

Abstract
Complex or quantitative traits are important in medicine, agriculture and evolution, yet, until recently, few of the polymorphisms that cause variation in these traits were known. Genome-wide association studies (GWAS), based on the ability to assay thousands of single nucleotide polymorphisms (SNPs), have revolutionized our understanding of the genetics of complex traits. We advocate the analysis of GWAS data by a statistical method that fits all SNP effects simultaneously, assuming that these effects are drawn from a prior distribution. We illustrate how this method can be used to predict future phenotypes, to map and identify the causal mutations, and to study the genetic architecture of complex traits. The genetic architecture of complex traits is even more complex than previously thought: in almost every trait studied there are thousands of polymorphisms that explain genetic variation. Methods of predicting future phenotypes, collectively known as genomic selection or genomic prediction, have been widely adopted in livestock and crop breeding, leading to increased rates of genetic improvement.
Keywords: genomic selection, complex traits, genome-wide association studies
1. Introduction
Complex or quantitative traits are important in medicine (e.g. diabetes), agriculture (e.g. yield of rice) and evolution (e.g. body size). These traits are called complex because they are controlled by many genes and by environmental factors. Although the genetics of quantitative traits has been studied for over 100 years, very few of the polymorphisms that cause variation in these traits were known until recently. The development of assays that could determine the genotype of an individual at thousands of single nucleotide polymorphisms (SNPs) has revolutionized the study of complex traits. The SNPs assayed might be neutral polymorphisms with no effect on the traits studied, but linkage disequilibrium (LD) between the SNPs and the causal polymorphisms generates an association between the traits and some of the SNPs. Genome-wide association studies (GWAS), which assay a genome-wide panel of SNPs, have discovered thousands of associations between SNPs and complex traits [1]. These GWAS are intended to map the causal polymorphisms to a region of the genome but do not identify them.
The data from GWAS can be used for three purposes. First, they can be used to predict future phenotypes. In human medicine, this might be the probability that a person will develop type 2 diabetes in the future. In agriculture, it is usually the phenotype of the offspring of animals or plants that we wish to predict so that those with the best breeding value can be selected as parents of the next generation. Although the SNPs used may have no causal relationship with the trait, they may still be useful for prediction due to their LD with causal variants. Second, GWAS data are used to map the causal variants to a region of the genome and hopefully to identify them. This increases our understanding of the biology of complex traits and may suggest methods of controlling them such as new drug targets. Third, GWAS data provide an overview of the genetic architecture of complex traits that is useful in medicine, agriculture and evolution. We would like to know how many polymorphisms control a trait, what are their effects and allele frequencies, the LD between them, and how they evolve.
Usually different methods of analysis of GWAS data have been used for each of these three purposes. For instance, mapping causal polymorphisms is usually done by fitting one SNP at a time in a regression model [2]. Conversely, predicting genetic value is most often done by assuming all SNPs have an effect drawn from the same normal distribution [3]. In this paper, we will argue that a new model, where SNPs are assumed to have an effect drawn from a mixture of normal distributions with increasing variances, can be used for genomic prediction, mapping of causal variants and inference on the genetic architecture of complex traits. The rest of the paper begins by introducing the statistical model and its use for prediction of phenotype. This is followed by describing the use of the same model for mapping of causal variants and understanding the genetic architecture of complex traits. We then discuss the limitations of the method, and the implications for our understanding of complex traits and for future research on prediction of phenotype and mapping of causal variants.
2. Prediction of genetic value
In many datasets, the number of SNPs (p) is greater than the number of individuals with records (n). Consequently, if the effects of the SNPs on the trait are treated as fixed effects in a multiple regression analysis, there is no unique solution. However, the total variance explained by all SNPs (a result of their effect sizes and allele frequencies) must be less than the total genetic variance, and this places a restriction on the effect sizes. More accurate predictions can be obtained by treating the effect sizes as random variables drawn from a distribution which is consistent with the total genetic variance [4]. In general, the best prediction of a random variable (g) from a set of predictors (x) is E(g|x), that is, the expected or average value of g conditional on the values observed for x [5]. (Here best means minimum mean-squared errors.) We will restrict discussion to a linear predictor of the form b′x, where b is a vector of regression coefficients. Then the best prediction rule implies that we estimate b by E(b|‘data’), where data might be the genotypes (x) and phenotypes (y) of individuals from a GWAS. In this formulation of the problem, the elements of b (bi) are the apparent effect of the SNP on the trait and are treated as random effects drawn from a distribution (p(b)). As there are typically thousands of SNPs, it is possible to imagine the distribution of SNP effects on a trait as simply the distribution of these thousands of effects: many may have no effect at all and some may have a large effect.
Therefore, 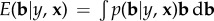 , which can be re-expressed using Bayes theorem as
, which can be re-expressed using Bayes theorem as 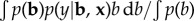
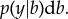 Here, p(y|b, x) is the likelihood of the phenotypes (y) given the genotypes of the individual (x) and the effects of the SNPs (b). This is the method invented by Meuwissen et al. [4] and called genomic selection or genomic prediction.
Here, p(y|b, x) is the likelihood of the phenotypes (y) given the genotypes of the individual (x) and the effects of the SNPs (b). This is the method invented by Meuwissen et al. [4] and called genomic selection or genomic prediction.
The statistical analysis resulting from applying this best prediction rule depends on the prior distribution chosen for b. Meuwissen and colleagues considered three possible prior distributions [4]. In one, b is assumed to be normally and independently distributed with a mean of 0 and a variance (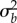 ) that is same for all SNPs: this method is an example of best linear unbiased prediction (BLUP). In the other two, Meuwissen et al. used a t distribution, and a mixture of zero and a t distribution. The method called ‘Bayes R’ by Erbe et al. [6] is a further development, which uses a prior distribution for b which is a mixture of four normal distributions each with zero mean but with variances of
) that is same for all SNPs: this method is an example of best linear unbiased prediction (BLUP). In the other two, Meuwissen et al. used a t distribution, and a mixture of zero and a t distribution. The method called ‘Bayes R’ by Erbe et al. [6] is a further development, which uses a prior distribution for b which is a mixture of four normal distributions each with zero mean but with variances of 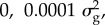
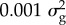 and
and 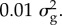 The mixing proportions are estimated from the data, so this is a flexible prior that can approximate many possible distributions for b (the SNP effects). In our applications of this model, we have assumed that the mixing proportions are drawn from a Dirichlet distribution with parameters (1, 1, 1, 1). This is a deliberately vague prior so that it has little impact on the final estimates of the mixing proportions which are driven mainly by the data.
The mixing proportions are estimated from the data, so this is a flexible prior that can approximate many possible distributions for b (the SNP effects). In our applications of this model, we have assumed that the mixing proportions are drawn from a Dirichlet distribution with parameters (1, 1, 1, 1). This is a deliberately vague prior so that it has little impact on the final estimates of the mixing proportions which are driven mainly by the data.
While BLUP is a linear method in that b is estimated by a linear combination of the phenotypic data y, the other methods are nonlinear in y. In this paper, we advocate the use of these nonlinear models with particular reference to Bayes R.
The BLUP prior corresponds to a ‘pseudo-infinitesimal’ model in which all polymorphic sites in the genome have an effect on every trait, and all effects are of similar magnitude and very small. For instance, if there are 1 million SNPs, each one is assumed to explain approximately 10−6 of the genetic variance (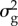 ). As a consequence of this assumption, all estimated SNP effects are shrunk severely towards 0 when BLUP is used. The other models allow the distribution of SNP effects to depart from this pseudo-infinitesimal distribution, with some SNPs having zero effect and some SNPs having a large effect on the trait.
). As a consequence of this assumption, all estimated SNP effects are shrunk severely towards 0 when BLUP is used. The other models allow the distribution of SNP effects to depart from this pseudo-infinitesimal distribution, with some SNPs having zero effect and some SNPs having a large effect on the trait.
Genomic prediction requires a reference or training population in which the individuals have both phenotypes and genotypes. Analysis of these data generates a prediction equation which can then be used to predict genetic value in individuals with genotypes but without phenotypes. In accordance with convention in genetic evaluation, if not in statistics, we will define the ‘accuracy’ of the prediction as the correlation between the predicted genetic value and the true genetic value among these individuals. The factors determining the accuracy when BLUP is used have been considered in theory by Daetwyler et al. [7] and Goddard [8]. The accuracy of predicting genetic values depends on the proportion of the genetic variance explained by the markers (referred to below as SNPs) and the accuracy with which the effect of those SNPs is estimated. Both components of accuracy depend on the LD within the genome. Low LD increases the number of individuals with records and the number of SNPs needed to achieve a given accuracy. Consequently, accuracy is typically lower in humans than within a breed of cattle, where long-distance LD exists due to small recent effective population size. In cattle, the proportion of genetic variance explained by SNPs is in the range 0.5–0.9, and in humans it is 0.3–0.5 for many traits [9–12].
In practice, the accuracy of genomic prediction using the nonlinear methods is equal to or higher than the accuracy of BLUP [12–14]. For example Kemper et al. [14] found a 5% increase in accuracy of genomic prediction for milk yield traits in dairy cattle using Bayes R compared with BLUP, and Moser et al. [12] found an increase in accuracy of genomic predictions of Bayes R over BLUP for Crohn's disease, rheumatoid arthritis, and type 1 diabetes, but not for bipolar disorder, coronary artery disease, hypertension or type 2 diabetes. However, the small advantage of nonlinear models over BLUP points to the high number of causal variants affecting most traits.
Genomic selection is now widely used in livestock (especially dairy cattle) and crops. It should double the rate of genetic improvement in dairy cattle [15].
3. Mapping and identification of causal polymorphisms
To map genes for a quantitative trait (QTL, or quantitative trait loci) to a position on the genome using GWAS data, the most common analysis is to fit one SNP at a time in a regression model [1]. While this is straightforward and computationally undemanding, there are several disadvantages to this approach. Many SNPs are likely to be in LD with a single QTL generating many SNPs associated with the trait. Common practice is to focus on the most highly associated SNP within a genomic region (e.g. 2 Mb). However, in livestock and many crops LD extends for a long distance, and so SNPs more than 2 Mb from the QTL may still show a significant association with the trait. A further complication is that there are typically so many QTL for each complex trait that an SNP may be in LD with more than one QTL. Therefore, it is difficult to tell how many QTL are indicated by the GWAS results.
A solution to this problem would be to fit all SNPs simultaneously. In this way, only the SNPs that are necessary to track each QTL should be included in the final model. Usually, there are more SNPs than subjects so there is no unique solution if the SNP effects are treated as fixed effects. A widely used alternative is to fit all SNP effects as random effects using the BLUP model discussed above and then to fit one SNP as a fixed effect [16–19]. However, this does not eliminate the problem and seems illogical—why fit one as fixed and all the rest as random effects? A better solution is to fit all SNP simultaneously as random effects, which is the same model as used for genomic prediction. However, the BLUP model estimates small effects for all SNPs and so does a poor job of mapping QTL. By contrast, the Bayes R model, in which many SNPs have no effect, gives a large effect to SNPs that best track the QTL.
Figure 1 compares partial results of a genome-wide analysis of fat percentage in bovine milk in a region around the FASN (fatty acid synthase) gene (data described in [20]), using either Bayes R or the common GWAS method (SNP fitted singly as a fixed effect using the linear mixed model). In the Bayes R model (figure 1a), a single SNP just upstream of the FASN gene has the highest posterior probability, while in the GWAS model (figure 1b) there are several SNP extending across to the CCDC57 (coiled-coil domain containing 57) gene region with almost equally significant effects. FASN has previously been suggested as a candidate gene affecting fat percentage of milk because FASN is a key enzyme in de novo fatty acid biosynthesis (e.g. [21,22]). In an RNAseq study, FASN was also found to be more highly expressed in lactating bovine mammary tissue than in 17 other bovine tissues [23]. Although there may be a second QTL effect due to a variant of the CCDC57 gene, there is less evidence of this from the Bayes R analysis which fits all SNPs simultaneously, while the evidence from the GWAS single SNP regression predicts almost equal significance with the variants close to FASN.
Figure 1.

Genome-wide analysis of bovine milk fat percentage showing results for a region around the FASN gene. (a) Posterior probability that an SNP has a non-zero effect from Bayes R where all SNPs are fitted in the model simultaneously. (b) −log10 p-value from GWAS single SNP regressions. The top Bayes R variant is annotated (with base pair position) and shown as a purple diamond, and the strength of LD (r2) between this and all other variants is colour coded. (Adapted from data published in [20].)
If there are multiple SNPs in high LD, the analysis may be unable to say which is the best one to include in the model. Then all these SNPs may receive a low posterior probability of a non-zero effect. However, when the results are combined across the SNPs, it is clear that a QTL resides in this region. One way to do this is to predict the genetic value of each individual based only on the SNPs in the region. If this local estimated genetic value has a high variance, it indicates a QTL in this region. An example is given in figure 2. It can be seen that Bayes R gives a sharper position for the QTL than BLUP.
Figure 2.

Local GEBV variance near the PAEP gene for FY, MY and PY using Bayes R and BLUP. Shown is the GEBV variance in overlapping 250 kb windows for Holstein and Jersey reference animals from SNP effects estimated from the multi-breed reference population. Traits: FY, fat yield; MY, milk yield; PY, protein yield. The position of PAEP on BTA11 is marked (*). Note the changed y-axis scale for each graph. Adapted from Kemper et al. [14]. (Online version in colour.)
By applying this Bayes R analysis to genome sequence data, rather than data from a panel of SNPs, we would hope to identify the causal polymorphisms directly. Unfortunately, if there are several variants in high LD, the analysis cannot tell which of them is causal. Also there is sampling error, such that the most significant variant or the one with the highest posterior probability may not be the causative mutation, particularly if the reference population is small. It is helpful in this situation to have independent information on the likelihood that a mutation in each site would affect the phenotype. Typically, in the analysis of GWAS, this information is used after the statistical analysis and in a subjective manner. We have modified Bayes R to include a priori information about the polymorphic sites (Bayes RC [20]). The sites are placed in categories and the mixing proportions for each category are estimated in the analysis. For instance, non-synonymous coding sites may be placed in one category and all other sites in another category. This provides objective evidence for the probability that coding sites are more likely to alter phenotype than non-coding sites. The Bayes RC approach can improve both the precision of QTL mapping and accuracy of genomic prediction provided there is good prior biological information available [20].
Figure 3 compares the results from Bayes RC, Bayes R and GWAS analyses of bovine milk protein percentage (data described in [20]) in the region of the LALBA gene. LALBA is a candidate gene because it codes for alpha-lactalbumin, a key regulator of lactose synthesis [24]. In this Bayes RC analysis (figure 3a), prior knowledge of candidate genes for milk production and variant annotation was used to allocate variants to categories (details in [20]). The genotype data included approximately 1 million genome-wide sequence coding variants and SNP from a high-density array. When the same data were analysed using Bayes R there were several suggestive QTL (figure 3b), but each had a much lower posterior probability than in the Bayes RC analysis, due to high LD between these sites. A GWAS analysis of the same data using single SNP regressions shows highly significant SNP across a wide region (figure 3c). Prior to analysis, variants in perfect LD or those far from genes were pruned out, so further analysis is required to determine the true causal variant.
Figure 3.

Comparison of results for three genome-wide analysis methods of bovine milk protein percentage in a region around the LALBA gene. (a) Bayes RC incorporates prior biological information on sites that are more likely to have an effect on the trait, while (b) Bayes R and (c) GWAS assume all sites are equally likely to affect the trait. The top Bayes RC variant is shown in purple (with base pair position), and the strength of LD (r2) between this and all other variants is colour coded (adapted from data published in [20]).
A polymorphism that affects one trait is also likely to affect other traits. Thus, a multi-trait analysis may increase power to find the causal sites. Additional traits are especially useful if the causal polymorphism has a large effect on the trait because this increases statistical power. Gene expression is a trait with large effects, especially mutations acting in cis to change the expression of the allele on the same chromosome as the mutation. Another benefit of cis eQTL is that they define the gene through whose expression the QTL has its effect, thus increasing our understanding of the pathway from gene to phenotype. Other genetically simpler traits with large effects might include individual proteins and product components. An example follows. Kemper et al. [25] found that a QTL that explained 0.001 of the genetic variance for milk yield co-segregated with a QTL that explained 0.1 of the genetic variance for phosphorus concentration in milk. The SNPs with a large effect on both traits mapped near a gene for a phosphorus anti-porter that transports glucose-6-phosphate in one direction and phosphorus in the other direction across cell membranes. Glucose is a substrate for lactose synthesis, which is the major osmolarity regulator in milk and hence drives milk volume. Thus, the allele that increases milk phosphorus concentration decreases milk volume. Gene expression data showed that the same sequence variant that increases phosphorus content in milk also increases expression of this gene.
4. Genetic architecture of complex traits
Traditional SNP regression analysis of GWAS reports the estimated effect of SNPs that are declared significant. However, this information does not give a good description of the genetic architecture of the trait. In SNP regression analysis, very stringent p-values (p < 5 × 10−8) are used to protect against testing as many as 1 000 000 SNPs for an effect. This has several consequences. The estimated effect of SNPs declared significant is grossly overestimated. A better estimate of the effect size can be obtained by estimating the effect of significant SNPs in an independent dataset. When this is done it is found that the significant SNPs explain a small proportion of the genetic variance estimated from pedigree or family relationships. This was called the ‘missing heritability’ paradox. Yang et al. [11] showed that if the combined effect of all SNPs was estimated, the proportion of genetic variance explained was much higher. For instance, the genetic variance for height in humans explained by significant SNPs was 5% of phenotypic variance, whereas all the SNPs together explained 45% of the phenotypic variance [11]. The explanation for this difference is simply that most SNP effects on height are too small to be significant given the stringent p-value used.
Even 45% is less than the 70% or more of the phenotypic variance estimated to be additive genetic variance by family studies, possibly because causal variants with low minor allele frequency (MAF) are not in high LD with any of the SNPs used. Yang et al. [26] found that imputed genome sequence explained 50% of the phenotypic variance for height. Sequence variants with MAF < 0.1 explained more variance than other MAF classes despite the fact that they were not accurately imputed. Accounting for this poor imputation of rare variants suggests that 60% of phenotypic variance is explained by genome sequence. It is also possible that the family studies overestimate the heritability due to confounding genetic effects with common family environment, or confounding additive and non-additive genetic variation. Thus, the genetic variance explained by sequence variants is almost equal to that estimated from family studies.
Bayes R provides an estimate of the number of causal variants affecting a trait and the distribution of their effects by approximating the distribution of effect sizes with a mixture of normal distributions. (We do not imply the SNP effects are literally drawn from a mixture of normal distributions, merely that this mixture can approximate almost any distribution that might describe the distribution of effect sizes.) For many traits in both humans and cattle, we find that there are thousands of SNPs with effect sizes drawn from a distribution with variance of 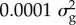 and a handful with variance
and a handful with variance 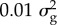 [12–14]. Thus, complex traits are more complex than was thought, with thousands of polymorphisms, each with very small effects, affecting each trait. Their allele frequencies are biased towards low MAF compared with a neutral model, but only slightly. That is, most of the variance is due to common variants [26].
[12–14]. Thus, complex traits are more complex than was thought, with thousands of polymorphisms, each with very small effects, affecting each trait. Their allele frequencies are biased towards low MAF compared with a neutral model, but only slightly. That is, most of the variance is due to common variants [26].
By allocating polymorphic sites into categories in our Bayes RC analysis, we can estimate the distribution of effect sizes for each category. Table 1 shows the results for milk yield in dairy cattle where SNPs were allocated to one of three categories (data described in [20]). Non-synonymous coding sites in a set of candidate genes affecting milk production had a higher proportion of non-zero effects than non-coding sites outside candidate gene regions. However, because non-coding sites are more numerous they explained most of the variance (table 1).
Table 1.
Proportion of SNP effects discovered in biologically defined SNP categories compared with the total variance explained, using a Bayes RC analysis of dairy cattle milk production. Prior to the analysis, SNPs were divided into categories based on prior knowledge of candidate genes and annotation of non-synonymous coding SNP. (Adapted from data published in [20].)
| SNP category | no. SNP per category (% of total) | proportion SNP effects per distributiona |
total variance explained per SNP category (%) | |||
|---|---|---|---|---|---|---|
| zero variance (%) |
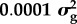 (%) (%) |
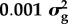 (%) (%) |
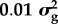 (%) (%) |
|||
| non-synonymous coding SNP in candidate genesb | 3768 (0.4%) | 95.6 | 3.9 | 0.38 | 0.10 | 8.6 |
| other SNP in or within 50 Kb of candidate genes | 57 722 (6%) | 99.0 | 1.0 | 0.03 | 0.008 | 15.7 |
| all other SNP | 847 905 (93%) | 99.6 | 0.4 | 0.01 | 0.0004 | 75.7 |
aBayes RC estimates SNP effects as a mixture of four normal distributions: one with zero mean and variance, and the others as N(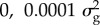 ), N(
), N(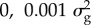 ), N(
), N(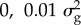 ), where
), where 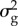 is the additive genetic variance for the trait.
is the additive genetic variance for the trait.
bThe candidate genes were a group of 790 genes selected based on differential expression in mammary gland, in experiments designed to manipulate milk production.
5. Discussion
Although the nonlinear models that we have advocated (such as Bayes R) give good predictions, they estimate or predict more variables (p) than there are subjects (n) and so there is justified concern that many other models could fit the data equally well. In Bayes R, we fix the variance for each component of the mixture to minimize the number of parameters to be estimated. There is no special reason for our choice of four components or variances of 0, 0.0001, 0.001 and 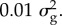 However, we have found that a mixture of these four components can approximate a wide range of distributions. A limitation might be the absence of a normal with even smaller variance in the mixture. Nevertheless, Moser et al. [12] found that the four-component mixture could still perform well when the data were simulated under a different model. The mixing proportions themselves are assumed to be drawn from a Dirichlet distribution with the prior being equivalent to one SNP in each component of the mixture. The estimated mixing proportions are very far from the prior because the number of SNPs in the distribution with zero or small variance is much larger than the number in the component of the mixture with larger variance. As the mixture requires only four parameters, it is not surprising that the data have some power to estimate these parameters. Nevertheless, we caution against too literal an interpretation of the estimates. This caution is partially due to inherent difficulties in estimating so many variables and partially due to the use of Markov chain Monte Carlo (MCMC) methods. It is difficult to know when an MCMC chain has converged. We typically run five independent chains and compare the results from the five chains. There are also differences between species. In humans, there is less long-distance LD than in cattle, and consequently traditional one-SNP-at-a-time regression leads to clearer mapping of QTL than in cattle. In addition, like most analyses, we have restricted ourselves to additive genetic models without dominance or epistasis. Despite these caveats, some generalizations emerge from the research that has covered multiple species and traits.
However, we have found that a mixture of these four components can approximate a wide range of distributions. A limitation might be the absence of a normal with even smaller variance in the mixture. Nevertheless, Moser et al. [12] found that the four-component mixture could still perform well when the data were simulated under a different model. The mixing proportions themselves are assumed to be drawn from a Dirichlet distribution with the prior being equivalent to one SNP in each component of the mixture. The estimated mixing proportions are very far from the prior because the number of SNPs in the distribution with zero or small variance is much larger than the number in the component of the mixture with larger variance. As the mixture requires only four parameters, it is not surprising that the data have some power to estimate these parameters. Nevertheless, we caution against too literal an interpretation of the estimates. This caution is partially due to inherent difficulties in estimating so many variables and partially due to the use of Markov chain Monte Carlo (MCMC) methods. It is difficult to know when an MCMC chain has converged. We typically run five independent chains and compare the results from the five chains. There are also differences between species. In humans, there is less long-distance LD than in cattle, and consequently traditional one-SNP-at-a-time regression leads to clearer mapping of QTL than in cattle. In addition, like most analyses, we have restricted ourselves to additive genetic models without dominance or epistasis. Despite these caveats, some generalizations emerge from the research that has covered multiple species and traits.
The availability of dense SNP panels and genome sequence on large numbers of individuals, who have also been recorded for a complex trait, has changed our understanding of the genetics of complex traits and led to great practical benefits in the genetic improvement of livestock and crops. It now appears that thousands of polymorphisms affect a typical complex trait. The effect of these QTL varies from large to very small, but most of the variance is due to QTL that individually explain a small proportion of the variance (e.g. less than 1%). Although a few mutations of large effect are likely to be at low MAF, most of the variance is due to QTL that have only slightly lower MAF on average than neutral SNPs.
This new understanding of the genetic architecture of complex traits has implications for prediction and for identification of causal polymorphisms. The results emphasize the need for large sample sizes for both purposes. Even with access to full genome sequence, most polymorphisms explain only a tiny fraction of the variance, and therefore large sample sizes are needed to reach the conventional significance level (p < 5 × 10−8).
The strategy for prediction depends on the Ne of the population. For populations with small recent Ne (livestock, some crops), a prediction equation derived by BLUP based on a moderately dense SNP panel works well. However, even here this approach has disadvantages. The prediction equation is generally not robust to minor changes in the population such as a change in breed, place or time. For instance, a prediction equation trained on one breed of livestock has little accuracy in another, closely related breed [6]. An alternative strategy is to use a nonlinear method and a multi-breed training population (or more genetically diverse population) in the hope that this will lead to more robust predictions.
For populations with large recent Ne, BLUP prediction of genetic value requires enormous training population size. As a population of sufficient size is often not available, some attempt is usually made to reduce the amount of the genome that is considered (e.g. by using coding variants only). However, a better alternative would be to aim for good coverage of the genome and to use nonlinear prediction methodology and the strategies discussed above to maximize the accuracy.
If the SNP panel does not explain all of the genetic variance, there is a limit to the accuracy that can be achieved. To overcome this limit, genome sequence data, which should contain the causal variants, can be used. However, to extract extra accuracy from genome sequence data requires a nonlinear statistical method [27,28].
To estimate the effect of all sequence variants simultaneously is a challenging task. We have illustrated above two strategies that can be used to help with this task. First, a QTL with a small effect on one trait may have a larger effect on another trait. Therefore, a multi-trait analysis increases power to find sequence variants which have an effect on any trait. Gene expression is a particularly useful type of trait because the effects of cis eQTL tend to be large and the result immediately indicates the gene through which the polymorphism acts.
Second, external information on sites in the genome that, if mutated, would have an effect on phenotype, such as the ENCODE data [29], are useful in deciding which sites are most likely to be causal. Variants that change the amino acid sequence of proteins are more likely to affect phenotype than random sites in the genome, and this is used in the Bayes RC method described above. However, evidence is mounting that the majority of mutations that give rise to variation in complex traits reside in regulatory elements that alter gene expression [30–32] (reviewed by Pai et al. [33]).
Cis-acting elements affect gene expression only on the same DNA molecule, thus acting in an allele-specific manner. Detecting allele-specific expression (ASE) is an alternative method of finding cis eQTL [34]. ASE occurs at heterozygous variants where one allele is more highly expressed in the mRNA than the other. Recently, Crowley et al. [35] and Chamberlain et al. [23] reported that 89% of mice and cattle genes, respectively, show ASE in at least one tissue. Therefore, it seems likely that cis eQTL are very common.
Sites in the genome that would affect gene expression if mutated can also be identified by genomic features such as histone marks and transcription factor binding sites, which are indicative of enhancers [36], transcription start sites [36] or promoters [37], any of which could be involved in gene regulation, and which therefore might affect phenotype if mutated. A consortium called Functional Annotation of ANimal Genomes (FAANG [38]) is now planning to annotate livestock genomes for such histone marks as well as other markers of open chromatin in a similar fashion to that in humans (the ENCODE consortium).
Authors' contributions
All authors contributed to the writing of this review and to the original research, which has been reported elsewhere.
Competing interests
We declare we have no competing interests.
Funding
This work was supported in part by Australian Research Council grant DP130100563.
References
- 1.Wood AR, et al. 2014. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186. ( 10.1038/ng.3097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. 2014. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 46, 100–106. ( 10.1038/ng.2876) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.VanRaden P, Van Tassell C, Wiggans G, Sonstegard T, Schnabel R, Taylor J, Schenkel F. 2009. Invited review: reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 92, 16–24. ( 10.3168/jds.2008-1514) [DOI] [PubMed] [Google Scholar]
- 4.Meuwissen T, Hayes B, Goddard M. 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goddard ME, Wray NR, Verbyla K, Visscher PM. 2009. Estimating effects and making predictions from genome-wide marker data. Stat. Sci. 24, 517–529. ( 10.1214/09-STS306) [DOI] [Google Scholar]
- 6.Erbe M, Hayes BJ, Matukumalli LK, Goswami S, Bowman PJ, Reich CM, Mason BA, Goddard ME. 2012. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 95, 4114–4129. ( 10.3168/jds.2011-5019) [DOI] [PubMed] [Google Scholar]
- 7.Daetwyler HD, Villanueva B, Woolliams JA. 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 3, e3395 ( 10.1371/journal.pone.0003395) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goddard M. 2009. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136, 245–257. ( 10.1007/s10709-008-9308-0) [DOI] [PubMed] [Google Scholar]
- 9.Haile-Mariam M, Nieuwhof GJ, Beard KT, Konstatinov KV, Hayes BJ. 2013. Comparison of heritabilities of dairy traits in Australian Holstein-Friesian cattle from genomic and pedigree data and implications for genomic evaluations. J. Anim. Breed. Genet. 130, 20–31. ( 10.1111/j.1439-0388.2013.01001.x) [DOI] [PubMed] [Google Scholar]
- 10.Román-Ponce S-I, Samoré AB, Dolezal MA, Bagnato A, Meuwissen TH. 2014. Estimates of missing heritability for complex traits in Brown Swiss cattle. Genet. Select. Evol. 46, 36 ( 10.1186/1297-9686-46-36) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, et al. 2010. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569. ( 10.1038/ng.608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moser G, Lee SH, Hayes BJ, Goddard ME, Wray NR, Visscher PM. 2015. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. PLoS Genet. 11, e1004969 ( 10.1371/journal.pgen.1004969) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolormaa S, et al. 2013. Accuracy of prediction of genomic breeding values for residual feed intake and carcass and meat quality traits in Bos taurus, Bos indicus, and composite beef cattle. J. Anim. Sci. 91, 3088–3104. ( 10.2527/jas.2012-5827) [DOI] [PubMed] [Google Scholar]
- 14.Kemper KE, Reich CM, Bowman P, vander Jagt CJ, Chamberlain AJ, Mason BA, Hayes BJ, Goddard ME. 2015. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet. Select. Evol. 47, 29 ( 10.1186/s12711-014-0074-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaeffer LR. 2006. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 123, 218–223. ( 10.1111/j.1439-0388.2006.00595.x) [DOI] [PubMed] [Google Scholar]
- 16.Kennedy BW, Quinton M, van Arendonk JA. 1992. Estimation of effects of single genes on quantitative traits. J. Anim. Sci. 70, 2000–2012. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, et al. 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. ( 10.1038/ng1702) [DOI] [PubMed] [Google Scholar]
- 18.Kang HM, Sul JH, Service SK, Zaitlen NA, Kong S-Y, Freimer NB, Sabatti C, Eskin E. 2010. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. ( 10.1038/ng.548) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X, Stephens M. 2012. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824. ( 10.1038/ng.2310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacLeod IM, Bowman PJ, Vander Jagt CJ, Haile-Mariam M, Kemper KE, Chamberlain AJ, Schrooten C, Hayes BJ, Goddard ME. 2016. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics 17, 144 ( 10.1186/s12864-016-2443-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris C, et al. 2007. Fatty acid synthase effects on bovine adipose fat and milk fat. Mamm. Genome 18, 64–74. ( 10.1007/s00335-006-0102-y) [DOI] [PubMed] [Google Scholar]
- 22.Bionaz M, Loor J. 2008. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genomics 9, 366 ( 10.1186/1471-2164-9-366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chamberlain AJ, Vander Jagt CJ, Hayes BJ, Khansefid M, Marett LC, Millen CA, Nguyen TTT, Goddard ME. 2015. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genomics 16, 1–20. ( 10.1186/s12864-015-2174-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heine WE, Klein PD, Reeds PJ. 1991. The importance of α-lactalbumin in infant nutrition. J. Nutr. 121, 277–283. [DOI] [PubMed] [Google Scholar]
- 25.Kemper KE, Littlejohn MD, Lopdell T, Hayes BJ, Visscher PM, Carrick MJ, Goddard ME. Submitted Leveraging genetically simple traits to identify small-effect variants for complex phenotypes. BMC Genomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, et al. 2015. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120. ( 10.1038/ng.3390) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meuwissen T, Goddard M. 2010. Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genetics 185, 623–631. ( 10.1534/genetics.110.116590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacLeod IM, Hayes BJ, Goddard ME. 2014. The effects of demography and long-term selection on the accuracy of genomic prediction with sequence data. Genetics 198, 1671–1684. ( 10.1534/genetics.114.168344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Consortium TEP. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. ( 10.1038/nature11247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guenther CA, Tasic B, Luo L, Bedell MA, Kingsley DM. 2014. A molecular basis for classic blond hair color in Europeans. Nat. Genet. 46, 748–752. ( 10.1038/ng.2991) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karim L, et al. 2011. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 43, 405–413. ( 10.1038/ng.814) [DOI] [PubMed] [Google Scholar]
- 32.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. 2009. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 10, 184–194. ( 10.1038/nrg2537) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pai AA, Pritchard JK, Gilad Y. 2015. The genetic and mechanistic basis for variation in gene regulation. PLoS Genet. 11, e1004857 ( 10.1371/journal.pgen.1004857) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pastinen T. 2010. Genome-wide allele-specific analysis: insights into regulatory variation. Nat. Rev. Genet. 11, 533–538. ( 10.1038/nrg2815) [DOI] [PubMed] [Google Scholar]
- 35.Crowley JJ, et al. 2015. Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat. Genet. 47, 353–360. ( 10.1038/ng.3222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawkins RD, et al. 2011. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 21, 1393–1409. ( 10.1038/cr.2011.146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visel A, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. ( 10.1038/nature07730) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersson L, et al. 2015. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 16, 57 ( 10.1186/s13059-015-0622-4) [DOI] [PMC free article] [PubMed] [Google Scholar]