceae, Lecanoromycetes). Mol. Phylogent. Evol. 56, 1089–1095. ( 10.1016/j.ympev.2010.05.013) [DOI] [PubMed] [Google Scholar]
- 108.O'Brien HE. 2013. PhotobiontDiversity.org: a searchable database of photobiont sequences. PhotobiontDiversity.org.
- 109.Yahr R, Vilgalys R, DePriest PT. 2006. Geographic variation in algal partners of Cladonia subtenuis (Cladoniaceae) highlights the dynamic nature of a lichen symbiosis. New Phytol. 171, 847–860. ( 10.1111/j.1469-8137.2006.01792.x) [DOI] [PubMed] [Google Scholar]
- 110.Muggia L, Pérez-Ortega S, Kopun T, Zellnig G, Grube M. 2014. Photobiont selectivity leads to ecological tolerance and evolutionary divergence in a polymorphic complex of lichenized fungi. Ann. Bot. 114, 463–475. ( 10.1093/aob/mcu146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leavitt SD, Kraichak E, Nelsen MP, Altermann S, Divakar PK, Alors D, Esslinger TL, Crespo A, Lumbsch T. 2015. Fungal specificity and selectivity for algae play a major role in determining lichen partnerships across diverse ecogeographic regions in the lichen-forming family Parmeliaceae (Ascomycota). Mol. Ecol. 24, 3779–3797. ( 10.1111/mec.13271) [DOI] [PubMed] [Google Scholar]
- 112.Kivlin SN, Hawkes CV, Treseder KK. 2011. Global diversity and distribution of arbuscular mycorrhizal fungi. Soil Biol. Biochem. 43, 2294–2303. ( 10.1016/j.soilbio.2011.07.012) [DOI] [Google Scholar]
- 113.Lonsdale D, Pautasso M, Holdenrieder O. 2008. Wood-decaying fungi in the forest: conservation needs and management options. Eur. J. Forest Res. 127, 1–22. ( 10.1007/s10342-007-0182-6) [DOI] [Google Scholar]
- 114.van der Linde S, Holden E, Parkin PI, Alexander IJ, Anderson IC. 2012. Now you see it, now you don't: the challenge of detecting, monitoring and conserving ectomycorrhizal fungi. Fungal Ecol. 5, 633–640. ( 10.1016/j.funeco.2012.04.002) [DOI] [Google Scholar]
- 115.Guidot A, Debaud J-C, Effosse A, Marmeisse R. 2004. Below-ground distribution and persistence of an ectomycorrhizal fungus. New Phytol. 161, 539–547. ( 10.1046/j.1469-8137.2003.00945.x) [DOI] [PubMed] [Google Scholar]
- 116.Ji Y, et al. 2013. Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol. Lett. 16, 1245–1257. ( 10.1111/ele.12162) [DOI] [PubMed] [Google Scholar]
- 117.Lawson Handley L. 2015. How will the ‘molecular revolution'contribute to biological recording? Biol. J. Linn. Soc. 115, 750–766. ( 10.1111/bij.12516) [DOI] [Google Scholar]
- 118.Lamarche J, et al. 2015. Molecular detection of 10 of the most unwanted alien forest pathogens in Canada using real-time PCR. PLoS ONE 10, e0134265 ( 10.1371/journal.pone.0134265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schaad NW, Frederick RD, Shaw J, Schneider WL, Hickson R, Petrillo MD, Luster DG. 2003. Advances in molecular-based diagnostics in meeting crop biosecurity and phytosanitary issues. Annu. Rev. Phytopathol. 41, 305–324. ( 10.1146/annurev.phyto.41.052002.095435) [DOI] [PubMed] [Google Scholar]
- 120.Queloz V, Grünig CR, Berndt R, Kowalski T, Sieber TN, Holdenrieder O. 2010. Cryptic speciation in Hymenoscyphus albidus. For. Pathol. 41, 133–142. ( 10.1111/j.1439-0329.2010.00645.x) [DOI] [Google Scholar]
- 121.Heuch J. 2014. What lessons need to be learnt from the outbreak of ash dieback disease, Chalara fraxinea in the United Kingdom? Arboricult. J. 36, 32–44. ( 10.1080/03071375.2014.913361) [DOI] [Google Scholar]
- 122.Korpelainen H, Pietiläinen M, Huotari T. 2015. Effective detection of indoor fungi by metabarcoding. Ann. Microbiol. 66, 1–4. ( 10.1007/s13213-015-1118-x) [DOI] [Google Scholar]
- 123.Pitkäranta M, Meklin T, Hyvärinen A, Paulin L, Auvinen P, Nevalainen A, Rintala H. 2008. Analysis of fungal flora in indoor dust by ribosomal DNA sequence analysis, quantitative PCR, and culture. Appl. Environ. Microbiol. 74, 233–244. ( 10.1128/aem.00692-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Findley K, et al. 2013. Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370. ( 10.1038/nature12171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv113 ( 10.1126/scitranslmed.3004404) [DOI] [PubMed] [Google Scholar]
- 126.Galimberti A, De Mattia F, Losa A, Bruni I, Federici S, Casiraghi M, Martellos S, Labra M. 2013. DNA barcoding as a new tool for food traceability. Food Res. Int. 50, 55–63. ( 10.1016/j.foodres.2012.09.036) [DOI] [Google Scholar]
- 127.López-González JA, Vargas-García MD, Lopez MJ, Suárez-Estrella F, Jurado MM, Moreno J. 2015. Biodiversity and succession of mycobiota associated to agricultural lignocellulosic waste-based composting. Bioresour. Technol. 187, 305–313. ( 10.1016/j.biortech.2015.03.124) [DOI] [PubMed] [Google Scholar]
- 128.Dentinger BT, Suz LM. 2014. What's for dinner? Undescribed species of porcini in a commercial packet. PeerJ 2, e570 ( 10.7717/peerj.570) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.O'Donnell K, Cigelnik E. 1997. Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol. Phylogent. Evol. 7, 103–116. ( 10.1006/mpev.1996.0376) [DOI] [PubMed] [Google Scholar]
- 130.Lindner DL, Banik MT. 2011. Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus. Mycologia 103, 731–740. ( 10.3852/10-331) [DOI] [PubMed] [Google Scholar]
- 131.Harder CB, Laessoe T, Froslev TG, Ekelund F, Rosendahl S, Kjøller R. 2013. A three-gene phylogeny of the Mycena pura complex reveals 11 phylogenetic species and shows ITS to be unreliable for species identification. Fungal Biol. 117, 764–775. ( 10.1016/j.funbio.2013.09.004) [DOI] [PubMed] [Google Scholar]
- 132.Vydryakova GA, Van DT, Shoukouhi P, Psurtseva NV, Bissett J. 2012. Intergenomic and intragenomic ITS sequence heterogeneity in Neonothopanus nambi (Agaricales) from Vietnam. Mycology 3, 89–99. [Google Scholar]
- 133.Hughes KW, Petersen RH, Lodge DJ, Bergemann SE, Baumgartner K, Tulloss RE, Lickey E, Cifuentes J. 2013. Evolutionary consequences of putative intra-and interspecific hybridization in agaric fungi. Mycologia 105, 1577–1594. ( 10.3852/13-041) [DOI] [PubMed] [Google Scholar]
- 134.Lindner DL, Carlsen T, Nilsson RH, Davey M, Schumacher T, Kauserud H. 2013. Employing 454 amplicon pyrosequencing to reveal intragenomic divergence in the internal transcribed spacer rDNA region in fungi. Ecol. Evol. 3, 1751–1764. ( 10.1002/ece3.586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li Y, Jiao L, Yao Y-J. 2013. Non-concerted ITS evolution in fungi, as revealed from the important medicinal fungus Ophiocordyceps sinensis. Mol. Phylogent. Evol. 68, 373–379. ( 10.1016/j.ympev.2013.04.010) [DOI] [PubMed] [Google Scholar]
- 136.Krüger M, Krüger C, Walker C, Stockinger H, Schüßler A. 2012. Phylogenetic reference data for systematics and phylotaxonomy of arbuscular mycorrhizal fungi from phylum to species level. New Phytol. 193, 970–984. ( 10.1111/j.1469-8137.2011.03962.x) [DOI] [PubMed] [Google Scholar]
- 137.Seifert KA. 2009. Progress towards DNA barcoding of fungi. Mol. Ecol. Res. 9, 83–89. ( 10.1111/j.1755-0998.2009.02635.x) [DOI] [PubMed] [Google Scholar]
- 138.Kauff F, Cox CJ, Lutzoni F. 2007. WASABI: an automated sequence processing system for multigene phylogenies. Syst. Biol. 56, 523–531. ( 10.1080/10635150701395340) [DOI] [PubMed] [Google Scholar]
- 139.O'Donnell K, et al. 2010. Internet-accessible DNA sequence database for identifying fusaria from human and animal infections. J. Clin. Microbiol. 48, 3708–3718. ( 10.1128/JCM.00989-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Debourgogne A, Gueidan C, de Hoog S, Lozniewski A, Machouart M. 2012. Comparison of two DNA sequence-based typing schemes for the Fusarium solani species complex and proposal of a new consensus method. J. Microbiol. Methods 91, 65–72. ( 10.1016/j.mimet.2012.07.012) [DOI] [PubMed] [Google Scholar]
- 141.Stielow JB, et al. 2015. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 35, 242–263. ( 10.3767/003158515X689135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Al-Hatmi AMS, Normand A-C, van Diepeningen AD, Hendrickx M, de Hoog GS, Piarroux R. 2015. Rapid identification of clinical members of Fusarium fujikuroi complex using MALDI-TOF MS. Fut. Microbiol. 10, 1939–1952. ( 10.2217/fmb.15.108) [DOI] [PubMed] [Google Scholar]
- 143.Al-Hatmi AMS, et al. 2016. Evaluation of two novel barcodes for species recognition of opportunistic pathogens in Fusarium. Fungal Biol. 120, 231–245. ( 10.1016/j.funbio.2015.08.006) [DOI] [PubMed] [Google Scholar]
- 144.Větrovský T, Kolařík M, Žifčáková L, Zelenka T, Baldrian P. 2015. The rpb2 gene represents a viable alternative molecular marker for the analysis of environmental fungal communities. Mol. Ecol. Resour. 16, 388–401. ( 10.1111/1755-0998.12456) [DOI] [PubMed] [Google Scholar]
- 145.Dentinger B, Gaya E, O'Brien H, Suz LM, Lachlan R, Díaz-Valderrama JR, Koch RA, Aime MC. 2015. Tales from the crypt: genome mining from fungarium specimens improves resolution of the mushroom tree of life. Biol. J. Linn. Soc. 117, 11–32. ( 10.1111/bij.12553) [DOI] [Google Scholar]
- 146.Liu Y, Leigh JW, Brinkmann H, Cushion MT, Rodriguez-Ezpeleta N, Philippe H, Lang BF. 2009. Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol. Biol. Evol. 26, 27–34. ( 10.1093/molbev/msn221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kurtzman CP. 2014. Use of gene sequence analyses and genome comparisons for yeast systematics. Int. J. Syst. Evol. Microbiol. 64, 325–332. ( 10.1099/ijs.0.054197-0) [DOI] [PubMed] [Google Scholar]
- 148.Grigoriev IV, et al. 2013. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699–D704. ( 10.1093/nar/gkt1183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Royal Botanic Gardens, Kew. 2015. A global resource for plant and fungal knowledge. Science strategy 2015–2020. Royal Botanic Gardens Kew; http://www.kew.org/sites/default/files/Kew%20Science%20Strategy%202015-2020%20Single%20pages.pdf. [Google Scholar]
- 150.Hibbett DS, Stajich JE, Spatafora JW. 2013. Toward genome-enabled mycology. Mycologia 105, 1339–1349. ( 10.3852/13-196) [DOI] [PubMed] [Google Scholar]
- 151.Collins RA, Cruickshank RH. 2014. Known knowns, known unknowns, unknown unknowns and unknown knowns in DNA barcoding: a comment on Dowton et al. Syst. Biol. 63, 1005–1009. ( 10.1093/sysbio/syu060) [DOI] [PubMed] [Google Scholar]
- 152.Fitzpatrick DA, Logue ME, Stajich JE, Butler G. 2006. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol. Biol. 6, 99 ( 10.1186/1471-2148-6-99) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ohm RA, et al. 2012. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 8, e1003037 ( 10.1371/journal.ppat.1003037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, Pati A. 2015. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771. ( 10.1093/nar/gkv657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Federhen S, et al. 2016. meeting report: GenBank microbial genomic taxonomy workshop (12–13 May, 2015). Stand. Genomic Sci. 11, 15 ( 10.1186/s40793-016-0134-1) [DOI] [Google Scholar]
- 156.Hittinger CT, et al. 2015. Genomics and the making of yeast biodiversity. Curr. Opin. Genet. Dev. 35, 100–109. ( 10.1016/j.gde.2015.10.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bruns TD, Arnold AE, Hughes KW. 2008. Fungal networks made of humans: UNITE, FESIN, and frontiers in fungal ecology. New Phytol. 177, 586–588. ( 10.1111/j.1469-8137.2008.02341.x) [DOI] [PubMed] [Google Scholar]
- 158.Blackwell M. 2011. The Fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 98, 426–438. ( 10.3732/ajb.1000298) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.