

Abstract
Inflammatory activation of microglia is a hallmark of several disorders of the central nervous system. In addition to protecting the brain against inflammatory insults, microglia are neuroprotective and play a significant role in maintaining neuronal connectivity, but the prolongation of an inflammatory status may limit the beneficial functions of these immune cells. The finding that estrogen receptors are present in monocyte-derived cells and that estrogens prevent and control the inflammatory response raise the question of the role that this sex steroid plays in the manifestation and progression of pathologies that have a clear sex difference in prevalence, such as multiple sclerosis, Parkinson's disease, and Alzheimer's disease. The present review aims to provide a critical review of the current literature on the actions of estrogen in microglia and on the involvement of estrogen receptors in the manifestation of selected neurological disorders. This current understanding highlights a research area that should be expanded to identify appropriate replacement therapies to slow the progression of such diseases.
Introduction
-
Microglia: the Immune Cells of the CNS
Microglia and brain development
Microglia in the adult, healthy brain
Energy metabolism and neuroinflammation
Microglia and aging
-
Mechanisms of Estrogen Actions in Microglia
Estrogens may modulate target cell activity by interacting with several receptors
Which ERs are expressed in microglia in the mature, adult brain?
Estrogen activity in microglia
-
Estrogens: Protective or Risk Factors in Brain Injury and Neurodegeneration?
Estrogens and stroke or hypoxic neuronal death
Demyelinating diseases
Neurodegenerative diseases
Concluding Remarks and Future Directions
I. Introduction
The nervous system is not readily accessible to peripheral immune cells, but evolution has favored the selection of microglia as the resident immune cells in the central nervous system (CNS) for the first line of protection against noxious stimuli, such as stress and pathogenic insults. To adapt to the needs of their environment, microglia are extremely plastic cells able to show an array of diversified phenotypes. Indeed, in response to a potential danger, microglia perform the following: 1) synthesize and release inflammatory molecules (eg, TNFα, reactive oxidative species, inflammatory cytokines, and chemokines); 2) alert the brain and other immune cells; 3) clear all debris in the parenchyma; and 4) provide nutrients to repair the damage induced in the cells surrounding the inflammatory battlefield. In addition, mounting evidence indicates that microglia play a major supporting role in neurogenesis and neuronal activity.
In the case of major injury, microglia attract peripheral immune cells to form an integrative network (with astroglia, neutrophils, lymphocytes, plasma cells, and macrophages) that provides the brain with a strong defensive system. This functional complex is finely regulated by a well-timed synthesis of inflammatory and anti-inflammatory molecules for the transient inception of the inflammatory response in the presence of insults and to return to a surveying stage as the immune emergency is resolved. Failure of such homeostatic mechanisms may have severe pathological consequences because an excessive, prolonged, or asynchronous immune activation plays a very active role in the onset and progression of pathologies ranging from chronic pain and epilepsy to neurodegeneration and psychiatric disorders (1–4).
An emerging theme in the study of microglia function is the sex-related differences highlighted by a growing number of studies in male and female vertebrates. The precise roles played by genetic, hormonal, or environmental cues in determining this sexual dimorphism remain to be clarified. Certainly, estrogens play a major role in controlling microglia activity. In this review, we will discuss recent advances in the understanding of microglial biology, with a particular focus on the influence of estrogens on their function and on the physiopathological relevance of this regulation. Furthermore, we will highlight the areas that need to be explored to verify the potential for estrogen receptor (ER) ligands in the attenuation of neuroinflammation in specific neuronal disorders.
II. Microglia: the Immune Cells of the CNS
A. Microglia and brain development
1. Microglia and structural organization of the developing brain
The existence of microglia was first described by Nissl in 1880. In the first decades of the twentieth century, the seminal work of Santiago Ramon y Cajal and his student Pio Del Rio Hortega formed the basis for determining the morphological and functional differences between microglia and other neural cells (5, 6). Microglia, unique among the major cell types in the CNS, are not derived from the ectoderm. In fact, during early fetal development, a major wave of myeloid precursors migrates from the embryonic yolk sac to the brain to become the resident microglia. Accordingly, genetic and cell lineage studies show that microglia originate from Pu.1-positive cells in both the mouse (7) and zebrafish (8), and fate-mapping experiments show colonization of the brain by CSF1R+ erythro-myeloid progenitors at embryonic day 8.5 (9, 10). The number of microglia precursors that migrate to the brain around embryonic day 8.0 (10) is finite and relatively small (8), but sufficient to proliferate, populate the entire brain, and self-maintain for an entire life span. The factors required for brain colonization have yet to be completely identified. In mice, colony-stimulating factor 1 (CSF1) is involved because mouse embryonic microglia express CSF1 receptor and Csf1 gene deletion results in a significant loss of microglia in adults (10, 11). It is conceivable that macrophages migrate in response to an inflammatory stimulus, as indicated by genetic studies conducted in zebrafish (12) and by the fact that in the developing brain, microglia are generally large, round amoeboid cells that produce elevated levels of cytokines and chemokines.
Microglia phagocytic activity contributes to the structural organization of the developing brain by eliminating redundant neurons and synaptic connections (13) (Figure 1). Microglia-dependent synaptic pruning is well documented in the developing hippocampus and thalamus, where in the presence of the complement system, these cells engulf PSD-95-containing postsynaptic dendritic spines driven by the fractalkine system (14). The phagocytic actions of microglia play a central role in the removal of apoptotic neurons (15), as well as inducing death in selected populations of viable neurons through a process called phagoptosis (16). The chemokine fractalkine (CX3CL1) released by the dying cells attracts phagocytic microglia, and the neurons to be engulfed are recognized by the phosphatidylserine (PS) exposed on the external surface. In addition, time-lapse microscopy in the brains of rodents and monkeys and in organotypic cortical slices showed that microglia phagocytose neural precursors in the cortex (17) and cerebellum (18). The criteria for the selection of the neurons to be eliminated during development require further investigation. However, the fact that microglia sense synaptic activity may suggest that the neurons eliminated are those not actively establishing synaptic contacts with their peers (19, 20). Finally, microglia promote the survival of neurons and the growth of their axons (21) through the secretion of neurotrophic factors, such as IGF-1, IL-1β, and interferon (IFN) γ (22). Such a function is maintained in the adult brain, as discussed in section B.
Figure 1.

Microglia are a dynamic mediator of synaptic development and homeostasis. Microglia in its surveying state senses the state of activity of neurons, is attracted to the dendritic spines through proteins of the complement and fractalkine, and participates in neuronal plasticity potentially through the release of proteases able to modulate the structure and functions of the synapses. Microglia possibly respond to the release of ATP, which may induce the shedding of lipid-rich vesicles that were reported to increase the frequency and amplitude of the excitatory postsynaptic potential.
2. Microglia colonization of the brain is sex dependent
The colonization of the developing mouse brain by microglia appears to occur differently in the two sexes because males were shown to have overall more microglia early in postnatal development (postnatal day [P] 3–4). This is in contrast with the fact that females have more microglia in selected brain areas later in development and in adulthood (23). The developing male hippocampus and cortex have a nearly 200-fold greater expression of the chemokine ligand CCL20 and 50-fold higher expression of the chemokine ligand CCL4 than those of females. Conceivably, these two chemokines play critical roles in driving the dimorphic perinatal colonization of brain regions relevant for cognition and memory, as well as a role in the highly sexual dimorphic preoptic area (23, 24). The cause of the elevated levels of chemokines in the developing male brain remains unknown, but the temporal correlation between microglial brain colonization and the surge of testicular activity (at embryonic day 17) suggests the involvement of sex hormones (25) (Figure 2). Additional investigations during brain maturation are necessary to learn whether sex hormone receptors have a sexual dimorphic expression. Microglia from P3 mice express ERα (26), and the mRNA content for this receptor subtype further increases in adult mice, suggesting that microglia sensitivity to estrogens increases with age (26). So far, no sexual differences were observed in ERα mRNA content at any age (26, 27). ERβ mRNA was detected in microglia primary cultures from P0 newborns (28), whereas the levels of this receptor are undetectable in microglia sorted “ex vivo” from mice at P3 and from adults (26, 27). The data provided so far on the expression of the progesterone receptor (PR) and the androgen receptor (AR) indicate that these receptors are not expressed in microglia in adult mice (27).
Figure 2.

Brain development: the sexually dimorphic activity of microglia. Primitive macrophages exit the yolk sac blood islands at the onset of circulation and colonize the neuroepithelium from E8.5 to give rise to microglia. The BBB starts to form from E13.5 and may isolate the developing brain from the contribution of fetal liver hematopoiesis. Embryonic microglia expand and colonize the whole CNS until adulthood. The high concentration of chemokines in the male brain facilitates microglia proliferation. E, embryonic day.
A sexually dimorphic behavior of microglia during development may be important because prenatal or perinatal infections may induce permanent neurological consequences. Indeed, excessive microglia activation during the developmental programming has been implicated in altered sexual behavior (29); dopamine-mediated functions and cognitive abilities (25, 30); a predisposition to mental disorders, such as schizophrenia and autism (31); and in neurological alterations that have a different occurrence in males and females (31). This argues for the necessity of further investigation on the role played by the endocrine-microglia communication in the structural organization of the brain.
B. Microglia in the adult, healthy brain
In the adult nervous system, microglia are distributed throughout the brain, with slight changes in concentrations and activity in each region (32, 33). In the human brain, microglia account for up to 16% of all non-neuronal CNS cells and reside mainly in the white matter. In the rodent brain, microglia are found more often in the gray matter, and their content is lower (5–12% of all glial cells) (32, 34). Observations in transgenic mice with a green fluorescent protein reporter located under the control of the promoter for the fractalkine receptor gene (Cx3cr1) (35) demonstrated that microglia cells never rest and are constantly patrolling the brain parenchyma. Even in the absence of inflammatory stimuli, the processes extending from the cell somata are in continuous motion and growth, retracting and protruding in filopodia-like membranes with a bulbous ending (34). In this “surveillance” state aimed at detecting acute or chronic injuries, microglia movements are regulated internally by K+/Cl− cotransporters that mediate the process of swelling (36) and by depolymerization and repolymerization of actin filaments. These changes are induced by environmental cues, such as glutamate acting through microglia 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl propanoic acid receptors (37), or purinergic molecules (38) and components of the complement system (5, 39). Through this activity, microglia monitor the microenvironment three-dimensionally (34) and fulfill their large number of housekeeping functions (eg, removal of cellular waste products and cell debris, remodeling of extracellular matrix, reshaping of synapses and neuronal connectivity) (40). In addition, microglia secrete growth factors (TGFβ, fibroblast growth factor, nerve growth factor) (41) and lipoproteins, thus participating in stem cell proliferation (42), neuronal dynamics, and maintenance of neuronal membranes.
When in the surveying state, which is characterized by a highly ramified phenotype, healthy microglia are not believed to secrete inflammatory molecules. Once challenged by physical or chemical insults (infection, trauma, oxygen deprivation), microglia are “activated” and acquire an amoeboid, macrophage-like morphology, becoming phagocytic and able to secrete a large variety of inflammatory molecules. Activated microglia move more rapidly and may cover relatively large distances in the brain parenchyma. Several other morphological features have been described for microglia. For instance, in chronic disorders, microglia might acquire a rod-like shape or become multinucleated and increase their dimensions in the presence of indigestible material. Finally, microglia might present processes that are short and stocky. This latter morphology is often observed in aging brains and is considered dystrophic (43).
All these phenotypes reflect a differential functional status with the expression of biochemical markers that are very useful for a more objective definition of the microglia functional status (Table 1). In their surveying stage, these cells express low levels of myeloid-monocytic markers, such as Fc receptor-cluster of differentiation (CD) 32 and CD64, integrins (CD11b and CD11c), major histocompatibility complex (MHC) classes I and II, and CD45 (44). In the presence of specific stimuli, such as tissue damage, the so-called “damage-associated molecular pattern” released by the injured cells induce microglia to transition to a proinflammatory state. In this “classical activation,” or M1 state, microglia retract their ramifications, potentiate phagocytic activity, and increase the expression of cell surface proteins relevant for the innate immune response, such as Toll-like receptors (TLRs), inflammasome, phagocytic and scavenger receptors, and receptors for advanced glycation of end products (45–54). The M1 state is also associated with the production of cytokines and chemokines, in particular CCL2 (also named monocyte chemotactic protein-1), which is responsible for the recruitment and migration of additional microglia to the insult site (55). In the M1 state, microglia also show increased expression of phagocytic oxidase (PHOX) (56) and inducible nitric oxide synthase (iNOS) (57), as well as the increased generation of nitric oxide (NO), the main cytotoxic mediator in acute and chronic inflammatory responses (58) (Table 1).
Table 1.
Molecular Characterization of Microglia Phenotypes
| Inflammatory (M1) | Alternative Activation (M2a) | Type II Alternative Activation (M2b) | Acquired Deactivation (M2c) | |
|---|---|---|---|---|
| Function | Killing of intracellular pathogens | Extinction of inflammatory response | Immunoregulation | |
| Pathogen phagocytosis | Killing of encapsulated parasites | Engulfment of apoptotic/dead cells | ||
| Extracellular matrix degradation | Extracellular matrix deposition | |||
| Tissue remodeling | ||||
| Markers | ↑ TLRs | ↑ Polyamines | ↑IL-10 | ↑ TGFβ |
| ↑ CR1, CR3, CR4 | ↓ IL-12 | ↓ IL-12 | ↑ IL-10 | |
| ↑ CD36, CD91 | ↓ iNOS | ↑CD16 | ↓ IL-12 | |
| ↑ RAGE | ↑ IL-1ra | ↑CD32 | ↑ Versican | |
| ↑ NFκB | ↑ CD163 | ↑CD64 | ↑ PTX3 | |
| ↑ TNF-α | ↑ CD206 | ↑ MHC-II | ↑ MARCO, | |
| ↑ IL-1β, IL-6 | ↑ MHC-II | ↓ MHC-II | ||
| ↑ IL-12, IL-23 | ↑ Arg-I, Ym-1, Fizz-1 | ↑ TIMP1 | ||
| ↑ CCL2 | ↑ TREM-2 | ↑ CD163 | ||
| ↑ iNOS, PHOX | ↑CD 33 | |||
| ↑ MHC-II | ||||
| ↑ MMPs | ||||
| ↑ TREM2 | ||||
| ↑ IL-6 | ||||
| ↑ CD14 | ||||
| ↑ CD40 | ||||
| ↑ CD74 | ||||
| ↑ CD68 | ||||
| Stimulus | IL-1β, TNFα, (IL-6) | IL-4, IL-13 | LPS, IL-1β | IL-10, TGFβ |
| Refs. | 309–312 | 59, 313, 314 | 315 | 59, 313 |
Abbreviations: RAGE, receptors for advanced glycation of end products; MMPs, matrix metalloproteinases; TIMP1, tissue inhibitors of metalloprotease 1.
This host defense mechanism, which is very effective in taming inflammation, may cause local collateral damage. Thus, upon removal of the inflammatory stimuli, an elaborate and organized response is required to replace lost and damaged cells and to restructure the damaged extracellular matrix, with the final aim to restore tissue homeostasis. At this point, microglia change their phenotype and promote the blockade of the immune response and the commencement of specific programs aimed at repairing the damaged tissue (59). This activity is carried out in concert with glia and neurons and includes the synthesis and secretion of specific anti-inflammatory cytokines, which are responsible for the transition of microglia to other functional phenotypes (60) (Table 1). In particular, IL-4 and IL-13 induce the “alternative activation” (or M2a phenotype) responsible for the resolution of the inflammatory phase by indirectly repressing the production of proinflammatory cytokines and the expression of iNOS (61, 62). Ligation of Ig Fcγ receptors (CD16, CD32, or CD64) by immune complexes on lipopolysaccharide (LPS) or IL-1β primed microglia results in the “type II alternative activation” (M2b phenotype), leading to down-regulated expression of IL-12, increased IL-10 secretion, and increased MHC-II expression (Table 1). The dampening of the inflammatory response is also associated with the “acquired deactivation” phenotype (or M2c phenotype), characterized by the production of IL-10 and TGFβ, and a strong repression of MHC-II (Table 1). These cytokines account for trophic effects and tissue-remodeling functions, including remodeling of the extracellular matrix (63), angiogenesis (64), and, in neurotrophic niches, neurogenesis (65). The acquired deactivation stage can also be induced by the presence of apoptotic cells because microglia recognize the PS exposed on the surface of apoptotic neurons (66). Soluble bridging molecules, such as the adapter protein growth arrest-specific 6 (67), bind to PS through their GLA domain (N-terminal 11 γ-carboxyglutamic acid residue), thus serving as eat-me signals that are recognized by receptor tyrosine kinases (68) on the microglial membrane (Tyro3, Axl, and Mer - TAM). In chronic inflammatory diseases associated with aging, this trophic function appears to be impaired (69), possibly in relation to a decline in growth arrest-specific 6 expression. These different stages of activation do not have well-defined boundaries, but they represent a continuum among each other, and similar to peripheral macrophages, they are classified on the basis of the genes they preferentially express (Table 1).
1. Microglia—astrocyte interactions
The anatomical changes induced by injury and disease in astrocytes were described more than 100 years ago. We now know that reactive astrocytes protect neural cells and tissues by several means that include the following: 1) the modulation of synaptic activity through the uptake of potentially toxic molecules, such as glutamate (70), or the blockage of transporters for inhibitory peptides, such as γ-aminobutyric acid (GABA) (71); 2) the release of glutathione and adenosine to control oxidative damage (72, 73); and 3) the degradation of protein aggregates in the brain parenchyma (74). Microglia activation and astrogliosis are commonly observed in the case of brain injury, infection, and neurodegenerative diseases; however, we lack the necessary insight into the functions of the bidirectional cross-talk occurring between these two cell types. The seminal work by Gan and colleagues (75) based on in vivo transcranial time-lapse, two-photon imaging demonstrated that after a small laser insult, microglia near the site of injury responded within a few minutes, and microglial processes converged to the site of injury driven by the ATP released from astrocytes, recognized by the P2Y12 G protein-coupled receptors expressed by the surveying microglia (76). Prior in vitro studies led to the hypothesis that astroglia may attenuate microglia reactivity or facilitate the resolution of the inflammatory response. This would occur through the synthesis and release of GABA, which decreases microglial production of inflammatory cytokines (77) and microglia expression of antioxidant molecules (eg, hemeoxygenase-1) regulated by the erythroid 2-related factor, Nrf2 (78). In turn, initial studies point to a microglia-mediated modulation of astrocytes through the release of purines (79) or other inflammatory molecules, such as prostaglandin D2, that are known to induce astrogliosis (80). The development of methodologies for the isolation and culture of pure populations of microglia or macroglia demonstrated that astrocytes are insensitive to inflammatory stimuli, and their ability to produce regulators of the proteolytic balance (tissue inhibitors of metalloproteases) in response to molecules, such as LPS, is mediated by microglia (81). This function was shown to be necessary for the survival of neurons after ischemic insult and in demyelinating diseases (82, 83). The finding of a reciprocal regulation between microglia and astrocytes in the control of neuroinflammation demands additional studies to better explain the extent to which impairments of this two-way communication are associated with the onset of CNS disorders.
2. Microglia—interactions with other immune cells
Another important function of microglia is the presentation of foreign antigens to T lymphocytes. In the healthy brain, antigen-presenting cells are represented by macrophages and dendritic cells in the meninges, choroid plexus, and perivascular spaces. Activated microglia up-regulate the expression of the molecules that are needed for an optimal antigen-presenting cell function (84). It is still controversial whether monocytes contribute to the adult microglial population. The current belief is that although monocytes may penetrate the adult brain and differentiate into microglia, these cells are short-lived and an unlikely source for maintaining the microglia population in steady-state conditions. However, during certain neuroinflammatory pathologies (eg, multiple sclerosis [MS] or Alzheimer's disease [AD]), the recruitment of circulating bone marrow progenitors can supplement, to some extent, the microglial population (85, 86).
3. Microglia—neuron interactions
The intimate relationship between neurons and microglia in the adult brain is believed to be the recapitulation of what was already described for microglial development. In the mature nervous system, the major form of communication between neurons and microglia is the fractalkine receptor, CX3CR1, but other proteins may be involved (eg, CD200 and receptors for neurotransmitters and neuropeptides). Microglia are the only brain cells that can express high levels of the CX3CR1, a G protein-coupled receptors activated by the CX3CL1 ligand, a transmembrane glycoprotein that may be released by neurons after proteolytic cleavage as a consequence of cytotoxic or other stimuli. The activation of the fractalkine receptor serves two main purposes: 1) modulation of synaptic pruning (14); and 2) constraint of microglia activation.
Recent molecular imaging studies with two-photon laser microscopy showed that surveying microglia stop and regularly interact with all synaptic elements (the presynaptic terminal, perisynaptic process, and synaptic cleft, but not the dendritic shaft); these contacts last for a highly variable period of time (20). It is important to note that the frequency of these contacts is relative to neuronal activity. For instance, in the case of ischemia induced by transient occlusion of the middle cerebral artery, the duration of microglia-neuron contacts are significantly prolonged (from minutes to an hour). During that period, several presynaptic terminals disappear, clearly suggesting that microglia may control spine densities in relation to neuronal activity (14, 20). The study of mice deficient in complement C1q or C3 showed defects in the elimination of CNS synapses (87), supporting the hypothesis of an involvement of the complement in microglia regulation of synaptic functions (Figure 1). In addition to the CX3CR1, microglia are equipped with a plethora of receptors for neurotransmitters and neuropeptides, such as GABA, glutamate, and substance P (88), enabling microglia to sense neuronal activity and synthesize and secrete inflammatory mediators, neurotrophic factors, or modulators of its own phagocytic activity (89). When damaged, neurons may attract microglia by releasing neurotransmitters, such as glutamate (90), or other signals such as growth factors (eg, fibroblast growth factor-2) (91). Once near neurons, microglia receptors sense neuronal activity and lead microglia to participate in plastic changes at the synapse through several mechanisms (41, 89). In the case of neuronal death, microglia are rapidly activated to clear the apoptotic cell debris, which could be harmful for the bystander neurons (51, 92).
Neurons, in turn, have the means to control the transition of microglia into the inflammatory phenotype and may increase the threshold of microglial sensitivity and reactions to neurotoxic stimuli. For instance, in CX3CR1-deficient mice (93, 94), the state of microglia activation in response to stimuli, such as LPS or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), was much higher than that in wild-type (wt) animals. Depending on the circumstances, neurons may facilitate the resolution of the inflammatory status and induce microglia to synthesize trophic factors relevant for neuronal health. Thus, the neuron-microglia reciprocal modulation must be kept under a tight balance because microglia may be relevant for a homeostatic neuronal signaling, by its excessive activity may damage neurons. This was noted by several in vitro studies that emphasized the damage-exacerbating effects of microglia-derived NO after prolonged stimulation with LPS (95–97), glucose stimulation, or ischemia (98, 99). High NO levels inhibit neuronal respiration, causing the release of glutamate (96, 97), and N-methyl-D-aspartate receptor-mediated neurotoxicity is potentiated by the presence of activated microglia. This mechanism may recapitulate physiological events necessary for brain development, which in the adult healthy brain are kept under control because neurons may limit microglia negative influences by releasing substances able to induce apoptosis of activated microglia (100, 101).
All together, these observations support the vision that microglia are a very significant complement to astrocytes in the regulation of neuronal synaptogenesis, transmission, and survival (40). However, at the same time, they underline the necessity of a continuous control of microglia functions for the activity of neurons.
C. Energy metabolism and neuroinflammation
Diet-induced obesity is associated with neuroinflammation (102), heightened cytokine levels in several brain regions (103, 104), hippocampal synaptic malfunctioning (105), altered neurogenesis (106), and cognitive impairment (103, 105). In rodents, within the first week of consuming a high-fat diet (HFD), markers of neuronal injury were observed in the arcuate nucleus of the hypothalamus and in the adjacent median eminence that were associated with reactive gliosis involving both microglia and astroglia (107). This effect was reversible and was generated by saturated, but not unsaturated fatty acids (107), indicating the existence of a selective mechanism. However, a continued exposure to a HFD led to a permanent activation of microglia (but not astroglia) in the mediobasal hypothalamus that was observed also in humans by means of magnetic resonance imaging (108). Exercise reduces neuroinflammation, and this could be due to an improved glucose tolerance (109). However, considering that the effects of HFD were circumscribed to the brain regions responsible for the control of energy homeostasis and that the peptides αMSH and neuropeptide Y induce cytokine and NO production by microglia, it is tempting to speculate that the activity of anorexigenic and orexigenic neurons in response to an unbalanced diet may be the trigger for microglia activation (78, 95, 110, 111). A better understanding of this phenomenon is relevant for human health because obesity and metabolic syndrome are important risk factors for the development of AD (112–115), and obese patients show deficits in learning, memory, and executive functioning (116).
Most interestingly, in animal studies, young females were shown to be refractory to diet-induced neuroinflammation; ERα knockout female mice fed a chronic HFD behaved similarly to wt males. This suggests that estrogens and their receptors may regulate the neuroinflammatory response, and in females, circulating estrogens may play a protective, anti-inflammatory role. What remains to be established are the mechanisms through which estrogens may modulate microglial inflammatory responses: estrogens may directly regulate the inflammatory genes, but may also regulate the production of compounds (such as neuropeptide Y) triggering inflammation in the hypothalamic peptidergic neurons, which are known to express ERα and to be susceptible to the actions of estrogen (117, 118).
D. Microglia and aging
Aging is the major risk factor for the development of neurodegenerative diseases, and several large-scale genetic studies have implicated microglial molecules in sporadic forms of neurodegenerative disease, thus giving strength to the hypotheses of a prominent role of neuroinflammation in the onset and progression of neurodegenerative disease. The general consensus is that, in the adult brain, microglia protect and defend other neural cells from pathological insults, but we know very little about microglia efficiency in a senescent brain. Therefore, the compelling question is whether senescent microglia are able to maintain their proliferative and brain-patrolling capacity together with their responsiveness to pathological insults. As previously mentioned, the microglia migrated from the embryonic yolk sac proliferate and self-renew throughout the entire life span. This suggests that microglia live for long periods and may somewhat undergo senescence by losing their efficiency and depriving the brain of its natural defense (119, 120). The finding that with aging there is an increased density of microglia in several brain regions (121, 122) suggests that the capacity to proliferate is maintained in time. This poses the question as to whether such a lifelong process leads to telomere shortening and the loss or gain of functions associated with the replicative senescence. In fact, we still know very little about the existence of the stem-like microglia progenitors proposed by Elmore et al (11), even if the heterogeneous distribution of microglia in the aging brain might support the view of subpopulations of cells throughout the parenchyma (122, 123). The few studies addressing the analysis of microglia morphology in aging brains show that aged human and rodent microglia are less ramified, with more tortuous processes carrying some bulbous swellings (43, 124). The motility of microglial processes appears to be diminished by age, as was indicated by in vivo imaging studies (125, 126) and by transcriptomic analysis, which found that young microglia express more motility genes than old microglia (127). Immuno-phenotyping and biochemical studies established that aging is associated with a general up-regulation of markers that are typical of the proinflammatory state, and microglia are more readily responsive to toxic insults (128–131). On the other hand, at least in rodents, markers of the microglia anti-inflammatory state were shown to be increased with age (124, 128). The studies conducted so far may at times appear contradictory because microglia might respond very differently in relation to the pathological context in which they are studied. Therefore, conclusive results could be drawn only from studies carried out in healthy aged brains (Figure 3). However, the ability to engulf and degrade the extracellular material resulting from the phagocytosis process remains underinvestigated in senescent microglia. Histological studies in the brains of aged, healthy humans or animals have shown accumulation of protein aggregates in the parenchyma. However, it is unclear whether this is the consequence of an age-dependent abnormal production of aberrant proteins or their lack of clearance due to a decreased microglia phagocytosis. It is likely that both mechanisms are occurring because it is conceivable that aging correlates with a decreased ability of microglia to proteolytically digest the engulfed protein aggregates and debris from the surrounding space with a consequent impairment of the phagocytotic process (132). In addition to brain local events triggering microglia activity, the generalized, systemic inflammation that accompanies aging in mammals may provide a significant contribution to inducing a proinflammatory, dysfunctional state of these cells that is also facilitated by the increased permeability of the brain–blood barrier described in aged organisms (84, 133–135).
Figure 3.

Morphological and functional elements point to alterations of microglia activity with age.
III. Mechanisms of Estrogen Actions in Microglia
A. Estrogens may modulate target cell activity by interacting with several receptors
1. Intracellular receptors ERα and ERβ—structure
In mammals, two isoforms of the ER have been described. They are referred to as α and β, each encoded by a separate gene (ESR1 and ESR2, respectively). The structure of the two receptors is very similar; however, the functions of the two may differ considerably in different cell systems (136) (Figure 4).
Figure 4.

Structure and PTM sites of the nuclear ERs. Like all steroid receptors, the ERs belong to a family of hormone-modulated transcription factors characterized by the presence of six functional domains: A and B, the N-terminal domain that contains the activation function 1 (AF-1) enabling the interaction with coregulators also in the absence of the ligand. C, The highly conserved DBD, responsible for the recognition of specific DNA sequences (named estrogen responsive elements or EREs) through the two Zn fingers. D, The hinge region, a flexible domain that connects DBD with the ligand binding domain (LBD) able to influence intracellular trafficking and subcellular distribution. E, The LBD responsible for ligand recognition that contains the ligand-dependent activation function 2 (AF-2); the LBD, contributes to the dimerization interface of the receptor in concert with the DBD. F, The C-terminal domain that participates in the binding to ligands. Both ERs undergo a large number of regulatory PTMs exemplified in the figure (human ERα and murine ERβ).
2. Intracellular receptors ERα and ERβ—functions
a. Inhibitory proteins.
In the absence of the cognate ligand, ERs are in a complex with proteins that prevent the receptor binding to the DNA (heat shock proteins, Hsp90, Hsp70, and other chaperons). The complex is mainly, but not exclusively, localized in the cell nucleus (137, 138). Upon binding the cognate ligands, these receptors undergo conformational changes that lead to the release of inhibitory proteins, thus unmasking the DNA binding domain (DBD).
b. Post-translational modifications.
ER activity, before and after DNA binding, is regulated by a constellation of post-translational modifications (PTMs). These modifications include the following: phosphorylation, acetylation, methylation, sumoylation, and palmitoylation (139). This large variety of PTMs regulates the half-life of the receptor proteins, as well as their cellular localization (140) and their ability to interact with DNA and other signaling proteins (141). Thus, PTMs are necessary to tune the receptor functions in relation to cues present in the host cell and in the whole organism. Moreover, initial studies have demonstrated that the state of PTM of ERs is highly plastic and significantly regulated by the hormonal milieu. To provide an example of the multiple consequences of PTM, ERα phosphorylation (which may occur at 10 different serine/threonine/tyrosine residues) is necessary for the receptor dimerization and the recruitment of specific transcription factors, such as p160 coregulators with chromatin-remodeling enzymes (142).
c. ER activation in the absence of natural or synthetic ligands.
Ligands are generally required to activate sex hormone receptor transcriptional activities. However, it is now well established that these receptors may also be activated in the absence of a ligand (unliganded activation). This phenomenon, initially proposed by O'Malley's group (143, 144) for the PR, was then supported by a large series of observations in other nuclear receptors including ERs. It is now well accepted that unliganded ERs can be activated by growth factors (such as epidermal and the insulin-like growth factors) (145–147) through the involvement of selected kinases (MAPK, protein kinase A, and p21 ras/ERK) (148–151). A series of biochemical and genetic studies suggested the existence of cell-specific phosphorylation sites required for unliganded receptor activation (eg, Ser 118 in COS-1 cells and Tyr 537 in neuroblastoma cells) (152).
d. Protein-protein interactions.
ER PTM involves their interaction with other proteins, such as calmodulin, cyclin D1, BRCA-1, and transcription factors (eg, c-fos, c-jun) that may modify receptor functions (153, 154). The interaction with coregulators is essential for the modulation of target gene transcription because it enables the recruitment of general transcription factors to the TATA box and histone modification to facilitate RNA pol II transcription of the target genes (155, 156).
e. ER intracellular signaling.
Once activated by ligands or by PTM, ERs regulate the activity of their target cells by several mechanisms that include the following: 1) dimerization that enables recognition and binding of estrogen response elements (EREs) in the promoter of target genes and interaction with coactivator and corepressors to promote/repress transcription; 2) binding to other nuclear transcription factors (eg, activator protein 1 [AP-1], nuclear factor-κB [NF-κB]) interfering with their transcriptional capacity; and 3) binding to cytoplasmic molecules involved in signal transduction (eg, Src, phosphatidylinositol-3-kinase [PI3K], signal transducers and activators of transcription) and alterations of their signaling (157).
3. Intracellular ERα and ERβ may translocate to the cell membrane to regulate specific cell functions
Monomers of the classical intracellular ERα and ERβ may be induced to migrate to the cell membrane and associate with caveolae (158) by serine palmitoylation (C451/447 for mouse/human ERα) (140). In the cell membrane, estrogen binding induces dimerization of the ER and rapid signaling through Gα and Gβγ proteins (159). The association with G proteins was shown to occur in a cell- and context-specific mode to provide the appropriate cell response to various stimuli. A rapid process of depalmitoylation regulates the length and extent of this signaling (160). The number of intracellular receptors that are palmitoylated and transported to the membrane is a fraction of the total (approximately 5–6% of all ERs), yet it is sufficient to have a significant impact on glucose and lipid metabolism in different cell types (161, 162). The recent generation of a mouse with a mutation at aa 451 (C451A) finally demonstrated that this receptor localization is essential at least for ovarian functions (163). Nevertheless, little is known so far about its functions in the CNS.
4. The pharmacology of intracellular ERα and ERβ
In the last 50 years, a number of synthetic ER ligands were generated and developed for clinical use. These ligands include the following: clomiphene, tamoxifen, toremifene, raloxifene, bazedoxifene, and ospemifene. The common characteristic of these ER modulators is that they were selected to circumvent the use of natural estrogens due to their potential effects on endometrial and breast cancers. These compounds were named selective ER modulators (SERMs) because they bind the ER, but their agonist-antagonist effect is tissue dependent. Thus, most of the SERMs have antagonistic actions on ER in reproductive tissues. The first and second generation SERMs, tamoxifen and raloxifene, are used to treat ER-positive breast cancer and postmenopausal osteoporosis, respectively. The third-generation SERM, bazedoxifene, effectively prevents osteoporosis while blocking estrogenic stimulation in breast and uterine tissues. Unfortunately, the specific estrogenic vs antiestrogenic effects of SERMs on neuronal, glial, or microglial cells have not been fully determined. The ability of these ligands to exert a tissue-specific action is attributed to the fact that by binding the ligand pocket of the ER, they induce conformational changes that are quite different than those of the 17β estradiol (E2)-ER complex, this possibly limits their ability to interact like natural estrogens with the coregulators (164). Selective ER down-regulators (or pure antiestrogens), an alternative to SERMs, are characterized by a different activity because they cause down-regulation and degradation of ERs. The prototype of a selective ER down-regulator is fulvestrant (ICI 182,780). More recently, the identification of microRNAs that can directly target ERs raised major interest, particularly in the cancer field. The mechanisms involved in the micro RNA-dependent modulation of ERs varies among the microRNAs isolated; most regulate the content of ERα indirectly; others, such as miR221–222, were shown to target ERα 3′-untranslated region and decrease ERα protein, but not mRNAs (165). A major advance in the field of synthetic ER ligands occurred recently with the identification of compounds that can discriminate the two ER isoforms and selectively bind to ERα (as its agonist PPT [1,3,5-tris (4-hydroxyphenyl)-4-propyl-1H-pyrazole] and antagonist side-chain pyrazoles) (166) or ERβ, eg, the selective agonist DPN [2,3-bis (4-hydroxyphenyl) propionitrile]. Unfortunately, the use of SERMs to target neural cells for clinical application is quite limited to date, due to still poor knowledge of the precise molecular targets of these molecules in the CNS and the lack of pharmacokinetic data reporting the levels of permeability across the blood-brain barrier (BBB) (167).
5. Membrane receptors—GPR30
With a structure completely different from the intracellular receptors previously described, another molecule, an orphan 7-transmembrane receptor, GPR30, was found to be able to recognize and bind estrogens. GPR30 is a G protein-coupled receptor that can bind 17β-estradiol in the nanomolar range, thus with an affinity for the hormone approximately 10-fold lower than the intracellular receptors (168). GPR30 is present in the cell membrane and in the endoplasmic reticulum. In different model systems, the activation of GPR30 is mediated by 17β-estradiol and has been associated with several functions, including Ca2+ mobilization (169–171), cAMP production (168), activation of protein kinases (171, 172), activation of specific ion channels (173), and modulation of gene expression (174). Yet, the mechanisms underlying the intracellular activities of GPR30 are still unclear. A recent study showed that upon activation, the GPR30 forms hetero-oligomer complexes with Ca2+-ATPase and inhibits its activity through tyrosine phosphorylation of the pump (175). It is likely that the receptor may interact with several intracellular signal transducers through mechanisms that may change depending on the cell type. GPR30 is expressed in macrophages and in microglia (both primary cultures from neonatal rat brain and BV-2 cells), where it was shown to inhibit the production of cytokines and oxidative stress-related genes after stimulation by LPS (176) or hypoxia (177). GPR30 pharmacology has been the subject of several studies. In breast cancer cells, GPR30 is activated by the ER antagonist ICI 182,780 (178), tamoxifen (179), and selective ligands for GPR30 have been identified and tested in several systems. The most investigated synthetic ligands for GPR30 are currently two steroids known as G-1 (agonist) (180) and G-15 (antagonist) (181, 182); both of these ligands can readily cross the BBB.
B. Which ERs are expressed in microglia in the mature, adult brain?
The presence of ER in microglia was reported by several authors, and a large number of reports highlighted the anti-inflammatory action of estrogens in microglia cells, cultures, and living animals. However, the abundance and type of ER expressed in microglia remains an object of discussion because of the large discrepancies in reports from different laboratories. Three major elements contribute to the inconsistencies in the literature.
1. Source of microglia and culture conditions
Microglia studies are carried out in retroviral immortalized cells: primary cell cultures from the neonatal or adult brain, pluripotent stem cells differentiated in vitro, and cells directly dissociated from embryonic, neonatal, or mature brains with a variety of methodologies. Functional (183) as well as more recent genome-wide transcriptomic studies (184) demonstrated substantial changes in the activity and transcriptome of fluorescence-activated cell sorting (FACS)-sorted or cultured microglia and in microglia in the different stages of activation. This is not surprising in view of the plasticity of these cells, and it suggests that the expression of ERs is likely to change depending on the model system utilized. Indeed, studies carried out in microglia cell lines showed that ERα and ERβ mRNA content changes significantly with the number of passages (185). Considering that the growth factors present in the serum and the estrogenic activity of phenol red may activate ERs, it is conceivable that the culture media may represent a further element affecting the expression of these receptors.
2. Microglia sex
Generally, when primary cultures are established, both sexes are utilized, and even for the available cell lines, the sex of origin (N9 and M4T.4 are male and BV-2 and C8-B4 are female) (185) is not taken into consideration.
3. Current technology for the quantitative analysis of ER gene expression
Our ability to verify the expression of ERs by immunohistochemical methodologies has been hampered by the minute dimensions of these cells, the low concentration of these receptors, and the lack of a reliable and constant source of high-affinity antibodies.
The analysis of the literature on whole-genome direct sequencing of microglia from adult brains clearly shows the presence of ERα, but not ERβ, mRNA (Table 2). The relative concentration of this mRNA is comparable to that for the mineralocorticoid receptor (MR) and is considerably lower than that for the mRNAs encoding the glucocorticoid receptor (GR), whereas no AR was found (Table 2). These data appear to be consistent with regard to the relative abundance of the different receptors, possibly because all studies utilized microglia dissociated from adult brains. Unfortunately, at present no data are available from the neonatal brain and primary cultures. In keeping with these findings are the results of the studies by Sierra et al (27), who first published a systematic analysis on the presence of selected steroid receptors in microglia isolated from the adult brain by cell sorting. Sierra et al (27) reported the presence of ERα, GR, and MR but no detectable expression of ERβ, AR, and PR. Staining with ERα antibodies revealed that this receptor is expressed mostly in the cytoplasm near the nucleus and in the cell processes (27, 186). Interestingly, not all microglia cells were stained, and the use of electron microscopy enabled visualization of labeling for ERα in microglia processes in close apposition to neuronal dendritic spines. Immunodetection of ERs in neonatal cultures of microglia showed both ERα and ERβ (138). The finding that ERα is low at P3 and increases rapidly to reach adult levels at P21 (26) suggested that the expression of these receptors may change in response to environmental cues. After treatment with LPS, ERα (together with GR and MR) is down-regulated, providing evidence that the functional status of microglia may influence the expression of these genes, which in adult animals does not appear to be influenced by the hormonal status or sex (27, 187). The high reproducibility of these results in different laboratories sheds light from earlier reports on the presence of ERβ or AR expression in microglia from healthy adult brains (188, 189). Nevertheless, ERβ may be expressed in microglia isolated from the spinal cord (190). Transformed cell lines (eg, the murine BV-2 and N9 cells) express ERβ, and ERα mRNA is not present (BV-2) (191) or is present at low levels (N9) (28, 192). Direct RNA sequencing of BV-2 transcriptome failed to detect mRNAs for all ERs (193); however, in this latter report, the low expression of GR suggests that the sensitivity of the assay used was not optimal when compared to other whole-genome RNA sequences.
Table 2.
Nuclear Receptor Expression in Microglia
| Source of Microglia | Method of Isolation | mRNA Content, RPKM |
Refs. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ERα | ERβ | GPR30 | AR | PR | GR | MR | |||
| BV-2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.0 | 0.0 | 193 | |
| Adult mouse whole brain | CD11b + magnetic separation | 1.5 | 0.0 | 1.5 | 0.1 | 0.0 | 42.2 | 3.5 | Maggi et al, unpublished data |
| Adult mouse whole brain | Percoll/FACS | 1.4 | 0.0 | 0.3 | 0.0 | 0.0 | 196 | 0.0 | 195 |
| Mouse brain cortex | FACS | 3.4 | 0.0 | 0.6 | 0.0 | 0.1 | 11.9 | 1.8 | 194 |
| Mouse brain cortex | Single cell RNAseq | 0.02 | 0.00 | 0.05 | 0.00 | 0.00 | 1.5 | 0.05 | 316 |
| Spinal cord | Percoll/CD11b-magnetic separation | 0.1 | 0.0 | n/a | 0.0 | 0.0 | 2.9 | 0.6 | 317 |
Abbreviation: RPKM, reads per kilobase of transcript per million mapped reads.
With regard to GPR30, the results of several whole-genome RNA sequencing data (194, 195) point to its expression in the adult brain microglia, and immunostaining experiments carried out in rat microglia from the neonatal brain showed its presence (176).
C. Estrogen activity in microglia
We are at an early stage in our understanding of estrogen influences on microglia activity, and the limited number of studies available have concentrated on the anti-inflammatory actions of estrogen. Very little is known with regard to the role of this hormone in the microglia functions reported above. For instance, considering the relevance of estrogens in shaping neuronal circuitries during the sexual differentiation of the CNS, it is surprising that the no investigator addressed the study of the role of estrogens in microglia-dependent synaptic pruning. Similarly, very little attention has been given to the effects of these hormones on microglia trophic and repair abilities, despite the well-known neuronal protective actions of estrogen. The following paragraphs will review the current literature on the anti-inflammatory effects that estrogen has in microglia.
1. Estrogen blockade of microglia activation after acute stimulation with inflammatory stimuli
There is a general consensus on the ability of estrogens to limit the microglia proinflammatory status after short exposure to bacterial lysates (196), viruses (197), unmethylated CpG oligonucleotides (198), or hypoxia (199, 200). The hypothesis of the anti-inflammatory potential of estrogen was based initially on the in vitro observation that 17β-estradiol prevented the morphological changes induced by LPS and the concomitant synthesis of proinflammatory molecules (such as matrix metalloproteinase 9, prostaglandin E2, iNOS with reactive oxygen species [ROS] production) (196). These findings were subsequently reinforced by investigations on the anti-inflammatory potential of the synthetic ligands of ERs, such as tamoxifen and raloxifene, and natural estrogens, such as genistein, daidzein, and kaempferol, that were shown to attenuate the β-amyloid peptide or LPS-induced microglia proinflammatory phenotype by inhibiting the synthesis of TNFα, IL-1β, monocyte chemotactic protein-1, or macrophage inflammation protein-2 (MIP2) in a dose-dependent manner (138) or the production of other inflammatory molecules, such as NO and ROS. These effects were blocked by prior treatment with ICI 182,780 (201–203). Most of these studies were performed in BV-2 cells or primary cultures of microglia, either alone or mixed with astrocytes. Which of the two intracellular isoforms of ER is responsible for the anti-inflammatory properties of estradiol remains controversial, and both ERα and ERβ may trigger anti-inflammatory responses in the presence of high concentrations of ligand. The use of isoform-specific modulators, such as PPT or DPN, showed that PPT-activated ERα was more effective than DPN-induced ERβ for the inhibition of microglial production of IL-1α, IL-1β, TNF-α, and cycloxigenase 2 (COX-2). This suggested that ERα plays a more significant role than ERβ in diminishing the inflammatory response of microglia. However, an increase in ERβ expression after treatment with 17β-estradiol or DPN in rat primary microglia provided greater attenuation of NO production, thus suggesting that ERβ also plays a role (204).
2. Estrogens and neuroinflammation—in vivo experiments
In vivo studies provided further, strong evidence on the capacity of estrogens to inhibit the neuroinflammatory processes. Ovariectomy (ovx) in rodents is clearly associated with more microglia with a proinflammatory morphology and up-regulation of a large number of markers of microglia reactivity (including the receptors for the recognition of inflammatory stimuli and for phagocytosis) (131, 205). The fact that the administration of estradiol before ovx blocked microglia activation suggested that the neuroinflammatory reaction after the surgery was mainly due to the lack of this hormone (131, 205, 206). Augmented expression of inflammatory markers was also observed in women in postmenopause, particularly in areas of the brain functionally related to the regions shown to be most responsive to inflammatory stimuli in rodents.
The extent of microglia activation induced by ovx is exacerbated by aging. In fact, in ovariectomized old mice, the expression of inflammatory mediators is much stronger than in intact mice of the same age. This indicates that low circulating estrogens increase the susceptibility of senescent microglia to inflammation (207). Most interestingly, studies with a mouse reporting on the transcriptional activity of ERs (ERE-Luc mouse) (208) showed that the receptor transcriptional activity in the hippocampus diminished significantly with aging, although the synthesis of ERα mRNA was increased and circulating levels of estrogens remained quite high (Figure 5). It is therefore plausible that with age a form of estrogen resistance is involved in the impaired ability of microglia to resolve inflammation, possibly leading to an ever increasing neuroinflammatory phenotype. Age is correlated with increased inflammation in males as well. Additional studies are necessary to understand the state of ER activity and local estrogen production to evaluate the contribution of these hormones to the activity of senescent microglia in males. The in vivo local or systemic administration of the endotoxin LPS is a commonly used model system for triggering a robust but transient inflammatory reaction in the rodent brain and is a valid tool to evaluate the effect of circulating estrogens in the prevention of acute microglia inflammatory activation. Intraventricular or intraparenchymal injection of LPS demonstrated that the estrogen anti-inflammatory actions occurred in all brain areas studied and required the presence of ERα (209). The systemic administration of LPS enabled the authors to demonstrate that a peripheral inflammation may trigger a response also in the CNS with a rapid activation of microglia and expression of inflammatory cytokines. The same experimental setting performed in intact and in ERα or ERβ knockout mice indicated that ERα was more effective than the ERβ isoform in dampening the local production of inflammatory molecules; ERβ was required for the suppression of BBB permeability (210). The lower number of immunoreactive microglia cells in mice treated with estrogens and LPS compared with mice treated with LPS alone was observed in both male and female mice (186). Similar to estrogens, the acute administration of tamoxifen and raloxifene to ovariectomized young and aged mice reduced microglia activation after LPS stimulation (186, 198) or brain and spinal cord injury (211–213); this pointed to the fact that these SERMs have an agonist activity on microglia ER. Long-term treatment with 17β-estradiol or raloxifene in old ovariectomized females significantly decreased the number of microglia cells in the hippocampus (207) compared to placebo, suggesting that estrogens and SERMs may be considered as protective treatments against age- and disease-related pathologies.
Figure 5.
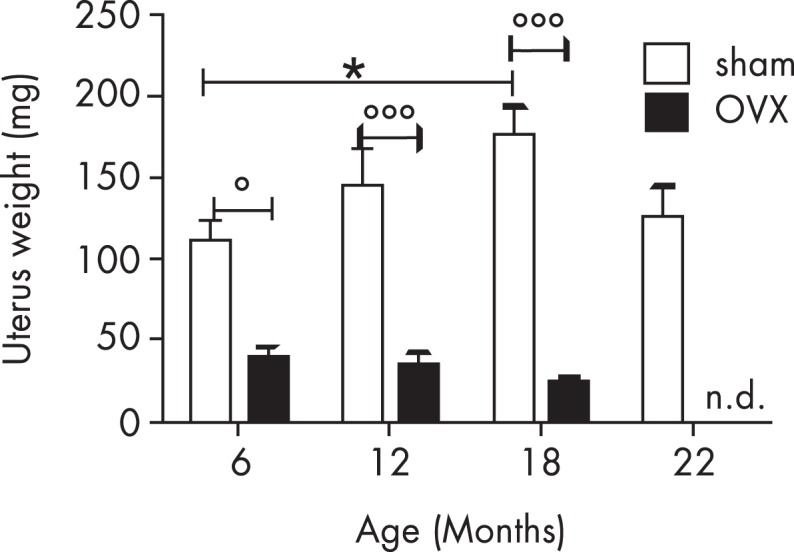
Aging effects on circulating estrogens. The uterus weight as a biomarker of circulating estrogens shows that, in mice, the activity of the ovaries does not decrease with age; actually at 18 months, when mice are not cycling, the plasma content of this hormone is higher than in young, fertile animals. Ovx clearly decreases the circulating levels of the hormone, showing that organs other than ovaries give a minimal contribution to steroidogenesis. °: P < 0.05; °°°: P < 0.001; *: P < 0.05; n.d., not detected.
3. Cellular mechanisms of ER-dependent anti-inflammatory activity
A detailed knowledge of how mammalian innate immunity is regulated has developed over the past 15 years. Membrane receptors and endosomal TLRs activated by a series of small molecules (derived from parasites, bacteria, fungi, and viruses) may dimerize to initiate a cytoplasmic response leading to the activation of the transcription factors responsible for the induction of proinflammatory cytokines, and type I IFN. Two important families of transcription factors activated downstream of TLR signaling are the NF-κB and IFN-regulatory factors. Other transcription factors, such as cAMP-responsive element-binding protein and AP-1, are also important (214). Currently, the understanding of the molecular mechanisms of estrogen anti-inflammatory actions is incomplete because of the multiplicity of responses elicited by estrogens in the neural, glial, and immune cells in the brain and the variability in the microglial experimental systems used (as explained in III B). Nevertheless, several lines of evidence indicate that estrogens and ERs control TLR signaling in myeloid cells (215, 216). EREs are present near genes encoding selected TLR (217), and studies in microglia and macrophages obtained from wt and genetically modified mice have demonstrated that deletion of the ERα DNA binding site blocks pathogen-associated molecular pattern/damage-associated molecular pattern-induced (PAMPs) up-regulation of TLR (218, 219). Aside from this activity, which suggests that estrogens increase the ability of microglia to respond to noxious stimuli, we also know that estrogens inhibit the production of inflammatory cytokines by interfering with TLR signaling through NF-κB and AP-1. Repeated studies have shown that p65 binding to its target genes is impaired by estrogens through a nongenomic pathway involving modulation of the PI3K-dependent pathway (220). The hypothesis that ERα inhibits NF-κB activity by inducing the synthesis of its inhibitory protein, IkBα, remains controversial. Similarly, AP-1 may be involved in the actions of estrogen because p85 PI3K signaling is involved in the estrogen-dependent blockade of TLR4 in macrophages. Estrogens can block the activity of p38 by interfering with its phosphorylation (221), but whether this occurs through direct binding to the intracellular ER or through other mechanisms has not been investigated. These results support the hypothesis that estrogens act by reducing the inflammatory response. However, more recent findings indicate the possibility that estrogens exert a more widespread effect on macrophage activity by controlling their ability to transition among different activation stages. Using time-lapse microscopy measurements of inflammatory cytokine production, one study demonstrated that estrogens may accelerate the resolution of LPS-induced inflammation by blocking IL-1β synthesis and increasing production of the anti-inflammatory IL-10 (222). The mechanism involved is of particular interest because in the absence of lymphoid cells that produce IL-4 to quench the inflammation, ERα would induce synthesis of suppressor of cytokine signaling 3 (SOCS3) through direct regulation of the Socs3 gene promoter in microglia. SOCS3 is a transcription factor that is instrumental for the synthesis of IL-10, the main cytokine involved in the onset of the acquired deactivation status (223). Thus, through this action, estrogen would augment the intrinsic ability of macrophages to end the proinflammatory phase (Figure 6). This anti-inflammatory activity of ERα would be even more valuable in the presence of other inflammatory cells that can terminate macrophage inflammation by secreting IL-4. In fact, this cytokine considerably increases the number of ERs in macrophages, therefore enhancing their anti-inflammatory potential. Thus, these studies indicate that the presence of the hormone estrogen and its ERα isoform facilitates both intrinsic and extrinsic programs for the resolution of inflammation and the direction of the LPS-stimulated immune cells toward the IL-10-dependent phenotype (acquired deactivation) responsible for tissue remodeling and restoration of homeostatic conditions (222).
Figure 6.

Estrogen and microglia functions. Estrogens regulate microglia inflammatory potential by interfering with the process of NF-κB activation (left) and by facilitating the transition to the stages where microglia exert neuroprotective functions (right), possibly including the maintenance and pruning of dysfunctional synapses (bottom).
This estrogen activity is particularly valuable in the case of chronic inflammation and in aging brains, where the maintenance of the microglial proinflammatory status may cause neuronal damage and could thus provide an explanation for the neuroprotective effects of estrogens demonstrated in models of neuronal injury and neurodegeneration (224, 225).
IV. Estrogens: Protective or Risk Factors in Brain Injury and Neurodegeneration?
Numerous studies using animal models in in vitro explant cultures or in observational studies and clinical trials involving humans have suggested that ovarian hormones play an important role in providing women protection against stroke and neurodegenerative diseases (157, 226–229). However, the mechanisms that enable such effects have not been fully elucidated. Dissecting the cell types targeted by estrogen has been slowed by the fact that ERs are expressed by all neural cells, and the neuroprotective effects likely result from receptor activation in more than one cell system. Furthermore, the cells involved may change depending on the nature of the disorder. This has broad implications for the selective targeting of ERs in the treatment of neurodegenerative conditions due to disease or injury, particularly in aging and in the postmenopause.
A. Estrogens and stroke or hypoxic neuronal death
In experimental models of stroke (middle cerebral artery occlusion [MCAO]), 17β-estradiol attenuated cell death resulting from ischemic injury and promoted neuronal survival and tissue integrity (157, 226). Because stroke activates a significant microglial reaction, the question raised by these findings was whether the neuroprotective effects of estrogens are dependent on their anti-inflammatory activity and their ability to modulate the synthesis of neuroprotective factors, such as IGF-1, in microglia (230). In this experimental model, the protective effect was observed only when estradiol was administered immediately rather than weeks after ischemia. This may suggest that estrogens should be present at high levels when microglia become activated; therefore, its primary target should be microglia. However, the role that microglia play in MCAO was clearly demonstrated using the Cre-loxP system, which selectively deletes ERα in cells of myeloid lineage (Cre recombinase under the control of the lysozyme M promoter) or in neurons (Cre recombinase under CAMKII promoter). The neuroprotective role of 17β-estradiol in MCAO was maintained only in mice that possessed monocytes without ERα, thus leading to the conclusions that neuronal ERα mediates the neuroprotective role of estrogens and that microglia ERα is dispensable, at least in stroke. The study was performed in male and female mice showing superimposable outcomes (231). However, it is worth noting that the results obtained with this model have been the subject of discussion because the recombination in microglia does not appear to be very efficient (232).
B. Demyelinating diseases
MS is a demyelinating disease characterized by a strong inflammatory component that is the main contributor to myelin sheath destruction and an ensuing progressive paralysis. The fact that MS affects women twice as often as men and that women may undergo clinical remission in the late stages of pregnancy suggests that sex hormones play a role in the development of this disorder (233). Indeed, clinical data and studies in animal models of MS (eg, experimental autoimmune encephalomyelitis [EAE]) support this hypothesis by demonstrating that estrogens ameliorate EAE severity in both males and females (234, 235). However, the mechanisms through which estrogen exerts its beneficial effects in MS requires further investigation because ERs are present in all neural cells affected by MS, including neurons, oligodendrocytes, Schwann cells (236), and microglia. Considering the strong neuroinflammatory component of this disease, estrogen could act in microglia by lessening its inflammatory reaction or minimizing the infiltration of circulating lymphocytes and monocytes. This has been investigated in the EAE model using genetic and pharmacological approaches. Most studies based on the administration of isoform-specific ligands of ERs (237) and ERα knockout mice as myeloid cell donors (238) indicated a key role for ERα in the protective effects of estrogen in EAE. However, ERβ also may play a role in demyelinating disorders (239, 240). The discrepancies in these previous results and conclusions may be due to differences in the experimental model used (EAE; or demyelination induced by Theiler's virus or cuprizone), the time at which the analysis was carried out, and the fact that the two receptors may have different functions. As suggested by the work of Brown et al (210), the major involvement of ERβ might be relative to its capacity to control the permeability of peripheral cells through the BBB. The activated ERβ might facilitate peripheral lymphocyte migration into the CNS by secreting the ILs that are necessary for dampening neuroinflammation. Indeed, studies performed in B cell-deficient mice have shown that IL-10 administration significantly improves the pathology (241). Finally, it is important to emphasize that not all MS animal models are applicable for studying the effects of sex hormones. For instance, cuprizone administration disrupts the estrous cycle, limiting the ability to establish sex differences (242).
C. Neurodegenerative diseases
1. Alzheimer's disease
Dementia is present in 16% of women and 11% of men aged over 71 years. This higher incidence in women was observed previously in age-matched groups, from 60–64 years up to 95 years of age. Therefore, it cannot be attributed to women having greater longevity. β-Amyloid accumulation is greater in women (243, 244) than in men (245, 246). This appears to be a characteristic feature of AD because no evidence of sex prevalence has been reported for mild cognitive impairment (MCI) or frontotemporal lobar degeneration. Most animal models of AD (Tg2576, APPswe/PSEN1E9, APP23, APPswexPS1, and 3xTg-AD) reproduce the same sex specificity of Aβ accumulation and show a poorer behavioral performance than those reported in humans (247–251). It remains to be established whether the lack of ovarian function plays a role in the sex-related differences in the incidence of AD. In the sporadic forms of AD, the association of homozygous single nucleotide polymorphisms of the genes ESR1 (rs9340799, rs2234693, rs2228480) and ESR2 (rs4986938) with apolipoprotein E4 (ApoE4) (the best established genetic risk factor for AD) (112, 252, 253) conferred an increased risk of cognitive impairment in both sexes, with a higher prevalence in women. The explanation for the sex dimorphic effect of this association may reside in the fact that estrogen affects cholesterol and lipid transport, and in the brain, estrogen regulates the expression of low-density lipoprotein receptor-related protein, which has been implicated in Aβ processing. These observations suggest that an impaired ER signaling may constitute a predisposing factor to AD, but by itself, it is not sufficient to increase the risk of developing AD. Nevertheless, an understanding of how a lack of estrogens can modify the course of the disease would be extremely valuable from both therapeutic and social standpoints.
For many years, ApoE4 has been considered the best known risk factor for AD pathology, and it accounts for only 10–20% of the sporadic AD risk. More recently, several independent genome-wide association studies have identified new common variants associated with sporadic AD (253). These findings have contributed to the diverging focus of the AD pathogenesis from the classical Aβ-centric view toward neuroinflammation (254–261). In fact, most of the genes associated with sporadic AD encode proteins relevant to immune cell functions (eg, CD33 [Refs. 257, 258, 262], CLU, BIN1, PICALM, CR1, CD2AP, EPHA1, ABCA7, MS4A4A/MS4A6E [Refs. 254–258], and TREM2 [Refs. 259, 260]). For instance, the R47H variant of the TREM2 (triggering receptor expressed on myeloid cells 2) gene was linked to the onset of AD with a probability comparable to that for ApoE4 (259–261). Epidemiological studies further argue for a relevant role of neuroinflammation in AD because pathologies characterized by high levels of inflammation, such as vascular disorders and metabolic diseases, increase the risk and prevalence of AD (112, 115, 116). For example, the risk for AD is augmented by 60% in patients with diabetes mellitus (114). Thus, the influence of estrogens on microglial functions may play a role in AD. In animal models of AD, 17β-estradiol increased microglia viability in vitro and in vivo, whereas in humans, 17β-estradiol enhanced the uptake of Aβ in human cortical microglia (263), possibly by increasing the expression of the complement protein C3 (264), which plays a pivotal role in cytokine-induced activation of microglial phagocytosis (265). Finally, estrogens were shown to up-regulate microglial proteasome activity through the p42/44 MAPK pathway, which is critical for a rapid and efficient turnover of oxidized or otherwise damaged proteins and therefore maintains microglial homeostasis in response to Aβ-induced activation and metabolic stress (266, 267). In APP23 mice overexpressing human amyloid precursor protein with the Swedish mutation, ovary ablation increased microglia activation at Aβ deposits and facilitated the progression of these cells toward a highly reactive state (206). Long-term administration of 17β-estradiol blocked this effect and decreased microglia reactivity compared to control animals. In the same study, estrogens were shown to inhibit Aβ-induced expression of the scavenger receptor in macrophage cells. Considering that estrogen facilitates the resolution of the inflammatory process (222), it may also play a role in down-regulating oxidative stress resulting from microglia hyperactivity. Despite the many lines of evidence indicating that estrogens have positive effects on the risk for AD, mixed results have been obtained with hormone replacement therapy (HRT) when it was used to counteract the development and progression of AD. There are reports pointing to a beneficial role of long-term HRT on the risk of AD and the age at onset in postmenopausal women (268–270), whereas other reports question the overall benefits (271–273) of HRT. These discrepancies may be due to preexisting genetic and hormonal differences, the time at which HRT was started (274), the timing of the early onset of a neurodegenerative disease, and the type of HRT (eg, presence/absence of progesterone). Progesterone treatment inhibited E2-mediated induction of neurotrophin expression and spatial memory performance (275). Similar effects were observed on Aβ accumulation after continuous progesterone treatment in adult female 3xTg-AD mice. However, the treatment had no effect by itself, but it counteracted the beneficial effect of E2 on Aβ accumulation (276). Nevertheless, the observation that cholinergic activity is decreased by continuous treatments in the hippocampus of ovariectomized female rats but is enhanced by cyclic treatment with 17β-estradiol and progesterone (277) suggests that treatments designed to mimic the natural hormonal fluctuations that occur during the ovarian cycle might have beneficial effects on AD-related disorders. Indeed, it has been described that in female 3×Tg-AD mice, cyclic progesterone delivery counteracted the increase in Aβ resulting from ovx and led to an enhancement of the Aβ-lowering effect of E2, along with significant improvements in working memory and visual attention (251). Moreover, progesterone treatment induced a reduction in tau hyperphosphorylation, thus suggesting a beneficial effect of cyclic treatment with progesterone in combination with E2 in lowering the hallmarks of AD.
2. Parkinson's disease
Sex is one of the strongest risk factors for Parkinson's disease (PD) because men have a 2-fold greater relative risk for developing PD than women of all ages. Furthermore, the phenotypic characteristics and symptomatology of the disease are also sexually dimorphic (278). Sex-specific differences have been reported in the gene expression profiles of neurons obtained from the substantia nigra (SN) of PD patients, which may indeed underlie the sexual dimorphism in the disease etiology, symptoms, and responses to therapy (279). The male prevalence of PD is also observed in PD animal models. Injections of neurotoxins (MPTP or methamphetamine in mice and 6-hydroxydopamine in rats) reduced the number of dopaminergic neurons in the SN and the dopamine levels in the striatum, with a higher potency observed in males using low doses of neurotoxins, possibly mimicking the early stages of PD (280–282). The sex prevalence in PD may be associated with intrinsic differences in the brain structures affected by the disease, as well as with sex-related environmental factors, because both events are related to estrogen. In fact, the organization of the SN-striatal dopaminergic (SNDA) system is sexually dimorphic, with males presenting a higher number of neuronal cells and regulatory networks, potentiated adaptive responses to psychomotor stimulants, and up-regulated expression of genes related to familial PD (279). Considering the role that estrogen plays in brain development, and particularly its influence on the dopaminergic system, the sex differences in the SNDA system may be determined by the actions of estrogen on brain cells and somehow favor PD development in men. On the other hand, strong evidence supports the hypothesis that estrogens contribute to the sex-related prevalence of PD in adults. This evidence includes the following: the inverse correlation between circulating estrogens and the severity of PD symptoms in women (283), the higher risk of PD in women with early natural or surgical menopause (284, 285), and the higher prevalence of PD in climacteric women in comparable age groups (283, 286). Furthermore, in animal studies of PD, estrogens were consistently shown to reduce the toxin-induced depletion of dopamine in the female striatum (280, 281).
So far, no firm hypothesis has been put forward to explain how estrogens affect the manifestation of PD in females. The current view suggests that accumulation of misfolded proteins, mitochondrial and endosomal dysfunction, and oxidative stress are the major biochemical mechanisms underlying the pathogenesis PD (287). However, a genetic association between a neuroinflammatory deficit and the pathogenesis of PD is still missing. As in the case of AD, microglia activation is strongly involved in the manifestation of PD, as well as a consequence or a cause of the selective vulnerability of neurons that produce dopamine, a highly reactive chemical that generates oxidative species and adds to the malfunctioning mitochondria, lysosomal, and protein aggregate toxicity. The following key features link microglia to the pathological hallmarks of PD: 1) higher microglial density in the midbrain relative to other brain areas (32, 288), likely as a result of the local oxidative environment; 2) microglia proliferation induced by dopaminergic toxicants specifically in the SN, which is a general sign of brain damage that is also observed in AD (289); the inflammatory response in the SN is different from other brain regions, such as the striatum or olfactory bulb because it occurs independently of IL-1β; 3) the delivery of LPS in the SN induces microglia activation, which selectively kills dopaminergic neurons and causes stable motor deficits (interestingly, serotoninergic neurons of the SN as well as neurons in the cortex and striatum are spared by this inflammatory insult) (290, 291); and 4) damaged dopaminergic neurons specifically release α-synuclein and neuromelanin aggregates that are potent triggers of microglia activation (292). Thus, microglia derangement can be considered as an “environmental” factor that may participate, either alone or in association with genetic variants, in the predisposition to PD.
Considering the sexual dimorphism in neuronal structures and functions affected in PD and the effects of estrogens on microglia, the logical question to be asked is whether estrogen may delay the onset and reduce the symptomatology of PD in females by targeting microglia. The fact that ER was not localized to dopaminergic neurons of the striatum, whereas ERα, ERβ, and GPR30 were expressed by microglia and interneurons in this brain region (293, 294) may point to the involvement of cells other than dopaminergic neurons that mediate hormonal neuroprotection, and microglia may certainly be taken into consideration. Although still limited, the available literature provides two main lines of evidence that strongly support the link between estrogen actions in microglia and neuroprotection in PD: the sexual diversity in microglia reactivity, which triggers neuronal death in males while providing neuroprotection in females, and the ruinous effects of menopause/ovx on neuroinflammation in females. In fact, a single peritoneal injection of LPS induces the selective loss of dopaminergic neurons in the SN and motor behavior deficits in male mice, whereas repeated injections of the endotoxin are necessary to induce neurotoxicity in females (295, 296). In line with this finding, estrogens reduce microglia activation after LPS administration through the inhibition of the Mac-1 receptor and PHOX protein complex, which regulate intracellular and extracellular ROS production (56, 209, 297). Accordingly, the genetic ablation of ERα in mice increases Mac-1 expression in microglia (209). Thus, these results strongly suggest a link between the activity of estrogen in microglia and the peculiar reactivity of these cells in females (209). Other approaches that more closely mimic the pathology of PD showed that the expression of inflammatory mediators, such as TNFα, IL-1β, IFNγ, and iNOS, is increased in the male SNDA system, and it is associated with an earlier and greater reduction in the striatal dopamine content in male mice compared to female mice (298). Accordingly, Morale et al (299) showed that the toxic potential of activated microglia after MPTP injury is inversely proportional to the levels of circulating estrogens. Thus, it appears that the link between estrogen actions in microglia and female microglia reactivity may prove beneficial for the manifestation of PD. More recently, it was shown that the expression of Mac-1 and other neuroinflammatory genes is increased by ovx in the forebrain of middle-aged female rats, and this effect was reverted by the chronic administration of estrogens (300). In agreement with these findings, increased expression of neuroinflammatory genes was observed in the forebrains of pre- and postmenopausal women (205). Finally, recent evidence highlighted the role that the renin-angiotensin system plays in microglia and showed this system to be more potentiated in males, leading to a higher neuronal loss in the SN (301), whereas its down-regulation by estrogens results in reduced oxidative stress, neuroinflammation, and neurodegeneration in females (302, 303).
In summary, the above-mentioned studies allow one to draw a tentative summary of the microglial pathways that may play a role in the beneficial effects of estrogens on neurodegeneration. These effects include the following: 1) the reduction of intra- and extramicroglial oxidative stress through the restoration of mitochondrial functions and potentiation of reductive enzymatic systems; 2) the modification of damage-activated intracellular signaling that provides a faster healing process through the adjustment of microglia reactivity; and 3) altogether, these observations support the view that communication between estrogen and microglia is one the mechanisms that reduce the risk of PD in premenopausal women.
V. Concluding Remarks and Future Directions
Given the involvement of the inflammatory process in neurodegeneration and the anti-inflammatory potential, it seems reasonable to further evaluate how the actions of estrogen in microglia might influence the onset and progression of neurodegenerative diseases. However, in undertaking these studies, we should consider the numerous factors that can constitute a confounding element in the interpretation of the results. A summary of the mechanisms that aging and the lack of estrogens may perturb in microglia is shown in Figure 7.
Figure 7.

Estrogen-dependent protective effects of microglia. Several biochemical processes promoted by microglia and regulated by estrogens protect neuronal functions: phagocytosis clears the debris and dysfunctional proteins (ie, β-amyloid) in the parenchyma; the production of antioxidant systems and enzymes (ie, the renin-angiotensin axis) limits the oxidative stress; the healing process is facilitated when damage-activated intracellular signaling pathways are activated; and synaptic maintenance participates in neuronal signaling. With aging, misfolded proteins, cell debris, and other inflammatory stimuli accumulate in the brain parenchyma, inducing a continuous stimulation of microglia that with senescence has a decreased phagocytic potential and ability to return to the surveying state. This initiates a vicious cycle with a progressive increase of the production of inflammatory products detrimental for neuronal health.
The main factor to be taken into consideration is the model system that is used to study the effects of the hormones; the major limitation of primary cultures of neonatal microglia or transformed cells is represented by their bias in the ER expression that may not reproduce what is occurring in microglia of the mature brain. Cultures of microglia isolated from the adult brain may be a better model. However, the low recovery of the current isolation procedure represents a major limitation and in addition, the culture would not provide these cells with the stimuli necessary for their continuous surveillance and response to the environment. Perhaps biochemical studies aimed at studying microglia responsiveness to physiopathological or pharmacological stimuli should be assessed in cells freshly isolated from the brain. The use of FACS and transgenic mice carrying appropriate reporters for microglia identification may help to overcome the low efficiency of current methodologies for microglia isolation. Alternatively, microglia may be studied in living organisms. In this case, a rigorous characterization of the model used and of the experimental setting is necessary to obtain reproducible and meaningful results because our understanding of estrogen physiology is still in its infancy. Several authors study estrogen in intact males to avoid the influence of high levels of this hormone in the circulation. Besides limiting the vision of the research to the sex likely less influenced by female sex hormones, this may give variability in the results due to the circadian synthesis of T and the presence/absence of aromatase converting the male sex hormone into estrogens. Studies in females rely on the ovx/hormone replacement paradigm, which limits the interpretation of the outcome, because we know very little of the endocrine compensatory reactions induced by the removal of the ovaries. Indeed, we may find totally different effects of pharmacological or hormone replacement that is dependent on the time of ovx. A better understanding of the physiological functions of estrogens in intact males at different hours of the day, and in intact females in the different phases of the cycle, is mandatory to obtain reproducible and meaningful observations. The selection of the correct route, dosage, and timing of estrogen administration is also challenging, as it is in all cells targeted by estrogen, including microglia, and the response may dramatically change, leading to conflicting and uninterpretable results. To this aim, the use of appropriate reporter animals (ie, animals genetically modified to produce easily measurable proteins in response to a selective stimulus) (304) should be encouraged because these animals allow for the spatiotemporal analysis of specific biochemical pathways in single, living animals, thus facilitating the interpretation of the physiological changes occurring over time. For example, the available reporter animals could facilitate the identification of the phase of the cycle and the cells actively responding to estrogens, whereas others would facilitate the identification of microglia in the activated or deactivated status, and these reporter systems could be bred with each other to obtain the analysis of multiple end points at the same time.
The other confounding element in the study of the effects of estrogen in microglia in vivo is represented by the fact that all neuronal cells are capable of expressing ERs. For instance, estrogens target neurons where they may exert a direct neuroprotective function. This represents a confounding element for defining the microglial contribution to neuronal health. Once more, the use of appropriate reporter animals would sharpen our vision and facilitate the study of microglial activity during the development of the pathology in a specific model of disease. These models would also be amenable to the study of the effect of age, nutritional cues, and dietary interventions. Indeed, prior results have shown that age and nutritional status have major, sex-dependent effects on microglia activity, which must be taken into consideration in future studies (Box 1).
Box 1.
Critical issues for the study of estrogen action in microglia and the definition of replacement therapies
| The impact on microglia physiology of circulating estrogens and their local |
| conversion from testosterone (via aromatase and 5α-R/3β-HSD) |
| During brain development: |
| • microglia proliferation and colonization of the brain |
| • brain masculinization of neuronal circuitries |
| In adult brain: |
| • control of the inflammatory response |
| • synaptic maintenance |
| In females, the physiological fluctuations of circulating and locally synthetized estrogens have significant repercussions on the homeostasis of dendritic spine and synaptic signaling possibly relevant for the control of behavior and energy homeostasis. Further studies on the consequences of short term (estrous cycle, pregnancy) or permanent endocrine changes (ER genetic polymorphisms, menopause, gonadectomy) are needed. |
| Criticisms in the selection of the experimental model |
| Changes of ER expression in microglia due to |
| • age and sex of the donors, |
| • environmental and nutritional cues, |
| • compensatory events induced by experimental gonadectomy |
| • different ER ligands in male (testosterone derivatives: estrogens vs. 3β-diols) |
| • pathologies |
| Functional differences between neonatal and adult microglia for primary cultures |
| Poor cell recovery for ex vivo preparations of microglia |
| Lack of appropriate reporter systems for in vivo studies |
| Estrogen pharmacology: |
| • differential effects of estrogen replacement associated with route, dosage and timing of administration |
| • complexity of ER activity, including ER multiplicity of receptors, ER interactions with cytoplasmic and nuclear signaling molecules, cell-specific expression of co-regulators |
| • poor knowledge on SERMs activity in microglia |
| • wide distribution of the expression of ERs and estrogen synthetizing enzymes in brain cells |
In conclusion, the large body of experimental evidence provided so far indicates that microglia represent another target for the neuroprotective actions of estrogen. Indeed, as a plausible factor driving microglia colonization of the nervous system, as well as a modulator of microglia reactivity in adult brain, estrogens appear to play a role in the neurodegenerative process, conditioning the incidence of these pathologies as well as the course of their progression. The lack of a direct, strong linkage between ER mutations and neurodegenerative diseases suggests that these sex hormones do not play a primary role in preventing the progression of the neurodegenerative program, which, in the sporadic forms of these disorders, is highly multifactorial. However, the impairment of estrogenic signaling in combination with a lack of other elements relevant for neuronal health may facilitate the initiation of the neurodegenerative process as shown by the studies on the correlation between ER signaling and ApoE4 (305). The fact that estrogens are not primary contributors, but only participate in the complex combination of the events necessary to trigger the neurodegenerative process, represents the main obstacle for the study of the effects and the definition of adequate replacement therapies.
The identification of further correlations between estrogen deficiencies and pathologies of the CNS characterized by the significant neuroinflammatory component may provide a means for the study of the efficacy of replacement therapies, but time and cost factors may be unsuitable for what is needed. Such therapies, aimed at reconstituting the natural defenses of the brain against neuroinflammation might be less amenable to undesired collateral effect than exogenous molecules such as sodium thiosulfate (306), mitoapocynin (307), or kolaviron (308) recently proposed for the reduction of neuroinflammation. In the near future, efforts should be mainly aimed at a better understanding of the physiology of estrogen actions in the microglia of males and females. This knowledge is vital for the design of appropriate hormone replacement therapies that can overcome the lack of the natural hormone in targets, sparing their potential negative effects in reproductive organs.
Acknowledgments
This work was supported by the European Union Grants ERC-2012-ADG322977 (WAYS), and the Seventh Framework Programme (FP7/2007-2013) under Grant Agreement no. 278850 (INMiND), by Fondazione CARIPLO Grants 2013-0786 and 2014-0686, and by the National Institutes of Health Award Number R01AG027713 from the National Institute On Aging.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AD
- Alzheimer's disease
- AP-1
- activator protein 1
- ApoE4
- apolipoprotein E4
- AR
- androgen receptor
- BBB
- blood-brain barrier
- CD
- cluster of differentiation
- CNS
- central nervous system
- CSF1
- colony-stimulating factor 1
- DBD
- DNA binding domain
- DPN
- 2,3-bis (4-hydroxyphenyl) propionitrile
- EAE
- experimental autoimmune encephalomyelitis
- E2
- 17β estradiol
- ER
- estrogen receptor
- ERE
- estrogen response element
- FACS
- fluorescence-activated cell sorting
- GABA
- γ-aminobutyric acid
- GPR30
- G protein-coupled receptor 30
- GR
- glucocorticoid receptor
- HFD
- high-fat diet
- HRT
- hormone replacement therapy
- IFN
- interferon
- iNOS
- inducible nitric oxide synthase
- LPS
- lipopolysaccharide
- MCAO
- middle cerebral artery occlusion
- MCI
- mild cognitive impairment
- MHC
- major histocompatibility complex
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MR
- mineralocorticoid receptor
- MS
- multiple sclerosis
- NF-κB
- nuclear factor-κB
- NO
- nitric oxide
- ovx
- ovariectomy
- P
- postnatal day
- PD
- Parkinson's disease
- PHOX
- phagocytic oxidase
- PI3K
- phosphatidylinositol-3-kinase
- PPT
- 1,3,5-tris (4-hydroxyphenyl)-4-propyl-1H-pyrazole
- PR
- progesterone receptor
- PS
- phosphatidylserine
- PTM
- post-translational modification
- ROS
- reactive oxygen species
- SERM
- selective ER modulator
- SN
- substantia nigra
- SNDA
- SN-striatal dopaminergic
- SOCS3
- suppressor of cytokine signaling 3
- TLR
- Toll-like receptor
- wt
- wild-type.
Reference
- 1. Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friedrich MJ. Research on psychiatric disorders targets inflammation. JAMA. 2014;312(5):474–476. [DOI] [PubMed] [Google Scholar]
- 4. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217–224. [DOI] [PubMed] [Google Scholar]
- 5. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. [DOI] [PubMed] [Google Scholar]
- 6. Rio-Hortega P. The microglia. Lancet. 1939;233(6036):1023–1026. [Google Scholar]
- 7. Beers DR, Henkel JS, Xiao Q, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103(43):16021–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herbomel P, Thisse B, Thisse C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development. 1999;126(17):3735–3745. [DOI] [PubMed] [Google Scholar]
- 9. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. 2016;17(1):2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elmore MR, Najafi AR, Koike MA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Casano AM, Peri F. Microglia: multitasking specialists of the brain. Dev Cell. 2015;32(4):469–477. [DOI] [PubMed] [Google Scholar]
- 13. Mallat M, Marín-Teva JL, Chéret C. Phagocytosis in the developing CNS: more than clearing the corpses. Curr Opin Neurobiol. 2005;15(1):101–107. [DOI] [PubMed] [Google Scholar]
- 14. Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. [DOI] [PubMed] [Google Scholar]
- 15. Ashwell K. Microglia and cell death in the developing mouse cerebellum. Brain Res Dev Brain Res. 1990;55(2):219–230. [DOI] [PubMed] [Google Scholar]
- 16. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15(4):209–216. [DOI] [PubMed] [Google Scholar]
- 17. Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10):4216–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41(4):535–547. [DOI] [PubMed] [Google Scholar]
- 19. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8(11):e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ueno M, Fujita Y, Tanaka T, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–551. [DOI] [PubMed] [Google Scholar]
- 22. Sato K. Effects of microglia on neurogenesis. Glia. 2015;63(8):1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. J Neurochem. 2012;120(6):948–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lenz KM, Nugent BM, McCarthy MM. Sexual differentiation of the rodent brain: dogma and beyond. Front Neurosci. 2012;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rana SA, Aavani T, Pittman QJ. Sex effects on neurodevelopmental outcomes of innate immune activation during prenatal and neonatal life. Horm Behav. 2012;62(3):228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res. 2013;91(9):1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia. 2008;56(6):659–674. [DOI] [PubMed] [Google Scholar]
- 28. Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145(4):584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. J Neurosci. 2013;33(7):2761–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol. 2012;33(3):267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwarz JM, Bilbo SD. Sex, glia, and development: interactions in health and disease. Horm Behav. 2012;62(3):243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. [DOI] [PubMed] [Google Scholar]
- 33. Pepe G, Calderazzi G, De Maglie M, Villa AM, Vegeto E. Heterogeneous induction of microglia M2a phenotype by central administration of interleukin-4. J Neuroinflammation. 2014;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. [DOI] [PubMed] [Google Scholar]
- 35. Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20(11):4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rappert A, Biber K, Nolte C, et al. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol. 2002;168(7):3221–3226. [DOI] [PubMed] [Google Scholar]
- 37. Noda M, Nakanishi H, Nabekura J, Akaike N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci. 2000;20(1):251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Färber K, Markworth S, Pannasch U, et al. The ectonucleotidase cd39/ENTPDase1 modulates purinergic-mediated microglial migration. Glia. 2008;56(3):331–341. [DOI] [PubMed] [Google Scholar]
- 39. Nolte C, Möller T, Walter T, Kettenmann H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience. 1996;73(4):1091–1107. [DOI] [PubMed] [Google Scholar]
- 40. Tremblay MÈ, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31(45):16064–16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morgan SC, Taylor DL, Pocock JM. Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and δ-Notch signalling cascades. J Neurochem. 2004;90(1):89–101. [DOI] [PubMed] [Google Scholar]
- 42. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. [DOI] [PubMed] [Google Scholar]
- 43. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45(2):208–212. [DOI] [PubMed] [Google Scholar]
- 44. Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56(6):732–740. [DOI] [PubMed] [Google Scholar]
- 45. Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175(7):4320–4330. [DOI] [PubMed] [Google Scholar]
- 46. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. [DOI] [PubMed] [Google Scholar]
- 47. Crehan H, Hardy J, Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–149. [DOI] [PubMed] [Google Scholar]
- 48. Schafer DP, Stevens B. Synapse elimination during development and disease: immune molecules take centre stage. Biochem Soc Trans. 2010;38(2):476–481. [DOI] [PubMed] [Google Scholar]
- 49. Yakubenko VP, Bhattacharjee A, Pluskota E, Cathcart MK. αMβ2 integrin activation prevents alternative activation of human and murine macrophages and impedes foam cell formation. Circ Res. 2011;108(5):544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stuart LM, Bell SA, Stewart CR, et al. CD36 signals to the actin cytoskeleton and regulates microglial migration via a p130Cas complex. J Biol Chem. 2007;282(37):27392–27401. [DOI] [PubMed] [Google Scholar]
- 51. Witting A, Müller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by microglia/brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem. 2000;75(3):1060–1070. [DOI] [PubMed] [Google Scholar]
- 52. Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286(9):7214–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rahmadi A, Steiner N, Münch G. Advanced glycation endproducts as gerontotoxins and biomarkers for carbonyl-based degenerative processes in Alzheimer's disease. Clin Chem Lab Med. 2011;49(3):385–391. [DOI] [PubMed] [Google Scholar]
- 54. Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA. 2010;107(15):7036–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–438. [DOI] [PubMed] [Google Scholar]
- 56. Qin L, Liu Y, Wang T, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279(2):1415–1421. [DOI] [PubMed] [Google Scholar]
- 57. Possel H, Noack H, Putzke J, Wolf G, Sies H. Selective upregulation of inducible nitric oxide synthase (iNOS) by lipopolysaccharide (LPS) and cytokines in microglia: in vitro and in vivo studies. Glia. 2000;32(1):51–59. [DOI] [PubMed] [Google Scholar]
- 58. Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587(2):250–256. [DOI] [PubMed] [Google Scholar]
- 59. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. [DOI] [PubMed] [Google Scholar]
- 60. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schwartz M, Butovsky O, Brück W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29(2):68–74. [DOI] [PubMed] [Google Scholar]
- 62. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175(1):342–349. [DOI] [PubMed] [Google Scholar]
- 63. Edwards DR, Murphy G, Reynolds JJ, et al. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. EMBO J. 1987;6(7):1899–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17(1):109–118. [DOI] [PubMed] [Google Scholar]
- 65. Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor β increases neurogenesis in the adult dentate gyrus. Eur J Neurosci. 2006;23(1):83–93. [DOI] [PubMed] [Google Scholar]
- 66. Fadok VA, Henson PM. Apoptosis: giving phosphatidylserine recognition an assist–with a twist. Curr Biol. 2003;13(16):R655–R657. [DOI] [PubMed] [Google Scholar]
- 67. Prieto AL, Weber JL, Tracy S, Heeb MJ, Lai C. Gas6, a ligand for the receptor protein-tyrosine kinase Tyro-3, is widely expressed in the central nervous system. Brain Res. 1999;816(2):646–661. [DOI] [PubMed] [Google Scholar]
- 68. Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem. 2000;127(3):411–417. [DOI] [PubMed] [Google Scholar]
- 69. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–686. [DOI] [PubMed] [Google Scholar]
- 71. Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468(7321):305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, Swanson RA. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem. 2001;77(6):1601–1610. [DOI] [PubMed] [Google Scholar]
- 73. Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28(50):13574–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10(7):719–726. [DOI] [PubMed] [Google Scholar]
- 75. Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. [DOI] [PubMed] [Google Scholar]
- 76. Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9(12):1512–1519. [DOI] [PubMed] [Google Scholar]
- 77. Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59(1):152–165. [DOI] [PubMed] [Google Scholar]
- 78. Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26(6):1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shinozaki Y, Nomura M, Iwatsuki K, Moriyama Y, Gachet C, Koizumi S. Microglia trigger astrocyte-mediated neuroprotection via purinergic gliotransmission. Sci Rep. 2014;4:4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mohri I, Taniike M, Taniguchi H, et al. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J Neurosci. 2006;26(16):4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Welser-Alves JV, Crocker SJ, Milner R. A dual role for microglia in promoting tissue inhibitor of metalloproteinase (TIMP) expression in glial cells in response to neuroinflammatory stimuli. J Neuroinflammation. 2011;8:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Magnoni S, Baker A, Thomson S, et al. Neuroprotective effect of adenoviral-mediated gene transfer of TIMP-1 and -2 in ischemic brain injury. Gene Ther. 2007;14(7):621–625. [DOI] [PubMed] [Google Scholar]
- 83. Crocker SJ, Whitmire JK, Frausto RF, et al. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am J Pathol. 2006;169(6):2104–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med (Berl). 2006;84(7):532–543. [DOI] [PubMed] [Google Scholar]
- 85. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–372. [DOI] [PubMed] [Google Scholar]
- 86. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–558. [DOI] [PubMed] [Google Scholar]
- 87. Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. [DOI] [PubMed] [Google Scholar]
- 88. Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30(10):527–535. [DOI] [PubMed] [Google Scholar]
- 89. Xanthos DN, Sandkühler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15(1):43–53. [DOI] [PubMed] [Google Scholar]
- 90. Liu GJ, Nagarajah R, Banati RB, Bennett MR. Glutamate induces directed chemotaxis of microglia. Eur J Neurosci. 2009;29(6):1108–1118. [DOI] [PubMed] [Google Scholar]
- 91. Noda M, Takii K, Parajuli B, et al. FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J Neuroinflammation. 2014;11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55(3):233–238. [DOI] [PubMed] [Google Scholar]
- 93. Cook DN, Chen SC, Sullivan LM, et al. Generation and analysis of mice lacking the chemokine fractalkine. Mol Cell Biol. 2001;21(9):3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–924. [DOI] [PubMed] [Google Scholar]
- 95. Contestabile A, Monti B, Polazzi E. Neuronal-glial interactions define the role of nitric oxide in neural functional processes. Curr Neuropharmacol. 2012;10(4):303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21(17):6480–6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Stewart VC, Heslegrave AJ, Brown GC, Clark JB, Heales SJ. Nitric oxide-dependent damage to neuronal mitochondria involves the NMDA receptor. Eur J Neurosci. 2002;15(3):458–464. [DOI] [PubMed] [Google Scholar]
- 98. Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. 2010;41:242–247. [DOI] [PubMed] [Google Scholar]
- 99. Kim WK, Seo DO, Choi JJ, Ko KH. Immunostimulated glial cells potentiate glucose deprivation-induced death of cultured rat cerebellar granule cells. J Neurotrauma. 1999;16(5):415–424. [DOI] [PubMed] [Google Scholar]
- 100. Polazzi E, Contestabile A. Neuron-conditioned media differentially affect the survival of activated or unstimulated microglia: evidence for neuronal control on apoptotic elimination of activated microglia. J Neuropathol Exp Neurol. 2003;62(4):351–362. [DOI] [PubMed] [Google Scholar]
- 101. Tian L, Ma L, Kaarela T, Li Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J Neuroinflammation. 2012;9:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Maric T, Woodside B, Luheshi GN. The effects of dietary saturated fat on basal hypothalamic neuroinflammation in rats. Brain Behav Immun. 2014;36:35–45. [DOI] [PubMed] [Google Scholar]
- 103. Pistell PJ, Morrison CD, Gupta S, et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010;219:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gao Y, Ottaway N, Schriever SC, et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia. 2014;62(1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Erion JR, Wosiski-Kuhn M, Dey A, et al. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci. 2014;34(7):2618–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Purkayastha S, Cai D. Disruption of neurogenesis by hypothalamic inflammation in obesity or aging. Rev Endocr Metab Disord. 2013;14(4):351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Milanski M, Degasperi G, Coope A, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29(2):359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Thaler JP, Yi CX, Schur EA, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yi CX, Al-Massadi O, Donelan E, et al. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol Behav. 2012;106(4):485–490. [DOI] [PubMed] [Google Scholar]
- 110. Ferreira R, Santos T, Viegas M, et al. Neuropeptide Y inhibits interleukin-1β-induced phagocytosis by microglial cells. J Neuroinflammation. 2011;8:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Delgado R, Carlin A, Airaghi L, et al. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J Leukoc Biol. 1998;63(6):740–745. [DOI] [PubMed] [Google Scholar]
- 112. Leduc V, Domenger D, De Beaumont L, Lalonde D, Belanger-Jasmin S, Poirier J. Function and comorbidities of apolipoprotein E in Alzheimer's disease. Int J Alzheimers Dis. 2011;2011:974361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71(14):1057–1064. [DOI] [PubMed] [Google Scholar]
- 114. Vagelatos NT, Eslick GD. Type 2 diabetes as a risk factor for Alzheimer's disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. 2013;35:152–160. [DOI] [PubMed] [Google Scholar]
- 115. Luchsinger JA, Gustafson DR. Adiposity, type 2 diabetes, and Alzheimer's disease. J Alzheimers Dis. 2009;16(4):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Elias MF, Elias PK, Sullivan LM, Wolf PA, D'Agostino RB. Obesity, diabetes and cognitive deficit: the Framingham Heart Study. Neurobiol Aging. 2005;26(suppl 1):11–16. [DOI] [PubMed] [Google Scholar]
- 117. Fontana R, Della Torre S, Meda C, Longo A, Eva C, Maggi A. Estrogen replacement therapy regulation of energy metabolism in female mouse hypothalamus. Endocrinology. 2014;155(6):2213–2221. [DOI] [PubMed] [Google Scholar]
- 118. Musso R, Maggi A, Eva C. 17 β-Estradiol stimulates mouse neuropeptide Y-Y(1) receptor gene transcription by binding to estrogen receptor α in neuroblastoma cells. Neuroendocrinology. 2000;72(6):360–367. [DOI] [PubMed] [Google Scholar]
- 119. Streit WJ, Xue QS. Human CNS immune senescence and neurodegeneration. Curr Opin Immunol. 2014;29:93–96. [DOI] [PubMed] [Google Scholar]
- 120. Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014;88(4):594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10(2):263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tremblay MÈ, Zettel ML, Ison JR, Allen PD, Majewska AK. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia. 2012;60(4):541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hefendehl JK, Neher JJ, Sühs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. 2014;13(1):60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55(4):412–424. [DOI] [PubMed] [Google Scholar]
- 125. Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, et al. Rapid microglial response around amyloid pathology after systemic anti-Aβ antibody administration in PDAPP mice. J Neurosci. 2008;28(52):14156–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer's disease. Nature. 2008;451(7179):720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Orre M, Kamphuis W, Osborn LM, et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging. 2014;35(1):1–14. [DOI] [PubMed] [Google Scholar]
- 128. Landreth GE, Reed-Geaghan EG. Toll-like receptors in Alzheimer's disease. Curr Top Microbiol Immunol. 2009;336:137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jimenez S, Baglietto-Vargas D, Caballero C, et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28(45):11650–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23(3):309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Benedusi V, Meda C, Della Torre S, Monteleone G, Vegeto E, Maggi A. A lack of ovarian function increases neuroinflammation in aged mice. Endocrinology. 2012;153(6):2777–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ritzel RM, Patel AR, Pan S, et al. Age- and location-related changes in microglial function. Neurobiol Aging. 2015;36(6):2153–2163. [DOI] [PubMed] [Google Scholar]
- 133. Dantzer R. Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci. 2001;933:222–234. [DOI] [PubMed] [Google Scholar]
- 134. Ericsson A, Arias C, Sawchenko PE. Evidence for an intramedullary prostaglandin-dependent mechanism in the activation of stress-related neuroendocrine circuitry by intravenous interleukin-1. J Neurosci. 1997;17(18):7166–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dahlman-Wright K, Cavailles V, Fuqua SA, et al. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev. 2006;58(4):773–781. [DOI] [PubMed] [Google Scholar]
- 137. King WJ, Greene GL. Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature. 1984;307(5953):745–747. [DOI] [PubMed] [Google Scholar]
- 138. Ishihara Y, Itoh K, Ishida A, Yamazaki T. Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J Steroid Biochem Mol Biol. 2015;145:85–93. [DOI] [PubMed] [Google Scholar]
- 139. Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60(9):520–528. [DOI] [PubMed] [Google Scholar]
- 140. Acconcia F, Ascenzi P, Bocedi A, et al. Palmitoylation-dependent estrogen receptor α membrane localization: regulation by 17β-estradiol. Mol Biol Cell. 2005;16(1):231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Maggi A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim Biophys Acta. 2011;1812(8):1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Atsriku C, Britton DJ, Held JM, et al. Systematic mapping of posttranslational modifications in human estrogen receptor-α with emphasis on novel phosphorylation sites. Mol Cell Proteomics. 2009;8(3):467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Power RF, Mani SK, Codina J, Conneely OM, O'Malley BW. Dopaminergic and ligand-independent activation of steroid hormone receptors. Science. 1991;254(5038):1636–1639. [DOI] [PubMed] [Google Scholar]
- 144. Denner LA, Weigel NL, Maxwell BL, Schrader WT, O'Malley BW. Regulation of progesterone receptor-mediated transcription by phosphorylation. Science. 1990;250(4988):1740–1743. [DOI] [PubMed] [Google Scholar]
- 145. Ignar-Trowbridge DM, Nelson KG, Bidwell MC, et al. Coupling of dual signaling pathways: epidermal growth factor action involves the estrogen receptor. Proc Natl Acad Sci USA. 1992;89(10):4658–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270(5241):1491–1494. [DOI] [PubMed] [Google Scholar]
- 147. Ma ZQ, Santagati S, Patrone C, Pollio G, Vegeto E, Maggi A. Insulin-like growth factors activate estrogen receptor to control the growth and differentiation of the human neuroblastoma cell line SK-ER3. Mol Endocrinol. 1994;8(7):910–918. [DOI] [PubMed] [Google Scholar]
- 148. Chen D, Riedl T, Washbrook E, et al. Activation of estrogen receptor α by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell. 2000;6(1):127–137. [PubMed] [Google Scholar]
- 149. Patrone C, Gianazza E, Santagati S, Agrati P, Maggi A. Divergent pathways regulate ligand-independent activation of ER α in SK-N-BE neuroblastoma and COS-1 renal carcinoma cells. Mol Endocrinol. 1998;12(6):835–841. [DOI] [PubMed] [Google Scholar]
- 150. Aronica SM, Katzenellenbogen BS. Stimulation of estrogen receptor-mediated transcription and alteration in the phosphorylation state of the rat uterine estrogen receptor by estrogen, cyclic adenosine monophosphate, and insulin-like growth factor-I. Mol Endocrinol. 1993;7(6):743–752. [DOI] [PubMed] [Google Scholar]
- 151. Al-Dhaheri MH, Rowan BG. Protein kinase A exhibits selective modulation of estradiol-dependent transcription in breast cancer cells that is associated with decreased ligand binding, altered estrogen receptor α promoter interaction, and changes in receptor phosphorylation. Mol Endocrinol. 2007;21(2):439–456. [DOI] [PubMed] [Google Scholar]
- 152. Sheeler CQ, Singleton DW, Khan SA. Mutation of serines 104, 106, and 118 inhibits dimerization of the human estrogen receptor in yeast. Endocr Res. 2003;29(2):237–255. [DOI] [PubMed] [Google Scholar]
- 153. Li L, Li Z, Howley PM, Sacks DB. E6AP and calmodulin reciprocally regulate estrogen receptor stability. J Biol Chem. 2006;281(4):1978–1985. [DOI] [PubMed] [Google Scholar]
- 154. Govind AP, Thampan RV. Proteins interacting with the mammalian estrogen receptor: proposal for an integrated model for estrogen receptor mediated regulation of transcription. J Cell Biochem. 2001;80(4):571–579. [DOI] [PubMed] [Google Scholar]
- 155. Spencer TE, Jenster G, Burcin MM, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389(6647):194–198. [DOI] [PubMed] [Google Scholar]
- 156. Lonard DM, O'Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8(10):598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Maggi A, Ciana P, Belcredito S, Vegeto E. Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu Rev Physiol. 2004;66:291–313. [DOI] [PubMed] [Google Scholar]
- 158. Kim HP, Lee JY, Jeong JK, Bae SW, Lee HK, Jo I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor α localized in caveolae. Biochem Biophys Res Commun. 1999;263(1):257–262. [DOI] [PubMed] [Google Scholar]
- 159. Levin ER. Extranuclear estrogen receptor's roles in physiology: lessons from mouse models. Am J Physiol Endocrinol Metab. 2014;307(2):E133–E140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Galluzzo P, Ascenzi P, Bulzomi P, Marino M. The nutritional flavanone naringenin triggers antiestrogenic effects by regulating estrogen receptor α-palmitoylation. Endocrinology. 2008;149(5):2567–2575. [DOI] [PubMed] [Google Scholar]
- 161. O'Mahony F, Razandi M, Pedram A, Harvey BJ, Levin ER. Estrogen modulates metabolic pathway adaptation to available glucose in breast cancer cells. Mol Endocrinol. 2012;26(12):2058–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pedram A, Razandi M, O'Mahony F, Harvey H, Harvey BJ, Levin ER. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci Signal. 2013;6(276):ra36. [DOI] [PubMed] [Google Scholar]
- 163. Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111(2):E283–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Maggi A, Vegeto E. Intracellular receptors. In: Clementi F, Fumagalli G, eds. General and Molecular Pharmacology: Principles of Drug Action. Hoboken, NJ: John Wiley, Sons, Inc; 2015:268–283. [Google Scholar]
- 165. Muluhngwi P, Klinge CM. Roles for miRNAs in endocrine resistance in breast cancer. Endocr Relat Cancer. 2015;22(5):R279–R300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS. Antagonists selective for estrogen receptor α. Endocrinology. 2002;143(3):941–947. [DOI] [PubMed] [Google Scholar]
- 167. Arevalo MA, Santos-Galindo M, Lagunas N, Azcoitia I, Garcia-Segura LM. Selective estrogen receptor modulators as brain therapeutic agents. J Mol Endocrinol. 2011;46(1):R1–R9. [DOI] [PubMed] [Google Scholar]
- 168. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146(2):624–632. [DOI] [PubMed] [Google Scholar]
- 169. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7(12):715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Liu L, Zhao Y, Xie K, Sun X, Gao Y, Wang Z. Estrogen-induced nongenomic calcium signaling inhibits lipopolysaccharide-stimulated tumor necrosis factor α production in macrophages. PLoS One. 2013;8(12):e83072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. [DOI] [PubMed] [Google Scholar]
- 172. Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660. [DOI] [PubMed] [Google Scholar]
- 173. Zhang C, Kelly MJ, Ronnekleiv OK. 17 beta-estradiol rapidly increases ATP-sensitive potassium channel activity in gonadotropin-releasing hormone neurons [corrected] via a protein kinase signaling pathway. Endocrinology. 2010;151(9):4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Maggiolini M, Vivacqua A, Fasanella G, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. 2004;279(26):27008–27016. [DOI] [PubMed] [Google Scholar]
- 175. Tran QK, VerMeer M, Burgard MA, Hassan AB, Giles J. Hetero-oligomeric complex between the G protein-coupled estrogen receptor 1 and the plasma membrane Ca2+-ATPase 4b. J Biol Chem. 2015;290(21):13293–13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Blasko E, Haskell CA, Leung S, et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol. 2009;214:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Habib P, Slowik A, Zendedel A, Johann S, Dang J, Beyer C. Regulation of hypoxia-induced inflammatory responses and M1–M2 phenotype switch of primary rat microglia by sex steroids. J Mol Neurosci. 2014;52(2):277–285. [DOI] [PubMed] [Google Scholar]
- 178. Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. [DOI] [PubMed] [Google Scholar]
- 179. Ignatov A, Ignatov T, Roessner A, Costa SD, Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat. 2010;123(1):87–96. [DOI] [PubMed] [Google Scholar]
- 180. Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2(4):207–212. [DOI] [PubMed] [Google Scholar]
- 181. Dennis MK, Burai R, Ramesh C, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5(6):421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Bai LY, Weng JR, Hu JL, Wang D, Sargeant AM, Chiu CF. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem Biol Interact. 2013;206(2):375–384. [DOI] [PubMed] [Google Scholar]
- 183. Moussaud S, Lamodière E, Savage C, Draheim HJ. Characterisation of K+ currents in the C8–B4 microglial cell line and their regulation by microglia activating stimuli. Cell Physiol Biochem. 2009;24:141–152. [DOI] [PubMed] [Google Scholar]
- 184. Wes PD, Holtman IR, Boddeke EW, Möller T, Eggen BJ. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia. 2016;64(2):197–213. [DOI] [PubMed] [Google Scholar]
- 185. Crain JM, Watters JJ. Estrogen and P2 purinergic receptor systems in microglia: therapeutic targets for neuroprotection. Open Drug Discov J. 2010;2:148–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Tapia-Gonzalez S, Carrero P, Pernia O, Garcia-Segura LM, Diz-Chaves Y. Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: potential role of microglial ERs. J Endocrinol. 2008;198(1):219–230. [DOI] [PubMed] [Google Scholar]
- 187. Sárvári M, Kalló I, Hrabovszky E, Solymosi N, Liposits Z. Ovariectomy and subsequent treatment with estrogen receptor agonists tune the innate immune system of the hippocampus in middle-aged female rats. PLoS One. 2014;9(2):e88540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. García-Ovejero D, Veiga S, García-Segura LM, Doncarlos LL. Glial expression of estrogen and androgen receptors after rat brain injury. J Comp Neurol. 2002;450(3):256–271. [DOI] [PubMed] [Google Scholar]
- 189. Mor G, Nilsen J, Horvath T, et al. Estrogen and microglia: a regulatory system that affects the brain. J Neurobiol. 1999;40(4):484–496. [DOI] [PubMed] [Google Scholar]
- 190. Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JÅ. Targeting estrogen receptor β in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2013;110(9):3543–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Dimayuga FO, Reed JL, Carnero GA, et al. Estrogen and brain inflammation: effects on microglial expression of MHC, costimulatory molecules and cytokines. J Neuroimmunol. 2005;161:123–136. [DOI] [PubMed] [Google Scholar]
- 192. Baker AE, Brautigam VM, Watters JJ. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor β. Endocrinology. 2004;145(11):5021–5032. [DOI] [PubMed] [Google Scholar]
- 193. Crotti A, Benner C, Kerman BE, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17(4):513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Vegeto E, Pollio G, Ciana P, Maggi A. Estrogen blocks inducible nitric oxide synthase accumulation in LPS-activated microglia cells. Exp Gerontol. 2000;35:1309–1316. [DOI] [PubMed] [Google Scholar]
- 197. Soucy G, Boivin G, Labrie F, Rivest S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J Immunol. 2005;174(10):6391–6398. [DOI] [PubMed] [Google Scholar]
- 198. Suuronen T, Nuutinen T, Huuskonen J, Ojala J, Thornell A, Salminen A. Anti-inflammatory effect of selective estrogen receptor modulators (SERMs) in microglial cells. Inflamm Res. 2005;54(5):194–203. [DOI] [PubMed] [Google Scholar]
- 199. Zhang L, Nair A, Krady K, et al. Estrogen stimulates microglia and brain recovery from hypoxia-ischemia in normoglycemic but not diabetic female mice. J Clin Invest. 2004;113(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Habib P, Dreymueller D, Ludwig A, Beyer C, Dang J. Sex steroid hormone-mediated functional regulation of microglia-like BV-2 cells during hypoxia. J Steroid Biochem Mol Biol. 2013;138:195–205. [DOI] [PubMed] [Google Scholar]
- 201. Jeong JW, Lee HH, Han MH, Kim GY, Kim WJ, Choi YH. Anti-inflammatory effects of genistein via suppression of the toll-like receptor 4-mediated signaling pathway in lipopolysaccharide-stimulated BV2 microglia. Chem Biol Interact. 2014;212:30–39. [DOI] [PubMed] [Google Scholar]
- 202. Chinta SJ, Ganesan A, Reis-Rodrigues P, Lithgow GJ, Andersen JK. Anti-inflammatory role of the isoflavone diadzein in lipopolysaccharide-stimulated microglia: implications for Parkinson's disease. Neurotox Res. 2013;23(2):145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Park SE, Sapkota K, Kim S, Kim H, Kim SJ. Kaempferol acts through mitogen-activated protein kinases and protein kinase B/AKT to elicit protection in a model of neuroinflammation in BV2 microglial cells. Br J Pharmacol. 2011;164(3):1008–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Smith JA, Das A, Butler JT, Ray SK, Banik NL. Estrogen or estrogen receptor agonist inhibits lipopolysaccharide induced microglial activation and death. Neurochem Res. 2011;36(9):1587–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Sárvári M, Hrabovszky E, Kalló I, et al. Menopause leads to elevated expression of macrophage-associated genes in the aging frontal cortex: rat and human studies identify strikingly similar changes. J Neuroinflammation. 2012;9:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Vegeto E, Belcredito S, Ghisletti S, Meda C, Etteri S, Maggi A. The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology. 2006;147(5):2263–2272. [DOI] [PubMed] [Google Scholar]
- 207. Lei DL, Long JM, Hengemihle J, et al. Effects of estrogen and raloxifene on neuroglia number and morphology in the hippocampus of aged female mice. Neuroscience. 2003;121(3):659–666. [DOI] [PubMed] [Google Scholar]
- 208. Ciana P, Raviscioni M, Mussi P, et al. In vivo imaging of transcriptionally active estrogen receptors. Nat Med. 2003;9(1):82–86. [DOI] [PubMed] [Google Scholar]
- 209. Vegeto E, Belcredito S, Etteri S, et al. Estrogen receptor-α mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci USA. 2003;100(16):9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Brown CM, Mulcahey TA, Filipek NC, Wise PM. Production of proinflammatory cytokines and chemokines during neuroinflammation: novel roles for estrogen receptors α and β. Endocrinology. 2010;151(10):4916–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Liu JL, Tian DS, Li ZW, et al. Tamoxifen alleviates irradiation-induced brain injury by attenuating microglial inflammatory response in vitro and in vivo. Brain Res. 2010;1316:101–111. [DOI] [PubMed] [Google Scholar]
- 212. Ismailoğlu O, Oral B, Görgülü A, Sütçü R, Demir N. Neuroprotective effects of tamoxifen on experimental spinal cord injury in rats. J Clin Neurosci. 2010;17(10):1306–1310. [DOI] [PubMed] [Google Scholar]
- 213. Barreto GE, Santos-Galindo M, Garcia-Segura LM. Selective estrogen receptor modulators regulate reactive microglia after penetrating brain injury. Front Aging Neurosci. 2014;6:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. O'Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–460. [DOI] [PubMed] [Google Scholar]
- 215. Rettew JA, Huet YM, Marriott I. Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology. 2009;150(8):3877–3884. [DOI] [PubMed] [Google Scholar]
- 216. Liu L, Zhao Y, Xie K, et al. Estrogen inhibits LPS-induced IL-6 production in macrophages partially via the nongenomic pathway. Immunol Invest. 2014;43(7):693–704. [DOI] [PubMed] [Google Scholar]
- 217. Li X, Li M, Bai X. Upregulation of TLR2 expression is induced by estrogen via an estrogen-response element (ERE). Arch Biochem Biophys. 2014;549:26–31. [DOI] [PubMed] [Google Scholar]
- 218. Cunningham MA, Wirth JR, Naga O, Eudaly J, Gilkeson GS. Estrogen receptor α binding to ERE is required for full Tlr7- and Tlr9-induced inflammation. SOJ Immunol. 2014;2(1):pii:07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Cunningham MA, Wirth JR, Freeman LR, Boger HA, Granholm AC, Gilkeson GS. Estrogen receptor α deficiency protects against development of cognitive impairment in murine lupus. J Neuroinflammation. 2014;11:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Ghisletti S, Meda C, Maggi A, Vegeto E. 17β-Estradiol inhibits inflammatory gene expression by controlling NF-κB intracellular localization. Mol Cell Biol. 2005;25(8):2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Calippe B, Douin-Echinard V, Delpy L, et al. 17β-Estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor α signaling in macrophages in vivo. J Immunol. 2010;185(2):1169–1176. [DOI] [PubMed] [Google Scholar]
- 222. Villa A, Rizzi N, Vegeto E, Ciana P, Maggi A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci Rep. 2015;5:15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Matthews J, Almlöf T, Kietz S, Leers J, Gustafsson JA. Estrogen receptor-α regulates SOCS-3 expression in human breast cancer cells. Biochem Biophys Res Commun. 2005;335(1):168–174. [DOI] [PubMed] [Google Scholar]
- 224. Gyenes A, Hoyk Z, Csakvari E, Siklos L, Parducz A. 17β-Estradiol attenuates injury-induced microglia activation in the oculomotor nucleus. Neuroscience. 2010;171(3):677–682. [DOI] [PubMed] [Google Scholar]
- 225. Selvamani A, Sathyan P, Miranda RC, Sohrabji F. An antagomir to microRNA Let7f promotes neuroprotection in an ischemic stroke model. PLoS One. 2012;7(2):e32662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wise PM, Dubal DB, Rau SW, Brown CM, Suzuki S. Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the Women's Health Initiative. Endocr Rev. 2005;26(3):308–312. [DOI] [PubMed] [Google Scholar]
- 227. Azcoitia I, Arevalo MA, De Nicola AF, Garcia-Segura LM. Neuroprotective actions of estradiol revisited. Trends Endocrinol Metab. 2011;22(12):467–473. [DOI] [PubMed] [Google Scholar]
- 228. Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol. 2008;29(4):507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Suzuki S, Brown CM, Dela Cruz CD, Yang E, Bridwell DA, Wise PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc Natl Acad Sci USA. 2007;104(14):6013–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Sohrabji F, Williams M. Stroke neuroprotection: oestrogen and insulin-like growth factor-1 interactions and the role of microglia. J Neuroendocrinol. 2013;25(11):1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Elzer JG, Muhammad S, Wintermantel TM, et al. Neuronal estrogen receptor-α mediates neuroprotection by 17beta-estradiol. J Cereb Blood Flow Metab. 2010;30(5):935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Wieghofer P, Knobeloch KP, Prinz M. Genetic targeting of microglia. Glia. 2015;63(1):1–22. [DOI] [PubMed] [Google Scholar]
- 233. Alonso A, Hernán MA. Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology. 2008;71(2):129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Luchetti S, van Eden CG, Schuurman K, van Strien ME, Swaab DF, Huitinga I. Gender differences in multiple sclerosis: induction of estrogen signaling in male and progesterone signaling in female lesions. J Neuropathol Exp Neurol. 2014;73(2):123–135. [DOI] [PubMed] [Google Scholar]
- 235. Laffont S, Garnier L, Lélu K, Guéry JC. Estrogen-mediated protection of experimental autoimmune encephalomyelitis: lessons from the dissection of estrogen receptor-signaling in vivo. Biomed J. 2015;38(3):194–205. [DOI] [PubMed] [Google Scholar]
- 236. Santagati S, Melcangi RC, Celotti F, Martini L, Maggi A. Estrogen receptor is expressed in different types of glial cells in culture. J Neurochem. 1994;63(6):2058–2064. [DOI] [PubMed] [Google Scholar]
- 237. Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol. 2005;185(2):243–252. [DOI] [PubMed] [Google Scholar]
- 238. Garidou L, Laffont S, Douin-Echinard V, et al. Estrogen receptor α signaling in inflammatory leukocytes is dispensable for 17β-estradiol-mediated inhibition of experimental autoimmune encephalomyelitis. J Immunol. 2004;173(4):2435–2442. [DOI] [PubMed] [Google Scholar]
- 239. Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)α and ERβ ligand treatment. Proc Natl Acad Sci USA. 2007;104(37):14813–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Moore SM, Khalaj AJ, Kumar S, et al. Multiple functional therapeutic effects of the estrogen receptor β agonist indazole-Cl in a mouse model of multiple sclerosis. Proc Natl Acad Sci USA. 2014;111(50):18061–18066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Zhang J, Lapato A, Bodhankar S, Vandenbark AA, Offner H. Treatment with IL-10 producing B cells in combination with E2 ameliorates EAE severity and decreases CNS inflammation in B cell-deficient mice. Metab Brain Dis. 2015;30(5):1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Taylor LC, Gilmore W, Ting JP, Matsushima GK. Cuprizone induces similar demyelination in male and female C57BL/6 mice and results in disruption of the estrous cycle. J Neurosci Res. 2010;88(2):391–402. [DOI] [PubMed] [Google Scholar]
- 243. Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005;62(6):685–691. [DOI] [PubMed] [Google Scholar]
- 244. Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci. 2004;1019:24–28. [DOI] [PubMed] [Google Scholar]
- 245. Proust-Lima C, Amieva H, Letenneur L, Orgogozo JM, Jacqmin-Gadda H, Dartigues JF. Gender and education impact on brain aging: a general cognitive factor approach. Psychol Aging. 2008;23(3):608–620. [DOI] [PubMed] [Google Scholar]
- 246. Henderson VW, Buckwalter JG. Cognitive deficits of men and women with Alzheimer's disease. Neurology. 1994;44(1):90–96. [DOI] [PubMed] [Google Scholar]
- 247. Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA. 2002;99(11):7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Halford RW, Russell DW. Reduction of cholesterol synthesis in the mouse brain does not affect amyloid formation in Alzheimer's disease, but does extend lifespan. Proc Natl Acad Sci USA. 2009;106(9):3502–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Sturchler-Pierrat C, Staufenbiel M. Pathogenic mechanisms of Alzheimer's disease analyzed in the APP23 transgenic mouse model. Ann NY Acad Sci. 2000;920:134–139. [DOI] [PubMed] [Google Scholar]
- 250. Anderson GL, Manson J, Wallace R, et al. Implementation of the Women's Health Initiative study design. Ann Epidemiol. 2003;13(9 suppl):S5–S17. [DOI] [PubMed] [Google Scholar]
- 251. Carroll JC, Rosario ER, Kreimer S, et al. Sex differences in β-amyloid accumulation in 3xTg-AD mice: role of neonatal sex steroid hormone exposure. Brain Res. 2010;1366:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Di Paolo G, Kim TW. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41(10):1094–1099. [DOI] [PubMed] [Google Scholar]
- 255. Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41(10):1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303(18):1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43(5):429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43(5):436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368(2):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368(2):107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging. 2013;34(6):1711.e15–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Bertram L, Lange C, Mullin K, et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83(5):623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Li R, Shen Y, Yang LB, Lue LF, Finch C, Rogers J. Estrogen enhances uptake of amyloid β-protein by microglia derived from the human cortex. J Neurochem. 2000;75(4):1447–1454. [DOI] [PubMed] [Google Scholar]
- 264. Fan JD, Wagner BL, McDonnell DP. Identification of the sequences within the human complement 3 promoter required for estrogen responsiveness provides insight into the mechanism of tamoxifen mixed agonist activity. Mol Endocrinol. 1996;10(12):1605–1616. [DOI] [PubMed] [Google Scholar]
- 265. Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28(25):6333–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Reed JL, Dimayuga FO, Davies LM, Keller JN, Bruce-Keller AJ. Estrogen increases proteasome activity in murine microglial cells. Neurosci Lett. 2004;367(1):60–65. [DOI] [PubMed] [Google Scholar]
- 267. Fitzpatrick JL, Mize AL, Wade CB, Harris JA, Shapiro RA, Dorsa DM. Estrogen-mediated neuroprotection against β-amyloid toxicity requires expression of estrogen receptor α or β and activation of the MAPK pathway. J Neurochem. 2002;82(3):674–682. [DOI] [PubMed] [Google Scholar]
- 268. Zandi PP, Carlson MC, Plassman BL, et al. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA. 2002;288(17):2123–2129. [DOI] [PubMed] [Google Scholar]
- 269. Waring SC, Rocca WA, Petersen RC, O'Brien PC, Tangalos EG, Kokmen E. Postmenopausal estrogen replacement therapy and risk of AD: a population-based study. Neurology. 1999;52(5):965–970. [DOI] [PubMed] [Google Scholar]
- 270. Tang MX, Jacobs D, Stern Y, et al. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348(9025):429–432. [DOI] [PubMed] [Google Scholar]
- 271. Almeida S, Fiegenbaum M, de Andrade FM, Osório-Wender MC, Hutz MH. ESR1 and APOE gene polymorphisms, serum lipids, and hormonal replacement therapy. Maturitas. 2006;54(2):119–126. [DOI] [PubMed] [Google Scholar]
- 272. Yaffe K. Estrogens, selective estrogen receptor modulators, and dementia: what is the evidence? Ann NY Acad Sci. 2001;949:215–222. [DOI] [PubMed] [Google Scholar]
- 273. Mulnard RA, Cotman CW, Kawas C, et al. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer's Disease Cooperative Study. JAMA. 2000;283(8):1007–1015. [DOI] [PubMed] [Google Scholar]
- 274. Shao H, Breitner JC, Whitmer RA, et al. Hormone therapy and Alzheimer disease dementia: new findings from the Cache County Study. Neurology. 2012;79(18):1846–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Bimonte-Nelson HA, Francis KR, Umphlet CD, Granholm AC. Progesterone reverses the spatial memory enhancements initiated by tonic and cyclic oestrogen therapy in middle-aged ovariectomized female rats. Eur J Neurosci. 2006;24(1):229–242. [DOI] [PubMed] [Google Scholar]
- 276. Carroll JC, Rosario ER, Chang L, et al. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007;27(48):13357–13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Gibbs RB. Effects of gonadal hormone replacement on measures of basal forebrain cholinergic function. Neuroscience. 2000;101(4):931–938. [DOI] [PubMed] [Google Scholar]
- 278. Haaxma CA, Bloem BR, Borm GF, et al. Gender differences in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2007;78(8):819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Cantuti-Castelvetri I, Keller-McGandy C, Bouzou B, et al. Effects of gender on nigral gene expression and Parkinson disease. Neurobiol Dis. 2007;26(3):606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Murray HE, Pillai AV, McArthur SR, et al. Dose- and sex-dependent effects of the neurotoxin 6-hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: differential actions of estrogen in males and females. Neuroscience. 2003;116(1):213–222. [DOI] [PubMed] [Google Scholar]
- 281. Miller DB, Ali SF, O'Callaghan JP, Laws SC. The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann NY Acad Sci. 1998;844:153–165. [PubMed] [Google Scholar]
- 282. Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol. 1996;50:275–331. [DOI] [PubMed] [Google Scholar]
- 283. Quinn NP, Marsden CD. Menstrual-related fluctuations in Parkinson's disease. Movement Disord. 1986;1(1):85–87. [DOI] [PubMed] [Google Scholar]
- 284. Benedetti MD, Maraganore DM, Bower JH, et al. Hysterectomy, menopause, and estrogen use preceding Parkinson's disease: an exploratory case-control study. Movement Disord. 2001;16(5):830–837. [DOI] [PubMed] [Google Scholar]
- 285. Rocca WA, Bower JH, Maraganore DM, et al. Increased risk of Parkinsonism in women who underwent oophorectomy before menopause. Neurology. 2008;70(3):200–209. [DOI] [PubMed] [Google Scholar]
- 286. Ragonese P, D'Amelio M, Savettieri G. Implications for estrogens in Parkinson's disease: an epidemiological approach. Ann NY Acad Sci. 2006;1089:373–382. [DOI] [PubMed] [Google Scholar]
- 287. Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson's disease. J Neuroimmune Pharmacol. 2013;8(1):189–201. [DOI] [PubMed] [Google Scholar]
- 288. Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20(16):6309–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Stott SR, Barker RA. Time course of dopamine neuron loss and glial response in the 6-OHDA striatal mouse model of Parkinson's disease. Eur J Neurosci. 2014;39(6):1042–1056. [DOI] [PubMed] [Google Scholar]
- 290. Castaño A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70(4):1584–1592. [DOI] [PubMed] [Google Scholar]
- 291. Hoban DB, Connaughton E, Connaughton C, et al. Further characterisation of the LPS model of Parkinson's disease: a comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain Behav Immun. 2013;27(1):91–100. [DOI] [PubMed] [Google Scholar]
- 292. Beach TG, Sue LI, Walker DG, et al. Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol. 2007;114(4):419–424. [DOI] [PubMed] [Google Scholar]
- 293. Almey A, Filardo EJ, Milner TA, Brake WG. Estrogen receptors are found in glia and at extranuclear neuronal sites in the dorsal striatum of female rats: evidence for cholinergic but not dopaminergic colocalization. Endocrinology. 2012;153(11):5373–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Thompson TL, Moss RL. Estrogen regulation of dopamine release in the nucleus accumbens: genomic- and nongenomic-mediated effects. J Neurochem. 1994;62(5):1750–1756. [DOI] [PubMed] [Google Scholar]
- 295. Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55(5):453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Liu Y, Qin L, Wilson B, et al. Endotoxin induces a delayed loss of TH-IR neurons in substantia nigra and motor behavioral deficits. Neurotoxicology. 2008;29(5):864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Vegeto E, Bonincontro C, Pollio G, et al. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci. 2001;21(6):1809–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Joniec I, Ciesielska A, Kurkowska-Jastrzebska I, Przybylkowski A, Czlonkowska A, Czlonkowski A. Age- and sex-differences in the nitric oxide synthase expression and dopamine concentration in the murine model of Parkinson's disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2009;1261:7–19. [DOI] [PubMed] [Google Scholar]
- 299. Morale MC, Serra PA, L'episcopo F, et al. Estrogen, neuroinflammation and neuroprotection in Parkinson's disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience. 2006;138(3):869–878. [DOI] [PubMed] [Google Scholar]
- 300. Sárvári M, Hrabovszky E, Kalló I, et al. Estrogens regulate neuroinflammatory genes via estrogen receptors α and β in the frontal cortex of middle-aged female rats. J Neuroinflammation. 2011;8:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Rodriguez-Perez AI, Valenzuela R, Joglar B, Garrido-Gil P, Guerra MJ, Labandeira-Garcia JL. Renin angiotensin system and gender differences in dopaminergic degeneration. Mol Neurodegener. 2011;6(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Rodriguez-Perez AI, Valenzuela R, Villar-Cheda B, Guerra MJ, Lanciego JL, Labandeira-Garcia JL. Estrogen and angiotensin interaction in the substantia nigra. Relevance to postmenopausal Parkinson's disease. Exp Neurol. 2010;224(2):517–526. [DOI] [PubMed] [Google Scholar]
- 303. Yung LM, Wong WT, Tian XY, et al. Inhibition of renin-angiotensin system reverses endothelial dysfunction and oxidative stress in estrogen deficient rats. PLoS One. 2011;6(3):e17437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Maggi A, Ciana P. Reporter mice and drug discovery and development. Nat Rev Drug Discov. 2005;4(3):249–255. [DOI] [PubMed] [Google Scholar]
- 305. Brown CM, Choi E, Xu Q, Vitek MP, Colton CA. The APOE4 genotype alters the response of microglia and macrophages to 17beta-estradiol. Neurobiol Aging. 2008;29(12):1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Lee M, McGeer EG, McGeer PL. Sodium thiosulfate attenuates glial-mediated neuroinflammation in degenerative neurological diseases. J Neuroinflammation. 2016;13:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Ghosh A, Langley MR, Harischandra DS, et al. Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of Parkinson's disease. J Neuroimmune Pharmacol. 2016;11(2):259–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Onasanwo SA, Velagapudi R, El-Bakoush A, Olajide OA. Inhibition of neuroinflammation in BV2 microglia by the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant protective mechanism. Mol Cell Biochem. 2016;414:23–36. [DOI] [PubMed] [Google Scholar]
- 309. Verreck FA, de Boer T, Langenberg DM, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. 2004;101(13):4560–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73(2):209–212. [DOI] [PubMed] [Google Scholar]
- 312. N′Diaye EN, Branda CS, Branda SS, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184(2):215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–185. [DOI] [PubMed] [Google Scholar]
- 314. Turnbull IR, Gilfillan S, Cella M, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177(6):3520–3524. [DOI] [PubMed] [Google Scholar]
- 315. Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Zeisel A, Muñoz-Manchado AB, Codeluppi S, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–1142. [DOI] [PubMed] [Google Scholar]
- 317. Chiu IM, Morimoto ET, Goodarzi H, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4(2):385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]