

Abstract
Plant-parasitic nematodes were found in 4 of the 12 clades of phylum Nematoda. These nematodes in different clades may have originated independently from their free-living fungivorous ancestors. However, the exact evolutionary process of these parasites is unclear. Here, we sequenced the genome sequence of a migratory plant nematode, Ditylenchus destructor. We performed comparative genomics among the free-living nematode, Caenorhabditis elegans and all the plant nematodes with genome sequences available. We found that, compared with C. elegans, the core developmental control processes underwent heavy reduction, though most signal transduction pathways were conserved. We also found D. destructor contained more homologies of the key genes in the above processes than the other plant nematodes. We suggest that Ditylenchus spp. may be an intermediate evolutionary history stage from free-living nematodes that feed on fungi to obligate plant-parasitic nematodes. Based on the facts that D. destructor can feed on fungi and has a relatively short life cycle, and that it has similar features to both C. elegans and sedentary plant-parasitic nematodes from clade 12, we propose it as a new model to study the biology, biocontrol of plant nematodes and the interaction between nematodes and plants.
Keywords: Ditylenchus destructor, plant-parasitic nematodes, biocontrol of plant nematodes, genomics of plant nematodes
1. Introduction
Plant-parasitic nematodes, which attack nearly all plant species, pose a great food safety threat worldwide and cause losses of more than $US80 billion per year. More than 4100 species of these nematodes have been described, and new species are continually being identified [1]. Three major types of plant-parasitic nematodes have been described based on their distinct life cycles. Sedentary endoparasitic nematodes use special feeding structures to obtain rich and continuous food sources from the plant host. Migratory plant endoparasitic nematodes do not induce feeding sites but feed while migrating between or through plant cells [2]. The third type of plant nematode is migratory ectoparasitic; these nematodes do not enter the plant and move in the soil to attack the roots [3]. The phylum Nematoda is divided into 12 clades based on small subunit ribosomal DNA (SSU rDNA) sequences [4]. Plant-parasitic nematodes were found in four of them, including clades 1, 2, 10 and 12. Plant-parasitic nematodes in different clades may have originated independently from their free-living fungivorous ancestors [5]. Two lines of evidence support this hypothesis. On the SSU rDNA sequence tree, the fungivorous nematode taxa are often located at the very base of the plant-parasitic nematode lineages [4,6]. Moreover, fungivorous nematodes usually possess onchiostyles (clade 1), spears (clade 2) or stomatostylets (clades 10 and 12) similar to their plant-parasitic relatives in the same clade [5]. However, the exact evolution of these parasites is unclear.
We focused on the plant nematodes from clade 12 because they contain eight of the top ten economically important nematodes, including Meloidogyne spp., Heterodera and Globodera spp., Pratylenchus spp., Radopholus spp., Ditylenchus spp., Rotylenchulus spp., Nacobbus spp. and Aphelenchoides spp [1]. To gain in-depth insights into the parasitic mechanism, genome sequences of nematodes with different life cycles were considered. Genome sequences are available for three sedentary endoparasitic nematodes (M. incognita, M. hapla and G. pallida) and one migratory endoparasite (Pratylenchus coffeae) in this clade [7–10]. In this study, we sequenced another migratory endoparasitic nematode (Ditylenchus destructor) that was positioned at the base of the most economically important plant nematodes, including root-knot and cyst nematodes, on the SSU rDNA tree [6]. This potato rot nematode can enter tubers through lenticels and then digest starch and pectin, leading to cell disintegration [11]. It can also cause additional damage during storage. Ditylenchus destructor is the second-ranking nematode pest of potatoes after the potato cyst nematode and is an internationally quarantined pest. Moreover, D. destructor can live on a very wide range of fungi and plants [12]. Unlike other plant-parasitic nematodes in clade 12, it can be cultured with fungi under laboratory conditions [13]. Using comparative genomics, we investigated the mechanisms underlying the parasitism of this nematode and the evolution of plant-parasitic nematodes.
Because some aspects of structure, development and reproduction are comparable between Caenorhabditis elegans and plant-parasitic nematodes, C. elegans is used as a model to study the biology of plant nematodes [14]. However, we found that C. elegans was not an ideal model when we focused on details of the interactions between plants and nematodes, the structure and development of these parasitic nematodes, and the biocontrol of plant nematodes. Additionally, we cannot use root-knot and cyst nematodes because their reproduction obligately relies on plants, and they are difficult to manipulate using molecular methods [14]. Ditylenchus destructor can feed on fungi or potatoes very easily and has a relatively short life cycle [12]. Therefore, we suggest its use as a model for biological and biocontrol studies of plant-parasitic nematodes. In this study, we consider this purpose based on its genomics.
2. Material and methods
(a). DNA/RNA extraction
Ditylenchus destructor Dd01 was isolated from sweet potatoes in Wuhan, China (electronic supplementary material, Supporting Methods). The nematodes were cultured for approximately 30 days on sweet potatoes. Both DNA and RNA were obtained from nematodes of mixed life stages. Total DNA was extracted using cetyltrimethylammonium bromide (CTAB)/Proteinase K. Total RNA was extracted using the HP Total RNA kit (Omega Bio-tek, USA) and treated with RNase-free DNase kit (Omega Bio-Tek).
(b). Genome sequencing and assembly
Short paired-end (160-, 380- and 800-bp) and mate-paired (3-, 5- and 8-kb) genomic DNA libraries were prepared and sequenced on an Illumina HiSeq 2500 platform (electronic supplementary material, table S1). High-quality genomic DNA extracted from D. destructor was prepared as a 20-kb library for P5-C3 chemistry. The PacBio RSII sequencing system generated 119 710 reads with a mean read length of 3.5 kb from one SMRT cell. RNA-seq was performed on an Illumina HiSeq 2000 platform; about 3 Gbp pair-end reads with length of 100 Gbp were obtained.
ALLPATHS-LG was used in the first step of the assembly [15]; reads from the three short paired-end and two mate-paired (3- and 5-kb) libraries were used in this step. In the second step, reads from the 8-kb mate-paired library were used to extend the scaffolds generated from ALLPATHS-LG using the tool SSPACE [16]. Then, subreads from PacBio were used to improve the assembly in PBJelly (http://sourceforge.net/projects/pb-jelly). The last step of the assembly was to use all of the paired- and mate-end reads to fill the gaps with GapFiller [17].
The assembly was cleared of contaminants as described by Cotton et al. [9] in two steps. Before gene model prediction, all scaffolds were searched against the NCBI non-redundant (nr) database with BlastX using an e-value of 10−5. Those that only had hits to bacterial sequence and for which no RNA-seq reads mapped were filtered out. After gene prediction, scaffolds with high GC content that did not have predicted gene models and scaffolds with no gene model with similarity to animals instead of plants, fungi or bacteria were removed.
(c). Genome annotation
RepeatScout [18] and RECON [19] were used to predict repeat sequences. RepeatModeler (http://repeatmasker.org/RepeatModeler.html) was used to classify consensus models of putative interspersed repeats. RepeatMasker (http://www.repeatmasker.org) was used to mask the repeats in the genome sequence.
MAKER v. 2.31.8 (http://www.yandell-lab.org/software/maker.html) was used to perform annotations as follows. Clean reads from the transcriptome with removal of adaptor sequences and low quality data were de novo assembled using the software Trinity (https://github.com/trinityrnaseq/trinityrnaseq) with the default settings to generate full-length cDNA sequences. These sequences were used to train the ab initio gene prediction programs SNAP [20] and Augustus [21]. The outputs of the above programs were used to re-train the tools themselves. Similarity-based gene predictions were generated using exonerated alignments of transcriptome sequences. Transcript fragments from the RNA-seq datasets were aligned and merged using TopHat [22] and Bowtie2 [23], and protein-based predictions were generated using GeneWise [24]. Gene predictions of ab initio and similarity methods were aggregated into a final set following three rounds of processing in MAKER software; the first two rounds improved the training of the ab initio tools, and a final round predicted proteins based on their similarities to all of the non-redundant protein sequences of the nematode phylum.
Functional annotation was performed using the InterProScan tool (5.14.53.0) [25] to scan the following 15 signature databases: COILS 2.2, Gene3D 3.5.0, PANTHER 8.1, Pfam-A 28.0, PIRSF 3.01, PRINTS 42.0, ProDom 2006.1, PROSITE 20.113, SMART 6.2, SignalP_EUK, SUPERFAMILY 1.75, TIGRFAMs 15.0 and TMHMM 2.0.
(d). Comparative genomics
Protein sequences of all available plant nematodes and C. elegans were clustered using the tool OrthoMCL [26]. The gene family-shared features among these nematodes were computed using an in-house Perl script. Protein sequences of each single-copy gene family were aligned using Muscle [27], and each alignment was trimmed with trimAl [28]. A maximum-likelihood core protein phylogenetic tree was constructed based on the concatenated alignment from all single-copy core protein families using PhyML [29] with the best model (LG + I + G) predicted by ProtTest [30]. Bootstrap support values were calculated from 1000 replicates.
(e). Expert functional annotation
The glycoside hydrolase (GH) and glycosyltransferase (GT) genes were identified and classified by searching query protein sequences against the CAZy database [31]. Then, target sequences were checked manually by identifying their conserved domains with the Pfam and CDD databases [32,33].
Protein sequences of effectors reported in the root-knot and cyst nematodes [34–37] were obtained from GenBank. Blast was used to predict putative homologous effectors from D. destructor. Signal peptides and transmembrane domains were predicted for each putative effector by SignalP (www.cbs.dtu.dk/services/SignalP) and TMHMM (www.cbs.dtu.dk/services/TMHMM), respectively. Only those with signal peptide but without transmembrane domain were considered.
Protein sequences associated with pressure resistance, core development and pathways from C. elegans were extracted according to their descriptions in WormBook (http://www.wormbook.org). Protein sequences for each of the processes and pathways were respectively used as original query sequences to search against all of the protein sequences investigated in this study by BlastP. Similar sequences were extracted and searched against all the protein sequences of C. elegans; only those having the highest scorer to the original query sequences were considered. Then, all candidate sequences were checked manually by identifying the conserved domains contained by the query sequences. If a query sequence did not contain a conserved domain, we filtered out the resulting sequences using an e-value of 10−5 and a coverage of 60%.
3. Results
(a). General features of the Ditylenchus destructor genome
The D. destructor strain studied in this work was isolated from sweet potatoes and caused serious damage to its host during storage (figure 1). We sequenced the D. destructor genome by combining the Illumina HiSeq 2500 and PacBio platforms to obtain approximately 200-fold coverage and produced a draft assembly of approximately 112 Mbp (N50 = 570 kb; table 1) with a mean GC content of 36.6%. The genome size was much larger than the genome sizes of other plant-parasitic nematodes [7–9,38]. The completeness was estimated to be 91% by CEGMA (core eukaryotic gene mapping approach) and showed a better assembly than the G. pallida [9] and M. incognita [7] genomes. Additionally, about 85% of the reads from RNA-seq were mapped to the genome sequence, confirming the completeness estimated by CEGMA. Genome repeats comprise approximately 23.4 Mbp (19.5%), of which only approximately 6 Mbp (28%) can be classified into known repeat types (electronic supplementary material, table S2). The features of the overall repeats are similar to the other plant-parasitic nematodes with genome sequences available [7–9,38].
Figure 1.

Photographs of injected sweet potatoes and D. destructor. (a) The outside phenotype of sweet potatoes six weeks after injection with D. destructor. (b) The inside phenotype of sweet potatoes six weeks after injection with D. destructor. (c) Nematodes extracted from sweet potatoes. (d) Light microscopy photographs of D. destructor. (e) The stylet (arrow) of D. destructor. (f) The male's tail of D. destructor. (g) The female's tail of D. destructor. (h) The J1 in a D. destructor egg.
Table 1.
Features of the Ditylenchus destructor draft genome. CDS, coding DNA sequence; CEGMA, core eukaryotic gene mapping approach.
| description | |
|---|---|
| genome size (bp) | 112 991 371 |
| number of scaffolds; contigs | 1761; 3818 |
| largest scaffold size (bp) | 3 556 246 |
| N counts in scaffold | 2 308 783 |
| N50 (bp); count > 1 kb in length | 570 381; 47 |
| N90 (bp); count > N90 length | 65 385; 260 |
| genome GC content (%) | 36.6 |
| repetitive sequences (%) | 19.5% |
| number of gene models | 13 938 |
| gene density (genes per Mb) | 124 |
| mean gene size (bp) | 4101 |
| mean CDS length (bp) | 1408 |
| mean exon number per gene | 8 |
| mean exon length (bp) | 145 |
| mean intron length (bp) | 325 |
| coding GC content (%) | 44% |
| CEGMA completeness: complete; partial (%) | 87.1; 91.1 |
A total of 13 938 protein-coding genes were predicted (table 1). The gene density was lower than the densities of other plant-parasitic nematodes and C. elegans. The mean gene length was 4101 bp, which was much longer than the lengths of other plant nematodes and C. elegans. The mean exon and intron lengths were 146 bp and 325 bp, respectively, and the mean exon number of each gene model was 8. The large intron length and number both contribute to the large lengths of each gene model.
All protein sequences of the plant-parasitic nematodes and C. elegans were clustered using the MCL algorithm [26]. A phylogenetic tree was constructed based on all single-copy core genes (figure 2). The tree had a topology similar to the SSU rDNA-based tree, with D. destructor positioned on the base branch of the root knot and cyst nematodes with 100% bootstrap support. A total of 3219 gene families were shared by all plant nematodes and C. elegans, and 329 gene families were orthologous among the four plant nematodes but not C. elegans (figure 3). Approximately 28% (3910) of the D. destructor genes had no orthologues in the other four nematodes. Ditylenchus destructor shared more orthologous genes with C. elegans than with other plant nematodes in clade 12.
Figure 2.
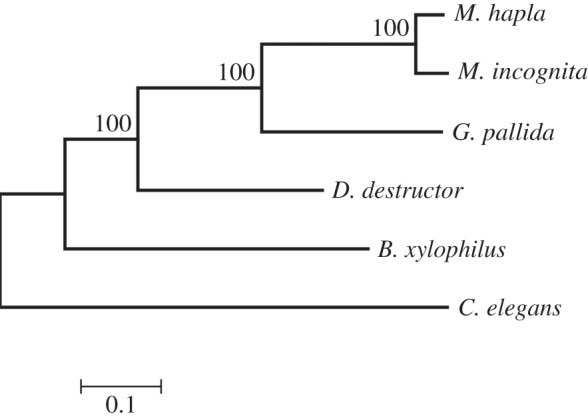
Phylogenomic analysis of nematodes. A maximum-likelihood phylogenetic tree was constructed based on a concatenated alignment of protein sequences for the 651 single-copy core genes by PhyML. Bootstrap support values were calculated from 1000 replicates. B. xylophilus, Bursaphelenchus xylophilus.
Figure 3.
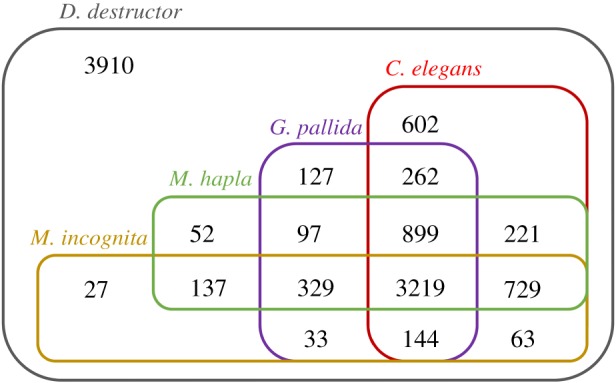
Homologs shared among plant-parasitic nematodes (D. destructor, G. pallida, M. hapla and M. incognita) and C. elegans. The homologous groups were computed by OrthoMCL with an inflation value of 1.5 after an all-against all Blast.
Among the 13 938 predicted genes in the D. destructor genome, 11 819 proteins were annotated based on the presence of characteristic protein domains identified by InterProScan [25]. Compared with other plant nematodes and C. elegans, no obvious extreme gene family expansions, like G protein-coupled receptor (GPCR) in M. halpa [8] and SPRY domain (domain in SP1a and the ryanodine receptor) proteins in G. pallida [9], were found. In the list of the top 25 Pfam hits featured from D. destructor compared with the other clade 12 plant nematodes, more proteins containing trypsin (PF00089), ABC transporter (PF00005), sugar (and other) transporter (PF00083) and short chain dehydrogenase (PF00106) domains (electronic supplementary material, table S3) were found. This result conforms to the capacity of D. destructor to metabolize carbohydrates and proteins from tubers of its major host, potatoes.
(b). Genomic insights into the plant parasitism mechanisms of Ditylenchus destructor
The cell wall is the first barrier that any endoparasitic plant nematode needs to overcome during plant invasion. Ditylenchus destructor has a cell wall modification gene content that is similar to the other three plant-parasitic nematodes in clade 12 (electronic supplementary material, table S4). Three cellulases from glycoside hydrolase family 5 (GH5) are present in D. destructor; each gene was positioned on the base of the other GH5 members from the root-knot and cyst nematodes in the protein sequence-based phylogenetic tree (electronic supplementary material, figure S1). Other cell wall-modifying genes identified in other plant nematodes were also present in D. destructor, including xylanases (GH30), arabinases (GH43) and pectate lyase (PL3). However, no genes homologous to polygalacturonase (GH28) or expansin were found in D. destructor, despite their frequent presence in other plant nematodes [39].
As the major host of D. destructor, potato-containing starch comprises more than 30% of its tubers [40]. To survive in this type of plant host, D. destructor harbours key enzymes for starch degradation. Although proteins belonging to GH13 have been predicted in all plant-parasitic nematodes, α-amylases (EC 3.2.1.1) were only identified from D. destructor and Bursaphelenchus xylohpilus, with a slight expansion in D. destructor (electronic supplementary material, table S4 and figure S2; figure 4). The α-amylase, which acts specifically on starch, cleaves the α-1,4 glycosidic bonds present from the inner part (endo-) of the amylose or amylopectin chain [41]. Phylogenetic analysis based on α-amylase protein sequences from nematodes and other organisms indicated a similar origin to other nematodes. These sequences were clustered together with those from animal parasitic and free-living nematodes and definitely apart from those of bacteria, fungi, insects and some higher animals (figure 4). The other two types of enzymes of GH13 predicted in this study included the branching enzyme (EC 2.4.1.18), which converts amylose into amylopectin, and oligo-1,6-glucosidase (EC 3.2.1.10), which releases an α-1,6-linked glucose from oligosaccharides produced from starch. All nematodes analysed in this study contained these enzymes. Another starch-degrading enzyme that belonged to the exo-amylase was also slightly enriched in D. destructor. Maltase-glucoamylase (GH31) cleaves both α-1,4 and α-1,6 bonds on the external glucose residues of amylose or amylopectin from the non-reducing end and thus produces only glucose (glucoamylase). Ditylenchus destructor contains nine of these proteins, which is more than even C. elegans (electronic supplementary material, table S4).
Figure 4.

Phylogenetic analysis of the α-amylase protein sequences from all nematodes and representatives of bacteria, fungi and other animals. Different branch colours represent different organisms (red for nematodes, blue for fungi, brown for bacteria, purple for invertebrates and green for vertebrates). Taxon names are coloured for different nematodes (dark green for plant nematodes, dark red for animal parasitic nematodes and blue for free-living nematodes).
Effectors play crucial roles in the interaction between nematodes and their plant hosts. Fifty-six proteins homologous to effectors of other plant nematodes were predicted in D. destructor, including 20 homologs to cyst nematodes and 36 homologs to root-knot nematodes (electronic supplementary material, table S5). Only five D. destructor proteins belonging to two families (FAR-1 (fatty-acid and retinol binding protein) and VAP-1 (venom allergen-like protein)) showed significant similarity to effector proteins of both root-knot and cyst nematodes. FAR-1 and VAP-1, both of which play important roles in the infection process, were also identified in several other plant nematodes, suggesting that they play similar roles in the nematode parasitic process. Most of the other putative effectors had limited information available concerning their roles in parasitism except that they were secreted during the infection process.
(c). Core processes and pathways involved in the control of nematode development
To gain more information about the evolution of the plant-parasitic nematodes, we focused on the diversity of the genes involved in the core processes and pathways identified in C. elegans.
Genes homologous to the C. elegans genes involved in the development control were surveyed in all plant-parasitic nematodes (table 2; electronic supplementary material, Supporting Results). With the exception of the genes involved in intestine development, less than half of the genes associated with each of the other processes had homologs in G. pallida, M. halpa and M. incognita. There were more homologous genes in the two migratory plant-parasitic nematodes B. xylophilus and D. destructor, but many key genes in these processes identified in C. elegans had no homologs in any of the plant-parasitic nematodes.
Table 2.
Numbers of genes associated with core processes in C. elegans and the five plant-parasitic nematodes.
| process/pathway | C. elegans | B. xylophilus | D. destructor | G. pallida | M. halpa | M. incognita |
|---|---|---|---|---|---|---|
| development control | ||||||
| intestine development | 24 | 19 | 20 | 15 | 16 | 16 |
| pharynx development | 25 | 11 | 15 | 12 | 7 | 11 |
| Q neuroblast development | 32 | 14 | 21 | 15 | 12 | 7 |
| embryo development | 48 | 22 | 21 | 19 | 21 | 17 |
| programmed cell death | 28 | 16 | 14 | 11 | 11 | 12 |
| translational control of maternal RNAs | 15 | 8 | 7 | 4 | 6 | 5 |
| signal transduction | ||||||
| RTK-Ras-ERK signalling | 58 | 44 | 48 | 40 | 42 | 37 |
| Eph receptor signalling | 25 | 20 | 22 | 17 | 19 | 13 |
| heterotrimeric G proteins | 25 | 16 | 15 | 12 | 12 | 14 |
| Hh signalling network | 63 | 28 | 32 | 18 | 21 | 20 |
| insulin/insulin-like growth factor signalling | 18 | 13 | 14 | 10 | 13 | 13 |
| notch signalling | 47 | 40 | 35 | 33 | 32 | 31 |
| nuclear hormone receptor | 284 | 68 | 51 | 54 | 23 | 18 |
| small GTPases | 34 | 29 | 29 | 22 | 24 | 21 |
| TGF-β signalling | 26 | 16 | 15 | 16 | 15 | 11 |
| chemoreceptor | 1276 | 324 | 197 | 120 | 68 | 108 |
| Wnt signalling | 38 | 31 | 33 | 26 | 26 | 21 |
| other core processes | ||||||
| autophagy | 19 | 13 | 10 | 11 | 14 | 10 |
| neurogenesis | 64 | 32 | 32 | 21 | 18 | 18 |
| sex determination | 23 | 7 | 11 | 8 | 5 | 7 |
| neuropeptide genes | 122 | 28 | 29 | 16 | 18 | 16 |
| RNAi pathway | 78 | 37 | 36 | 27 | 28 | 26 |
During development of the intestine, the actin protein ACT-5 has been reported to play a key role in the microvilli, and act-5 knockout resulted in lethality shortly after hatching [42]. However, no homologous gene was found in any of the plant-parasitic nematodes, suggesting a different microvilli structure (electronic supplementary material, table S6). Two genes involved in pharynx development (skn-1 and pha-4) were absent in plant nematodes despite the finding that mutations in one of these genes could result in an absent pharynx [43,44]. Moreover, only some of the genes that played no crucial roles in this process were conserved, such as gei-17, htz-1, lag-1, myo-2, pha-2, ubc-9 and ubc-39 (electronic supplementary material, table S7).
Programmed cell death (PCD) plays a fundamental role in animal development, and genes involved in the three phases of this process have been well studied in C. elegans [45]. Although more genes were conserved in D. destructor than in the other three clade 12 nematodes, most key genes in the first two stages were lost in all of the plant nematodes (electronic supplementary material, table S8). During the specification phase, only one transcriptional regulator (ces-2) was conserved in all nematodes. In the killing phase, no homologous genes were found in any of the plant nematodes for three (eag-1, ced-9 and ced-4) out of the four genes in the core machinery involved in the activation of the apoptotic programme. Most genes in the execution phase were conserved in all plant nematodes.
(d). Core processes and nematode signal transduction pathways
In contrast with the genes involved in developmental control, the major signalling systems were conserved among all nematodes and most key genes in these pathways identified in C. elegans were conserved in all plant nematodes (table 2; electronic supplementary material, Supporting Results). Compared with the three sedentary parasitic plant nematodes, the two migratory plant-parasitic nematodes contain more genes that are homologous with the free-living nematode C. elegans. The main differences among these plant nematodes and C. elegans arise from genes encoding receptors, such as the nuclear hormone receptors (NHRs) and GPCRs. Additionally, most of these types of genes identified in C. elegans have no homologs in any of the plant nematodes (electronic supplementary material, tables S9, S10 and Supporting Results).
In response to harsh environmental conditions, C. elegans larvae undergo dauer arrest at the second moult. There are four pathways that play critical roles during the formation of this situation: the guanylyl cyclase, TGFβ-like, insulin-like and hormonal signalling pathways. The last three pathways exhibit limited conservation in the plant nematodes; only a few genes in each pathway have homologs in these nematodes (electronic supplementary material, table S11), with relatively more conservation in D. destructor and B. xylophilus. Although many of the genes involved in TGF-β signalling are conserved in all plant nematodes, genes associated with the regulation of dauer development are almost all lost, with daf-4 being the only exception (electronic supplementary material, tables S11 and S12). The insulin-like and steroid hormone pathways are in similar situations. Sex determination in C. elegans has been well studied. Only limited genes involved in this process are conserved in all nematodes, including mag-1, mog-1, mog-4, mog-6 and tra-3 (table 2; electronic supplementary material, table S13). Ditylenchus destructor possesses more genes that are similar to C. elegans than G. pallida, M. incognita and M. halpa. For example, D. destructor contains the mab-23 gene, which is associated with male differentiation and behaviour.
RNA interference (RNAi) represents an important gene regulation pathway implemented by double-strand (dsRNA)-based gene silencing. RNAi can be used as a molecular tool for functional research in eukaryotic organisms. For the control of plant nematodes, it can be applied in transgenic plants to promote resistance against parasites [46]. Caenorhabditis elegans has 78 RNAi effectors, but all plant nematodes contain less than half of these effectors [9,38]. The two migratory plant nematodes have more RNAi effectors than the sedentary plant nematodes (table 2; electronic supplementary material, table S14). Most of the small RNA biosynthetic proteins have homologs in all plant nematodes, and genes involved in dsRNA uptake and argonaute formation are more conserved in D. destructor and B. xylophilus. Genes homologous to rrf-1, rrf-3 and csr-1 were found in D. destructor and B. xylophilus but not in the other plant nematodes. For all of these processes, approximately half or more of the effectors identified in C. elegans have no homologous members in the plant nematodes.
4. Discussion
We present the draft genome sequence of D. destructor, a migratory plant nematode from clade 12. In addition to plant cell wall degradation enzymes, the α-amylase enzyme is important for the capacity of D. destructor to parasitize plants. Unlike the cell wall modification-associated genes, the α-amylase gene was not acquired by horizontal gene transfer (HGT) and might have been inherited from the nematode ancestor. Moreover, the presence of fungal cell wall degradation enzymes coincides with the finding that D. destructor is a fungi feeder (electronic supplementary material, table S4). Indeed, most members of the genus Ditylenchus are fungivorous [47].
On the SSU-based phylogenetic tree, the genus Ditylenchus is on the base of the most important sedentary nematodes, the root-knot and cyst nematodes. The phylogenetic tree of all single-copy core proteins of the plant nematodes and C. elegans supports that D. destructor is positioned more closely to the root [6]. We suggest that Ditylenchus spp. are more ancient than sedentary endoparasitic nematodes and may represent an intermediate stage during the evolution of plant nematodes. Some members of this genus, particularly Ditylenchus dipsaci, have much larger host ranges than other plant nematodes, which supports the ancient origin of this genus [2,48]. For each of the four clades containing plant-parasitic nematodes, information concerning feeding habits and puncturing devices supports the hypothesis that plant-parasitic nematodes have originated from fungivorous ancestors [5]. Most members of the genus Ditylenchus feed on fungi, although other members (including D. destructor) feed on both fungi and plants. This finding also supports the hypothesis that Ditylenchus is more ancient than other sedentary endoparasitic nematodes in the same clade.
Comparative genomics among different plant nematodes in clade 12 and C. elegans supports the ancient position of Ditylenchus spp. and its intermediate situation between free-living and sedentary nematodes. First, D. destructor exhibits no genome reduction, and no gene family associated with plant parasitism has been expanded, in contrast with the other plant nematodes. This result is in accordance with its two different lifestyles (free-living and facultative plant parasitism). Second, some effectors from root-knot and cyst nematodes have homologies in D. destructor, but there is little overlap between the two sedentary nematodes [9]. These common effectors may originate from nematodes such as Ditylenchus spp. Root-knot and cyst nematodes inherited these effectors separately from a Ditylenchus-like ancestor [5]. Third, the key effector, the cellulase gene, from D. destructor shows a more ancient situation than the effectors of the two sedentary nematodes. Other cell modification-associated genes have similar features, such as the arabinase (GH43) gene. The member in D. destructor was positioned more closely to the roots of the phylogenetic trees (electronic supplementary material, figures S3). Fourth, genes associated with response to and protection against environmental pressures were more conserved in D. destructor (electronic supplementary material, Supporting Results). As a facultative fungivorous nematode, D. destructor faces more challenges owing to diverse environmental pressures in the soil than obligate plant-parasitic nematodes. Fifth, core development processes are more conserved in D. destructor. Compared to the free-living nematode C. elegans, most genes involved in core development have undergone substantial losses in all plant nematodes. However, D. destructor usually possesses relatively more conserved members in terms of both gene families and sequence identities. From free-living to sedentary plant-parasitic styles, nematodes exhibit large transformations in their core development, and D. destructor may represent an intermediate situation in this process. Sixth, in contrast with pathways associated with developmental control, signal transduction pathways are conserved in all plant-parasitic nematodes. Additionally, many of these pathways are conserved among animals from different phyla [49,50]. However, we could find obvious diversity in the plant nematodes when compared to C. elegans, and we found more homologous genes in D. destructor than in the root-knot and cyst nematodes. Taken together, our results suggest that sedentary lifestyles, for example formation of root-knot and cyst, may represent advanced states of plant parasitism because the nematodes do not kill the plant and can continuously absorb nutrients from the plant host [51]. If we consider the fungi-feeding lifestyles as the primary state, the migratory lifestyles can represent an intermediate state. Migratory nematodes, such as Ditylenchus spp., can cause the death of their plant hosts, after which they need to relocate from the host to the soil to feed on fungi and to find a new appropriate plant host [2].
Some migratory plant nematodes were phylogenetically close to sedentary nematodes. For example, Pratylenchus spp. were clustered together with Meloidogyne spp. on the phylogenetic tree based on SSU rDNA sequences [5]. These nematodes maintain life migratory lifestyles, but lost the capability of free-living feeding on fungi. Information from the genomics indicated that some processes associated with developmental control (for example, sex determination) were not conserved, whereas those involved in signal transduction were more conserved, such as the dauer pathway and RNAi pathway [10]. This can be considered as indirect evidence for the more ancient position of Ditylenchus spp. during the evolutionary history.
We focused on the biocontrol of plant nematodes to facilitate future studies related to plant protection. Most of our previous studies have used C. elegans as a model, but C. elegans is neither a plant nematode nor a true parasite [52,53]. Therefore, we focused on the biology and genetics of D. destructor to enable its use as a model organism to study the interactions between plant nematodes and plants. Ditylenchus destructor shared many genomic features with both the root-knot and cyst nematodes and the model organism C. elegans. Based on the genomics study, we suggest that some basic features, especially those associated with signal transduction of plant nematodes, can be well studied using C. elegans as a model because they are conserved in all nematodes. However, to study other features, such as those involved in specific organism development and the interactions between nematodes and plants, D. destructor is a superior model because most of these features are plant nematode-specific. Ditylenchus destructor can be manipulated by RNAi [54]. We suggest that D. destructor can be used as a model nematode to study the interactions between plant nematodes and their hosts and the biocontrol of economically important agricultural nematodes.
Supplementary Material
Data accessibility
The sequence data are accessible via the National Center for Biotechnology Information under accession code LSTP00000000 (version LSTP01000000; BioProject: PRJNA312427).
Author contributions
J.Z., D.P., L.R. and M.S. designed and planned the study. L.C., F.C. and M.X. isolated the nematodes and extracted the DNA and RNA. J.Z., S.J. and H.L. analysed the data. J.Z. and M.S. wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by grants from the China 948 Program of the Ministry of Agriculture (2016-X21, 2011-G25) and the National Natural Science Foundation of China (31500003 and 31270137).
References
- 1.Jones JT, et al. 2013. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant. Pathol. 14, 946–961. ( 10.1111/mpp.12057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moens M, Perry RN. 2009. Migratory plant endoparasitic nematodes: a group rich in contrasts and divergence. Annu. Rev. Phytopathol. 47, 313–332. ( 10.1146/annurev-phyto-080508-081846) [DOI] [PubMed] [Google Scholar]
- 3.Wyss U, Lehmann H, Jank-Ladwig R. 1980. Ultrastructure of modified root-tip cells in Ficus carica, induced by the ectoparasitic nematode Xiphinema index. J. Cell Sci. 41, 193–208. [DOI] [PubMed] [Google Scholar]
- 4.Holterman M, van der Wurff A, van den Elsen S, van Megen H, Bongers T, Holovachov O, Bakker J, Helder J. 2006. Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades. Mol. Biol. Evol. 23, 1792–1800. ( 10.1093/molbev/msl044) [DOI] [PubMed] [Google Scholar]
- 5.Quist CW, Smant G, Helder J. 2015. Evolution of plant parasitism in the phylum Nematoda. Annu. Rev. Phytopathol. 53, 289–310. ( 10.1146/annurev-phyto-080614-120057) [DOI] [PubMed] [Google Scholar]
- 6.van Megen H, van den Elsen S, Holterman M, Karssen G, Mooyman P, Bongers T, Holovachov O, Bakker J, Helder J. 2009. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 11, 927–950. ( 10.1163/156854109X456862) [DOI] [Google Scholar]
- 7.Abad P, et al. 2008. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 26, 909–915. ( 10.1038/nbt.1482) [DOI] [PubMed] [Google Scholar]
- 8.Opperman CH, et al. 2008. Sequence and genetic map of Meloidogyne hapla: a compact nematode genome for plant parasitism. Proc. Natl Acad. Sci. USA 105, 14 802–14 807. ( 10.1073/pnas.0805946105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotton JA, et al. 2014. The genome and life-stage specific transcriptomes of Globodera pallida elucidate key aspects of plant parasitism by a cyst nematode. Genome Biol. 15, R43 ( 10.1186/gb-2014-15-3-r43) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burke M, et al. 2015. The plant parasite Pratylenchus coffeae carries a minimal nematode genome. Nematology 17, 17 ( 10.1163/15685411-00002901) [DOI] [Google Scholar]
- 11.Henderson VE. 1951. Some host relationships of the potato-rot nematode, Ditylenchus destructor Thorne, 1945. Nature 167, 952 ( 10.1038/167952a0) [DOI] [PubMed] [Google Scholar]
- 12.Faulkner L, Darling H. 1961. Pathological histology, hosts, and culture of the potato rot nematode. Phytopathology 51, 778–786. [Google Scholar]
- 13.Peng H, Gao BL, Kong LA, Yu Q, Huang WK, He XF, Long HB, Peng DL. 2013. Exploring the host parasitism of the migratory plant-parasitic nematode Ditylenchus destuctor by expressed sequence tags analysis. PLoS ONE 8, e69579 ( 10.1371/journal.pone.0069579) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosso MN, Jones JT, Abad P. 2009. RNAi and functional genomics in plant parasitic nematodes. Annu. Rev. Phytopathol. 47, 207–232. ( 10.1146/annurev.phyto.112408.132605) [DOI] [PubMed] [Google Scholar]
- 15.Gnerre S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518. ( 10.1073/pnas.1017351108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. ( 10.1093/bioinformatics/btq683) [DOI] [PubMed] [Google Scholar]
- 17.Nadalin F, Vezzi F, Policriti A. 2012. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13(Suppl 14), S8 ( 10.1186/1471-2105-13-S14-S8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AL, Jones NC, Pevzner PA. 2005. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), 351–i358 ( 10.1093/bioinformatics/bti1018) [DOI] [PubMed] [Google Scholar]
- 19.Bao Z, Eddy SR. 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276. ( 10.1101/gr.88502) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5, 59 ( 10.1186/1471-2105-5-59) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanke M, Schoffmann O, Morgenstern B, Waack S. 2006. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 ( 10.1186/1471-2105-7-62) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. ( 10.1093/bioinformatics/btp120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. ( 10.1038/nmeth.1923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birney E, Clamp M, Durbin R. 2004. GeneWise and Genomewise. Genome Res. 14, 988–995. ( 10.1101/gr.1865504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. ( 10.1093/bioinformatics/btu031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. ( 10.1101/gr.1224503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. ( 10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. ( 10.1093/bioinformatics/btp348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. ( 10.1093/sysbio/syq010) [DOI] [PubMed] [Google Scholar]
- 30.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. ( 10.1093/bioinformatics/btr088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42(Database issue), D490–D495. ( 10.1093/nar/gkt1178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finn RD, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42(Database issue), D222–D230. ( 10.1093/nar/gkt1223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchler-Bauer A, et al. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res. 43(Database issue), D222–D226. ( 10.1093/nar/gku1221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Truong NM, Nguyen CN, Abad P, Quentin M, Favery B. 2015. Chapter twelve. Function of root-knot nematode effectors and their targets in plant parasitism. In Advances in Botanical Research vol. 73 (eds Carolina Escobar, Carmen Fenoll), pp. 293–324. Salt Lake City, UT: Academic Press. [Google Scholar]
- 35.Gardner M, Verma A, Mitchum MG. 2015. Chapter Eleven. Emerging roles of cyst nematode effectors in exploiting plant cellular processes. In Advances in Botanical Research vol. 73 (eds Carolina Escobar, Carmen Fenoll), pp. 259–291. Salt Lake City, UT: Academic Press. [Google Scholar]
- 36.Rutter WB, Hewezi T, Abubucker S, Maier TR, Huang G, Mitreva M, Hussey RS, Baum TJ. 2014. Mining novel effector proteins from the esophageal gland cells of Meloidogyne incognita. Mol. Plant Microbe Interact. 27, 965–974. ( 10.1094/MPMI-03-14-0076-R) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorpe P, et al. 2014. Genomic characterisation of the effector complement of the potato cyst nematode Globodera pallida. BMC Genomics 15, 923 ( 10.1186/1471-2164-15-923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikuchi T, et al. 2011. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 7, e1002219 ( 10.1371/journal.ppat.1002219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danchin EG, Rosso MN, Vieira P, de Almeida-Engler J, Coutinho PM, Henrissat B, Abad P. 2010. Multiple lateral gene transfers and duplications have promoted plant parasitism ability in nematodes. Proc. Natl Acad. Sci. USA 107, 17 651–17 656. ( 10.1073/pnas.1008486107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoover R. 2000. Composition, molecular structure, and physicochemical properties of tuber and root starches: a review. Carbohydr. Polym. 45, 253–267. ( 10.1016/S0144-8617(00)00260-5) [DOI] [Google Scholar]
- 41.Van Der Maarel MJ, Van Der Veen B, Uitdehaag JC, Leemhuis H, Dijkhuizen L. 2002. Properties and applications of starch-converting enzymes of the α-amylase family. J. Biotechnol. 94, 137–155. ( 10.1016/S0168-1656(01)00407-2) [DOI] [PubMed] [Google Scholar]
- 42.MacQueen AJ, Baggett JJ, Perumov N, Bauer RA, Januszewski T, Schriefer L, Waddle JA. 2005. ACT-5 is an essential Caenorhabditis elegans actin required for intestinal microvilli formation. Mol. Biol. Cell 16, 3247–3259. ( 10.1091/mbc.E04-12-1061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowerman B, Draper BW, Mello CC, Priess JR. 1993. The maternal gene skn-1 encodes a protein that is distributed unequally in early C. elegans embryos. Cell 74, 443–452. ( 10.1016/0092-8674(93)80046-H) [DOI] [PubMed] [Google Scholar]
- 44.Kiefer JC, Smith PA, Mango SE. 2007. PHA-4/FoxA cooperates with TAM-1/TRIM to regulate cell fate restriction in the C. elegans foregut. Dev. Biol. 303, 611–624. ( 10.1016/j.ydbio.2006.11.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lord CE, Gunawardena AH. 2012. Programmed cell death in C. elegans, mammals and plants. Eur. J. Cell Biol. 91, 603–613. ( 10.1016/j.ejcb.2012.02.002) [DOI] [PubMed] [Google Scholar]
- 46.Gheysen G, Vanholme B. 2007. RNAi from plants to nematodes. Trends Biotechnol. 25, 89–92. ( 10.1016/j.tibtech.2007.01.007) [DOI] [PubMed] [Google Scholar]
- 47.D S, MW B. 1991 Stem and bulb nematodes, Ditylenchus spp. In Manual of agricultural nematology (ed. WR Nickle), pp. 423–464. New York, NY: Marcel Dekker, Inc.
- 48.Subbotin SA, Madani M, Krall E, Sturhan D, Moens M. 2005. Molecular diagnostics, taxonomy, and phylogeny of the stem nematode Ditylenchus dipsaci species complex based on the sequences of the internal transcribed spacer-rDNA. Phytopathology 95, 1308–1315. ( 10.1094/PHYTO-95-1308) [DOI] [PubMed] [Google Scholar]
- 49.Pires-daSilva A, Sommer RJ. 2003. The evolution of signalling pathways in animal development. Nat. Rev. Genet. 4, 39–49. ( 10.1038/nrg977) [DOI] [PubMed] [Google Scholar]
- 50.Gazave E, Lapebie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, Borchiellini C, Vervoort M, Renard E. 2009. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol. Biol. 9, 249 ( 10.1186/1471-2148-9-249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jasmer DP, Goverse A, Smant G. 2003. Parasitic nematode interactions with mammals and plants. Annu. Rev. Phytopathol. 41, 245–270. ( 10.1146/annurev.phyto.41.052102.104023) [DOI] [PubMed] [Google Scholar]
- 52.Zhang F, et al. 2016. Bacillus thuringiensis crystal protein Cry6Aa triggers Caenorhabditis elegans necrosis pathway mediated by aspartic protease (ASP-1). PLoS Pathog.. 12, e1005389 ( 10.1371/journal.ppat.1005389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruan L, et al. 2015. A two-domain protein triggers heat shock pathway and necrosis pathway both in model plant and nematode. Environ. Microbiol. 17, 4547–4565. ( 10.1111/1462-2920.12968) [DOI] [PubMed] [Google Scholar]
- 54.Peng H, Peng D, Long H, He W, Qiao F, Wang G, Huang W. 2014. Characterisation and functional importance of β-1,4-endoglucanases from the potato rot nematode, Ditylenchus destructor. Nematology 16, 505–517. ( 10.1163/15685411-00002783) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data are accessible via the National Center for Biotechnology Information under accession code LSTP00000000 (version LSTP01000000; BioProject: PRJNA312427).