

Abstract
SHOX deficiency is the most frequent genetic growth disorder associated with isolated and syndromic forms of short stature. Caused by mutations in the homeobox gene SHOX, its varied clinical manifestations include isolated short stature, Léri-Weill dyschondrosteosis, and Langer mesomelic dysplasia. In addition, SHOX deficiency contributes to the skeletal features in Turner syndrome. Causative SHOX mutations have allowed downstream pathology to be linked to defined molecular lesions. Expression levels of SHOX are tightly regulated, and almost half of the pathogenic mutations have affected enhancers. Clinical severity of SHOX deficiency varies between genders and ranges from normal stature to profound mesomelic skeletal dysplasia. Treatment options for children with SHOX deficiency are available. Two decades of research support the concept of SHOX as a transcription factor that integrates diverse aspects of bone development, growth plate biology, and apoptosis. Due to its absence in mouse, the animal models of choice have become chicken and zebrafish. These models, therefore, together with micromass cultures and primary cell lines, have been used to address SHOX function. Pathway and network analyses have identified interactors, target genes, and regulators. Here, we summarize recent data and give insight into the critical molecular and cellular functions of SHOX in the etiopathogenesis of short stature and limb development.
Introduction
-
The SHOX Gene
Identification of SHOX
SHOX gene structure
SHOX gene expression
Mechanisms underlying SHOX regulation at the transcriptional and post-transcriptional level
-
The SHOX Protein
SHOX is a transcription factor
SHOX functional domains
-
SHOX-Related Pathways
SHOX is expressed in the growth plate
SHOX is a modulator of cell proliferation and apoptosis
Transcriptional targets
Upstream regulators
Possible roles of SHOX in bone development
-
Clinical Implications of SHOX Deficiency
Léri-Weill dyschondrosteosis
Langer mesomelic dysplasia
Turner syndrome
Sex chromosome aneuploidies
Idiopathic short stature
SHOX enhancer deletions in LWD and ISS
Treatment of SHOX deficiency
Clinical indicators of SHOX deficiency
Conclusions
I. Introduction
Adult body height depends substantially on the length of the long bones. Bone development and longitudinal growth are highly complex developmental processes that are influenced by multiple environmental and genetic factors. For example, malnutrition and untreated infectious disease hinder growth. However, environmental factors account for a relatively small percentage of the height variation within a population. Instead, most of the variation is due to genetic and possibly epigenetic factors. Not surprisingly, defects in the genes involved in bone development can produce diseases with varied skeletal defects, altered bone growth, and stature below or above the mean.
Short stature is a descriptive term indicating height that is significantly below the average of the general population for that person's age and sex. More precisely, short stature is statistically defined as 2 SD below the mean population height for age and sex (less than the third percentile) or, when evaluating shortness in relation to family background, more than 2 SD below the midparental height (1). It affects approximately 3% of children worldwide and is therefore a condition for which clinical attention is frequently sought during childhood. This is appropriate because growth failure may be an early sign of serious renal, gastrointestinal, endocrine, or genetic disease.
More than 150 genes are known to be involved in the etiology of syndromes characterized by short stature (2–4, 81). Genetic abnormalities associated with short stature include major chromosomal rearrangements, large-scale deletions or loss of a whole chromosome, and point mutations, small deletions or insertions, or copy number variation in key genes involved in bone development. Examples include mutations to SOX9 (MIM: 608160) (6), COL2A1 (7), and FGFR3 (MIM: 134934) (8, 9). (For a list of genes whose mutations are associated with short stature, we redirect the reader to the following reviews: Refs. 2, 4, 5, 10, and 11.) However, such known mutations appear to explain only a small percentage of growth failure cases; a high percentage of clinical conditions with short stature cases remains idiopathic. The identification of underlying gene defects in such cases will be crucial for more accurate diagnosis of growth disorders and for the development of novel tailored clinical interventions.
II. The SHOX Gene
A. Identification of SHOX
Numerous studies indicate a fundamental role of human sex chromosomes in height determination. For example, complete loss of the X chromosome causes the short stature and other abnormalities found in patients with Turner syndrome (TS) (MIM: 313000) (12). Deletions of the short arm of the X chromosome and the short arm of the Y chromosome (12–14) including the pseudoautosomal region 1 (PAR1) have also been consistently linked with short stature (15).
On the basis of these studies, PAR1 was proposed as a candidate region containing a key genetic locus involved in growth determination. Recombination frequency in this region in males is very high (16, 17). Genes residing in this region escape X inactivation (the process by which one of the X chromosomes is rendered genetically silent), and therefore two active copies of the genes are required for normal physiological function. Consequently, short stature phenotypes were considered to arise as a result of haploinsufficiency of the critical short stature gene in PAR1 (12, 14, 15). Deletion mapping of short stature patients with chromosomal aberrations narrowed down a critical interval, in proximity to the telomeric end of the short arms of the sex chromosomes, that was deleted in these patients (18). Subsequent cDNA and exon amplification within this region identified a novel homeobox gene, which was termed SHOX (MIM: 312865) for short stature homeobox-containing gene on chromosome X, and suggested to segregate with the short stature phenotype observed in these patients (12, 19). At about the same time, another group independently identified the same gene using a complementary approach based on a yeast artificial chromosome encompassing the 700-kb critical interval. The gene was named PHOG, for pseudoautosomal homeobox-containing osteogenic gene (20). Rao et al (19) provided the final proof for a role of SHOX in linear growth by screening individuals with idiopathic short stature (ISS) and identifying a point mutation (c.583CvT) leading to a premature stop codon (p.Arg195*) in exon 5 of SHOX in one patient. Pedigree analysis of the patient's family showed that the mutation cosegregated with the short stature phenotype in all affected members of the family (19).
After its discovery, a number of studies linked SHOX mutations to the short stature and the skeletal defects associated with Léri-Weill dyschondrosteosis (LWD) (MIM: 127300), Langer mesomelic dysplasia (MIM: 249700), and TS (MIM: 313000). Moreover, the association between SHOX deficiency and the short stature phenotype in a significant fraction of short individuals previously described as idiopathic became apparent, making mutations of this gene the most common genetic defect leading to short stature in humans. The clinical implication of SHOX mutations, therapeutic interventions, and clinical indicators of SHOX deficiency will be discussed in more detail below.
B. SHOX gene structure
The SHOX gene (MIM: 312865) spans approximately 40 kb, about 500 kb from the telomeres of sex chromosomes. Initial characterization of SHOX revealed seven exons encoding two alternatively spliced transcripts termed SHOXa and SHOXb. The two transcripts, which are identical at the 5′ end but differ in the final exon (6a vs 6b) at the 3′ end (19), are translated into distinct protein isoforms of 292 (SHOXa) and 225 (SHOXb) amino acids. However, the SHOX genomic structure has been extended recently to include four additional exons (2a, 7–1, 7–2, and 7–3) that encode novel SHOX isoforms (Figure 1 and below) (21).
Figure 1.

The SHOX gene. The SHOX gene maps to 505–527 kb from the telomere of the sex chromosomes on Xp22.33 and Yp11.32 and spans approximately 40 kb. It is composed of nine exons that produce two main transcripts, SHOXa and SHOXb, of different length. The two transcripts contain a DNA sequence called a homeobox that encodes the homeodomain, a conserved DNA-binding domain that characterizes the family of the homeodomain-containing transcription factors. Alternative SHOX isoforms are also formed by alternative splicing of the exons. The function of these isoforms is not entirely clear, but they may be involved in the spatiotemporal regulation of SHOX expression and activity (see also Section II C). Exon 7 variants are found to be exclusively expressed in fetal neuro-tissues arguing for a specific role of these variants during brain development. The different mRNAs are predicted to lead to peptides of different length. Tel, telomere.
Sequence alignments identified SHOX as a member of the Paired-like homeobox-containing genes (19, 20). This family codes for transcription factors containing a characteristic 60-amino acid DNA-binding domain called the homeodomain. Homeobox genes regulate pattern formation and organogenesis during both vertebrate and invertebrate embryogenesis and development (22, 23). They can function as both transcriptional activators and repressors in regulating the temporal and spatial expression of different target genes. SHOX has high similarity to the human SHOX2 gene and its mouse ortholog Shox2 (24). SHOX is present in most vertebrate species including chimpanzee, dog, chicken, frog, and fish, with the notable exception of rodents, where the gene was lost during evolution (25).
C. SHOX gene expression
1. Human
Expression studies revealed a clear difference in the expression pattern of the SHOX a and b isoforms. Whereas both are expressed predominantly in bone marrow fibroblasts, SHOXa is also expressed in several other tissues (19). More recently, analysis of the SHOX expression pattern in embryonic, fetal, and adult tissues—by real-time PCR (RT-PCR) coupled with sequence validation—revealed SHOX expression in multiple fetal (muscle, skin, intestine, eye, brain, spinal cord) and adult tissues, with the strongest expression in placenta, skeletal muscle, bone marrow, and adipose tissue (21). In addition, weak expression was detected in fetal and adult brain regions including the cerebellum, thalamus, and basal ganglia, implying a yet uncharacterized role for SHOX in brain development (21). In particular, the newly identified exon 2a was detected in several fetal and adult brain tissues, with the strongest expression in fetal eye and brain, and in adult bone marrow and skeletal muscle, whereas the three exon 7-containing splice variants were only detected at higher abundance in fetal brain tissues. The exact role of these novel SHOX isoforms remains to be deciphered, but their involvement has been postulated in the developmental and tissue-specific regulation of SHOX expression (21).
In situ hybridization studies on human embryos revealed a spatiotemporally restricted expression pattern of SHOX. Expression in the developing limbs is detected at Carnegie stage (CS) 14 as a broad band across the middle part of the limb (CS14 roughly corresponds to 5 weeks after conception). During condensation and chondrification, the expression becomes more pronounced around the precartilaginous anlagen of the elbow. When the various bones of the arm can be identified (at CS21), SHOX expression is still mainly confined to the middle portion of the arm, around the distal ends of the humerus, radius, and ulna, and in a few bones of the wrist. Analogously, the results of SHOX in situ hybridization on the lower limbs resembled the expression pattern described for the upper limb development. SHOX is also present in the first and second pharyngeal arches (25) that give rise to the maxilla, mandible, and some bones of the ear. Remarkably, TS and LWD patients with SHOX deficiency display skeletal defects in the same anatomical structures in which SHOX is expressed—forearm and lower legs as well as the maxilla, mandible, and external ear tract—providing clinical evidence for the role of SHOX in development of these structures (25). SHOX2 is also expressed in limbs, but in a more proximal position than SHOX (25). In addition, SHOX2 transcripts are detected in the developing heart (24–26).
2. Chicken
Similar to the human embryo expression pattern, chicken embryo Shox expression is observed in the central regions of early limb buds and is restricted to the distal two-thirds of the limb in later stages of development (27). These areas correspond to those affected in humans with SHOX deficiency. Shox is also expressed in connective tissue around cartilage and muscle, in a layer of cells below the dermis, and in the branchial arches, nervous system, and vasculature. The expression pattern of Shox2 in chicken limb buds also resembles that in humans and is confined mostly to the proximal third of the limb bud. Also similar to human embryos, chicken embryo Shox2 expression overlaps that of Shox only in the proximal limb bud (27). Given these similarities, the chick embryo has become a model system for studying the functions of both Shox and Shox2 in limb development (28–31).
3. Zebrafish
Studies on zebrafish, which also express both Shox and Shox2, confirmed an important role for Shox in embryonic growth and bone formation because morpholino-mediated silencing of Shox resulted in significant growth retardation (reduced somite number and body length) and decreased ossification in anterior vertebrae and a subset of craniofacial bones (32). Shox was expressed in a variety of zebrafish tissues and organs from embryonic to adult stage, including blood, heart, hatching gland, pharyngeal arch, olfactory epithelium, and fin bud. The predominant domains of Shox expression were mandibular arch, pectoral fins, anterior notochord, rhombencephalon, and mesencephalon, suggesting that Shox is involved in both bone and neural development (32, 33). Given that the osteogenic role of Shox appears to be conserved throughout evolution from fish to human, the zebrafish model may be a valuable tool to characterize SHOX functions by facilitating identification of SHOX gene expression regulators, SHOX-interacting proteins, and SHOX target genes and should therefore be explored further.
4. Rodents
Shox does not exist in rodents. Because rodents do not harbor Shox, but harbor only Shox2, Shox2 may have assumed the functions of both SHOX and SHOX2 (MIM: 602504) in these animals (34). Mice homozygous for the deleted Shox2 allele die during embryogenesis between embryonic day 11.5 (E11.5) and E14.5 (35, 36) or at E17.5 (37) due to aberrant formation of the sinoatrial node. These abnormalities lead to cardiovascular defects such as severe pacemaking and conduction deficiencies, indicating that Shox2 plays a critical role in heart development. The lethal mouse phenotype caused by Shox2 deletion provides a potential explanation for why homozygous SHOX2 deficiency has not been observed in any known human syndrome. Studies with conditional Shox2 knockout mouse models revealed that Shox2 also plays a critical role in the normal development of the proximal vertebrate limb because its genetic ablation results in severe shortened stylopodial elements (humerus and femur) (34). Shox2 is expressed in growth plate chondrocytes and in the perichondrium, and its removal from limb results in delayed chondrogenesis responsible for the dwarf phenotype observed (38). Shox2 regulates chondrocyte maturation by modulating Runx2 transcription (34, 39), a function that exerts in concert with Hox family members (38) (see also section IV D).
Shox2/SHOX2 expression in chicken and human is mainly found to the stylopodial elements and juxtaposes the expression pattern of Shox/SHOX that is instead mainly found in the zeugopodial elements (radius/ulna and fibula/tibia) (25, 27). Shox2 in mice is instead expressed in both the developing stylopodial and zeugopodial elements (34, 39). Therefore, it has been proposed that the extended expression of Shox2 is one of the mechanisms that mice have evolved to compensate for the lack of Shox in the developing forelimb (38).
A number of studies indicate that Shox2 is also required for the control of other development processes, including the development of nerves and muscles in the forelimb (40), palate (37), temporomandibular joint (41), facial motor nucleus and facial nerves (42), mechanosensory neurons of the dorsal root ganglia (43), and dorsal cerebellum (42). In addition, SHOX2 is also expressed in human and mouse sc adipocytes, where it is involved in the regulation of lipolysis by controlling the expression of the β3 adrenergic receptor encoding gene. Specific disruption of Shox2 in adypocytes protected mice from high fat diet-induced obesity (44).
D. Mechanisms underlying SHOX regulation at the transcriptional and post-transcriptional level
SHOX expression is restricted to certain compartments during specific phases of development, implying a tight spatiotemporal regulation. Several layers of complexity have emerged for regulating SHOX gene expression, including alternative promoters, alternative exons, and cis-regulatory sequences (enhancers) positioned up- and downstream of the transcription unit.
1. Alternative promoters
Two alternative promoters, P1 and P2, were reported to control SHOX expression at the transcriptional level. The two promoters generate two classes of mRNA that encode identical proteins but differ in their 5′ untranslated region (UTR) by the presence of seven AUG codons upstream of the SHOX open reading frame. The transcripts containing these seven AUG elements are translated with reduced efficiency, providing an interesting mechanism of regulation of SHOX protein levels not only at the transcriptional but also at the translational level (45). The two promoters may be alternatively used in response to different physiological situations, thereby contributing to the fine-tuned regulation of the levels and tissue specificity of SHOX expression. However, the circumstances under which one promoter is preferred over the other and the molecular mechanisms controlling their individual activity remain to be deciphered.
2. Enhancer elements
Cis-acting regulatory elements often influence the spatiotemporal expression of genes involved in embryonic development and differentiation. These noncoding DNA sequences enhance or repress gene transcription and may be located near the protein-coding region or at a considerable distance from it (up to 2 million base pairs away) (46). Generally, these regulatory elements are conserved throughout evolution because of their importance in transcriptional regulation (47). Disruption leading to haploinsufficiency has been reported in a number of human syndromes (48). Several conserved noncoding elements (CNEs), four downstream (31, 49, 50) and three upstream (with a distance of 200 and 250 kb of the SHOX gene, respectively) (30), have been identified within PAR1 (Figure 2). These elements are conserved in all species in which SHOX is present. CNE-4 and CNE-5 also reside near the SHOX paralog SHOX2 and were presumably duplicated together with the coding sequence (33). They already exist in fugu, the most evolutionary distant vertebrate, suggesting functional significance.
Figure 2.

Mechanisms regulating SHOX gene expression. A, Alternative promoters. Two promoters, P1 and P2, control SHOX expression. These promoters produce transcripts that differ in their 5′-UTR: P1 produces transcripts containing seven untranslated AUG codons, whereas P2 transcripts lack these regulative elements. The two types of mRNA are translated with different efficiency, thereby contributing to the fine-tuned regulation of the levels and tissue specificity of SHOX expression. B, Enhancer elements. Evolutionarily conserved regions in PAR1 identified in different studies. CNE, highly evolutionarily conserved noncoding DNA elements (30, 31, 162); ECR, evolutionarily conserved sequence (29); ECS, evolutionarily conserved sequence (5, 49). The upper horizontal line indicates the physical distance from Xp/Yp telomere (Tel;hg19,build37). Genomic positions: SHOXa (NM_000451.3), chrX:585, 079-607558; CNE-2, chrX:516, 610-517229; CNE-3, chrX:460, 279-460664; CNE-5, chrX:398, 357-398906; CNE-4, chrX:714, 085-714740; CNE-5, chrX:750, 825-751850; ECR1, xhrX:780, 580-781235; and CNE-9/ECS4, chrX:834, 746-835548 (Tel;hg19,build37). C, Splice variants. Different isoforms are generated by the SHOX gene through alternative splicing. Addition of exon 7 can be either attached directly to exon 5 and therefore become a part of the open reading frame or elongates the 3′-UTR of the SHOX transcript. The different 3′-UTR may be subjected to alternative microRNA-mediated regulation. Insertion of exon 2a leads to a premature stop codon in exon 3, which could lead to mRNA degradation. Exon III and IV contain the homeobox. Light gray boxes indicate untranslated regions; dark gray boxes depict open reading frame.
The function of these CNEs as enhancers of SHOX transcription has been demonstrated in human cells (49, 50), in chicken limb buds (30, 31, 50), and in zebrafish (33), suggesting that they may play a similar role in the regulation of SHOX expression during bone development in different species. In addition, several CNEs have also been shown to regulate shox expression in zebrafish brain and muscle tissue (33). In Section V.E, we will discuss the involvement of these regions in SHOX-related diseases.
3. Alternative exons
We have already cited the existence of several SHOX splice variants generated by alternative usage of SHOX exons. The mRNAs encoding these SHOX isoforms differ in the 3′-UTR region, and therefore their expression may be subject to diverse microRNA regulation. In addition, they encode SHOX isoforms (eg, SHOXb) that contain the same homeodomain of SHOXa but lack the protein transactivation domain (encoded by exon 6a) and are therefore unable to activate transcription. These isoforms might act as negative regulators of SHOXa by competing for the same DNA consensus sequences or by dimerizing with the active protein. The mechanisms regulating SHOX gene expression are summarized in Figure 2.
III. The SHOX Protein
The two proteins, SHOXa and SHOXb, share many of the features described below because SHOXb is identical to SHOXa in its first 211 amino acids and lacks only the C-terminal portion. Because all functional studies performed on SHOX so far have focused on SHOXa, this section refers to this isoform (hereafter referred to as SHOX).
A. SHOX is a transcription factor
SHOX is characterized by the presence of a homeodomain that is identical to the homeodomains of both SHOX2 and its mouse ortholog Shox2 (24). The homeodomain is composed of three helices. Helices I and II are antiparallel to each other, and helices II and III form a helix-turn-helix motif that is separated from helix I by a loop (22, 51). Helix III of the homeodomain (also called the recognition helix) contacts the DNA major groove, whereas its flexible N terminus inserts into the minor DNA groove (22, 52, 53).
First insights into the biological role of SHOX derived from studies of osteogenic (U2OS-TRex) and embryonic kidney (HEK 293-TRex) stable cell lines in which SHOX expression was under a Tet-on/Tet-off inducible system. The activities of wild-type SHOX were compared with those of a C-terminally truncated version (SHOX L185X, termed SHOX-STM) that resembled a SHOX mutant previously identified in individuals with LWD or ISS (c.583C>T) (19, 25, 54–58).
Transient transfection experiments showed that both wild-type SHOX and SHOX-STM localized in an unevenly distributed focal pattern within the nucleus of all cell lines analyzed (human U2OS and HEK-293, simian Cos7, and murine NIH 3T3) (51). Within the nucleus, colocalization studies indicated that SHOX did not colocalize with nucleoli, centromeres, or the basic transcription factor TFIIH. Therefore, the nature and composition of SHOX foci remain to be characterized. Experiments with a green fluorescent protein (GFP)-tagged version of SHOX (SHOX-GFP) also confirmed SHOX nuclear localization, which is consistent with its role as a transcription factor.
After screening an oligonucleotide random library (using systematic evolution of ligands by exponential enrichment), putative SHOX recognition sequences were isolated and amplified using PCR. SHOX binding to these DNA sequences was confirmed by electromobility shift assays. Similar to other paired-like homeodomain proteins (59), SHOX preferentially binds to palindromic motifs of the type 5′-TAAT(N)2–3ATTA, referred to as P2 or P3 elements, according to the number of nucleotides that separate the palindromic half-sites. Many homeodomain proteins, especially those of the paired-like class, exert their function by a cooperative dimerization during DNA binding (60). Consistent with this, SHOX is able to bind to DNA as both a monomer and a homodimer (51). The formation of SHOX homodimers was also confirmed by yeast two-hybrid system studies using SHOX as both bait and prey (51). Finally, SHOX-responsive elements were cloned in front of a Simian virus 40 minimal promoter that controlled expression of a luciferase reporter gene. Transient transfection of this plasmid in SHOX or SHOX-STM U2OS and HEK293 stable cell lines demonstrated that SHOX acts as a transcriptional activator of the luciferase reporter gene in osteosarcoma U2OS cells but not in HEK293 cells, suggesting that cell type-specific cofactors present in osteogenic cells are required for the transcriptional activity of SHOX (51).
B. SHOX functional domains
Missense mutations within the homeodomain that lead to amino-acid substitutions have been described in individuals with LWD or ISS. Nine different SHOX missense mutations within the homeodomain of LWD or ISS patients were analyzed functionally and shown to cause decreased SHOX biological function by affecting DNA binding, dimerization, and/or nuclear translocation (61). These studies helped to unravel important functional domains of the SHOX protein (Figure 3A) and provided an explanation at the molecular level for the clinical conditions present in patients with LWD and ISS (61–63).
Figure 3.

SHOX at a glance. A, Functional domains. Schematic view of the SHOX protein and its main functional domains. HD, homeodomain; NLS, nuclear localization signal; OAR, transactivation domain; N, N terminus; C, C terminus. B, SHOX interactome. The cellular factors known to interact with SHOX or mediate its cellular functions are depicted as a network.
1. Nuclear localization signal
Mutations in the amino acids A170 and R173 (p.A170P, p.R173C, and p.R173H), which have been found in three different families with LWD (61, 62, 64–67), resulted in an aberrant localization of SHOX in the cytoplasm (61, 68). Study of these mutant SHOX proteins led to discovery of the SHOX nuclear localization signal, which resides within the recognition helix of the homeodomain and represents a basic, nonclassical signal defined by the five amino acids AKCRK (68). All three LWD missense mutations altered this SHOX nuclear localization signal. Insertion of the AKCRK motif adjacent to the mutated amino acids restored the ability of SHOX proteins to translocate into the nucleus. Because SHOX must first translocate into the nucleus to exert its function as a transcription factor, these studies establish the impairment of nuclear localization as a mechanism underlying SHOX-related diseases (68).
2. SHOX dimerization domain
Several different SHOX homeodomain missense mutations found in patients with LWD and ISS were shown to diminish the dimerization ability of the SHOX protein (ie, p.L132V, p.R168W, p.A170P, and p.R173H), thus indicating involvement of the homeodomain in dimerization (61). Such mutations are thought to impair SHOX transcriptional activity by impairing the ability of the protein to form dimers.
3. SHOX transactivation domain
A truncated version of the SHOX protein (p.L185X) was unable to activate transcription in osteosarcoma U2OS cells, indicating that the C-terminal portion of the protein, which harbors an OAR (otp, aristaless, and rax) transactivation domain (69–71), is necessary for the transcriptional activity of SHOX. Because the SHOXb isoform lacks this region, it was suggested that this protein is inactive as a transcriptional activator. On the other hand, by sharing the same homeodomain, SHOXb can bind to the same DNA sequences as SHOXa and may therefore form heterodimers with SHOXa and modulate its activity (51).
In addition, one SHOX missense mutation within the homeodomain (p.R153L), which was reported to segregate with disease in at least four independent families, was also unable to activate transcription despite the mutated protein's ability to enter the nucleus, bind to DNA, and dimerize with similar efficiency to wild-type protein (61). These results suggest that the homeodomain itself contributes to the transactivation activity of SHOX.
4. SHOX phosphorylation site
SHOX is multiphosphorylated in vivo exclusively on serine residues, with Ser106 being the major SHOX phosphorylation site (63). Phosphorylation modulates the biological function of SHOX because substitution of A at S106 impaired its transcriptional activation capacity without affecting its nuclear localization and DNA-binding ability (63).
Ser106 and its adjacent residues Glu107, Asp108, and Glu109 represent a canonical phosphorylation consensus site (SEDE) of casein kinase II (CKII) that preferentially phosphorylates serine in acidic residue-rich regions. Consistent with this observation, CKII was shown to be involved in SHOX phosphorylation because the kinase efficiently phosphorylated SHOX on Ser106 in vitro and CKII-specific inhibitors strongly reduced SHOX phosphorylation in SHOX-expressing cells in vivo. Two SHOX variants harboring missense mutations in Ser106 (c.317>G leading to p.S106W) or in the CKII phosphorylation consensus site (c.325G>T leading to p.E109Q) have been detected in LWD individuals (http://www.shox.uni-hd.de). Most likely these mutants are defective in phosphorylation and thereby transcriptionally inactive.
IV. SHOX-Related Pathways
A. SHOX is expressed in the growth plate
Adult body height depends substantially on the length of the long bones. Long bone elongation occurs in the growth plate, a thin layer of cartilage entrapped between the epiphyseal and metaphyseal bone surrounded by the perichondrium, a layer of dense connective tissue that separates the developing skeletal elements from the surrounding mesenchyme (72).
The growth plate is a highly organized structure that can be subdivided into three distinct layers: the resting, proliferative, and hypertrophic zones (73). Each zone contains chondrocytes at a different stage of differentiation. The resting zone closest to the epiphysis consists of undifferentiated resting chondrocytes, directly derived from mesenchymal stem cells, displaying a typically round phenotype. These cells are rich in lipid, implying nutrient storage potential. They border the proliferative zone and function as precursor cells, generating new clones of rapidly proliferating chondrocytes that undergo several rounds of cell division in a column-wise orientation along the longitudinal axis of the growth plate. Proliferating chondrocytes secrete large amounts of matrix components, such as collagen type II, IX, and XI (MIM: 120140, 120120, 120280), and the proteoglycan aggrecan (MIM: 155760), whose formation is stimulated by up-regulation of the transcription factor SOX9 (MIM: 608160) (74).
At a certain stage, chondrocytes of the proliferative zone stop dividing and become hypertrophic, increasing their size 6- to 10-fold. They secrete large amounts of extracellular matrix rich in type X collagen (MIM: 120110) (72). Furthermore, these terminally differentiated chondrocytes express additional molecular markers such as vascular endothelial growth factor (MIM: 192240) and matrix metalloproteinase 13 (MIM: 600108). Vascular endothelial growth factor stimulates vascularization, whereas matrix metalloproteinase 13 serves to degrade the extracellular matrix proteins, thus facilitating vascular invasion (75).
The growth plate is the engine of longitudinal bone elongation—a coordinated process of mesenchymal condensation and chondrocyte proliferation, maturation, and hypertrophy, followed by vascular invasion and migration into the growth plate of osteoblasts and other bone marrow cell types, which together produce longitudinal bone growth (73). The list of factors that regulate growth plate physiology has been greatly enlarged, along with the fast technological development of the recent years in particular with the introduction of whole-genome single nucleotide polymorphism (SNP) arrays, array-comparative genomic hybridization, and whole-exome sequencing for the detection of gene variants that affect bone development and linear growth (2, 76–81). Studies of transgenic mice have provided functional insights into the role of some of these factors as molecular drivers of endochondral bone ossification. Multiple hormones, paracrine factors, extracellular matrix molecules, and intracellular proteins govern in a coordinated fashion the activity of growth plate chondrocytes through a wide variety of mechanisms. In this section, we will mainly focus on those factors and mechanisms that have been directly or indirectly linked with SHOX and SHOX pathways (Figure 3B and Table 1). For other regulatory systems and signaling cascades that regulate the complex process of endochondral ossification, we redirect the readers to excellent reviews on this topic (4, 72, 73, 77, 82). In general, among the hormones that modulate linear growth are the GH, IGFs, thyroid hormone, glucocorticoids, estrogens, and androgens (4, 83–85). Chondrocytes in the growth plate and to a lesser extent cells in the adjacent perichondrium modulate longitudinal bone growth by secreting an array of paracrine signaling molecules—such as retinoic acid (RA) (86), Indian hedgehog (MIM: 600726) (73, 87), PTHrP (MIM: 158470) (73), bone morphogenetic proteins (BMPs) (MIM: 604444) (88, 89), wingless-type mouse mammary tumor virus (MMTV)-integration site family members (90, 91), fibroblast growth factors (FGFs) (92), C-type natriuretic peptide (CNP) (MIM: 600296) (93–95), and proinflammatory cytokines such as TNF, IL-1β, and IL-6 (84, 96). These molecules, together with their receptors, control growth plate physiology by activating multiple signaling pathways (82). Furthermore, chondrocytes secrete cartilage extracellular matrix rich of collagens (eg, collagen type II and X), noncollagenous proteins (eg, byclan and decorin), and proteoglycans (eg, aggrecan), which also play important roles in growth plate regulation (97). A variety of transcription factors are also pivotal for chondrocyte differentiation (82). Notable examples include: SRY (sex-determining region Y) (MIM: 480000), SRY-box 9 (SOX9) (MIM: 608160), runt-related transcription factor 2 (RUNX2) (MIM: 600201), forkhead box A2 (FOXA2) (MIM: 600288), and members of the nuclear factor κB family (4, 73, 82). The coordinated network of signaling cascades that govern the temporal- and site-specific expression of these genes is only beginning to be elucidated (82). Not surprisingly, alterations of many of the genes involved in growth plate regulation can produce diseases characterized by skeletal defects, altered bone growth, and stature below or above the mean, indicating that linear growth disorders are disorders of growth plate chondrocytes recently reviewed in Baron et al (4) and Wit et al (3).
Table 1.
SHOX Interacting Proteins, Cellular Targets, and Modulators of Activity
| Category | Gene/Cell Factor | Abbreviation | Model System | Interaction With SHOX | First Author, Year (Ref.) |
|---|---|---|---|---|---|
| Upstream regulators | Retinoic acid | RA | Chicken embryos | Regulates Shox expression negatively | Tiecke, 2006 (27) |
| Bone morphogenetic protein 4 | BMP4 | Chicken embryos | Regulates Shox expression negatively | Tiecke, 2006 (27) | |
| Fibroblast growth factor 1 | FGF1 | Chicken embryos | Regulates Shox expression negatively | Tiecke, 2006 (27) | |
| Homeobox A9 | HOXA9 | Human cell lines, chicken micromass chicken buds | Binds to the SHOX promoter, regulates expression negatively | Durand, 2012 (126) | |
| Physical interacting proteins | Casein kinase 2, α 1 polypeptide | CSNK2A1 | Human cell lines | Involved in SHOX phosphorylation | Marchini, 2006 (63) |
| SRY (sex determining region Y)-box 5 | SOX 5 | Human cell lines and growth plate | Forms a complex with SHOX together with SOX6 and SOX9 | Aza-Carmona, 2011 (113) | |
| SRY (sex determining region Y)-box 6 | SOX6 | Human cell lines and growth plate | Forms a complex with SHOX together with SOX5 and SOX9 | Aza-Carmona, 2011 (113) | |
| SRY (sex determining region Y)-box 9 | SOX9 | Human cell lines and growth plate | Forms a complex with SHOX together with SOX5 and SOX6 | Aza-Carmona, 2011 (113) | |
| Short stature homeobox 2 | SHOX2 | Human cell lines | Forms heterodimers with SHOX | Aza-Carmona, 2014 (118) | |
| Transcriptional direct targets | Natriuretic peptide B | NPPB | Human cell lines and growth plate | Up-regulated by SHOX, co-expression | Marchini, 2007 (103) |
| Fibroblast growth factor receptor 3 | FGFR3 | Human cell lines and chicken micromass | Down-regulated | Decker, 2011 (28) | |
| Connective tissue growth factor | CTGF | Human cell lines and growth plate | Up-regulated, co-expression | Beiser, 2014 (119) | |
| Cellular mediators | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | CDKN1A | Human cell lines | Up-regulated in SHOX-expressing cells; involved in SHOX-induced cell cycle arrest | Marchini, 2004 (98) |
| Cyclin-dependent kinase inhibitor 1B (p27, Kip1) | CDKN1B | Human cell lines | Up-regulated in SHOX-expressing cells; involved in SHOX-induced cell cycle arrest | Marchini, 2004 (98) | |
| Tumor protein p53 | TP53 | Human cell lines | Up-regulated in SHOX-expressing cells | Marchini, 2004 (98) | |
| Retinoblastoma-like2 (p130) | RBL2 | Human cell lines | Up-regulated in SHOX-expressing cells | Marchini, 2004 (98) | |
| Cathepsin B | CTSB | Human cell lines | Mediators of SHOX-induced cell death | Hristov, 2014 (101) | |
| Reactive oxygen species | ROS | Human cell lines | Mediators of SHOX-induced cell death | Hristov, 2014 (101) | |
| Reactive nitrogen species | RNS | Human cell lines | Mediators of SHOX-induced cell death | Hristov, 2014 (101) |
To provide further direct evidence of an involvement of SHOX in human bone development, the SHOX expression pattern in fetal (22 weeks gestation) and pubertal (12, 13, and 15 years) human growth plate sections was analyzed by immunohistochemistry. SHOX was detected in the growth plate, particularly in terminally differentiated hypertrophic chondrocytes and, to a lesser extent, in chondrocytes of the resting and proliferative zones. In contrast, SHOX was not expressed in osteoblasts and osteoclasts, suggesting that it does not play a role in these bone cells (98). Munns et al (99) also observed SHOX in human growth plate chondrocytes from 12 weeks gestation until late childhood. These same authors described a highly disordered organization of radial growth plates in LWD patients undergoing surgery for Madelung deformity with disruption of the normal parallel columnar arrangement of chondrocytes (100), indicating abnormal endochondral ossification.
The presence of SHOX in the human growth plate, and in particular in chondrocytes of the hypertrophic region, suggests that SHOX may be involved in the developmental pathway that regulates chondrocyte proliferation and maturation.
B. SHOX is a modulator of cell proliferation and apoptosis
The first hints at the in vivo function of SHOX came from cell culture studies. In human osteosarcoma U2OS stable cell lines expressing SHOX in an inducible manner, SHOX-expressing cells were observed to grow more slowly than noninduced cells and stopped proliferating 4 days after SHOX induction. SHOX-expressing cells displayed dramatic morphological changes such as an enlarged and more differentiated phenotype with typical protrusions and multinucleation; a large fraction of cells contained two or more nuclei of equal size, most likely due to defective cytokinesis. After prolonged SHOX expression, a consistent fraction of cells detached from the culture dish and died. Flow cytometry analysis showed that SHOX-expressing U2OS cells were arrested in the G2/M phase of the cell cycle. This cell cycle arrest was associated with increased levels of the cyclin kinase inhibitors p21Cip1 and p27Kip1 and with alterations in the expression of other cell cycle regulatory factors such as p53, pRB, p107, and p130 (98).
The reduction in cell number after prolonged SHOX expression indicates that SHOX negatively affects cell viability. SHOX expression in U2OS cells triggers the intrinsic pathway of apoptosis characterized by mitochondrial outer membrane polarization (MOMP) and caspase activation (98, 101).
SHOX-induced cell cycle arrest and apoptosis were also confirmed in the pRB- and p53-deficient osteosarcoma Saos-2 cell line, indicating that these two proteins are not required for SHOX-induced cell cycle arrest and apoptosis (our unpublished results). Similar results were also obtained using normal nontransformed human cell cultures such as primary oral fibroblasts and primary chondrocytes. Conversely, the overexpression of SHOX harboring a missense mutation (p.R153L, detected in LWD), a C-terminal deletion (p.R185X, resembling a mutated SHOX found in LWD and ISS patients), or mutation within the phosphorylation site (p.S106A phosphorylation-defective mutant) did not affect cell viability. Because these three mutants are also inefficient in transcriptional activation, SHOX-mediated gene transcription was postulated to be necessary to trigger cell cycle arrest and apoptosis (61, 63, 98, 101).
In addition to triggering the intrinsic pathway of apoptosis, which is characterized by MOMP and caspase activation, SHOX expression was also associated with more acidic vesicles, and particularly lysosomes. SHOX induced partial rupture of lysosomal membrane integrity, leading to relocation of the active proteolytic form of cathepsin B from lysosomes to cytoplasm. Large amounts of cathepsin B (MIM: 116810) were also found in the culture medium, suggesting active secretion of the protease. By contrast, cathepsin L protein levels did not vary upon SHOX induction (101). Treatment of the cells with a specific cathepsin inhibitor (Ca074-Me) significantly protected the cells from SHOX-induced apoptosis, strongly suggesting that cathepsins (in particular cathepsin B) play a key role in this event. This finding is in agreement with other studies showing that cathepsins may participate in the induction of apoptosis (102). Furthermore, SHOX expression was found to be associated with oxidative stress characterized by intracellular accumulation of reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS and RNS are important mediators of SHOX-induced cell death because antioxidant treatment with N-acetyl-L-cysteine, reduced glutathione, or FeTPPS significantly reduced both SHOX-mediated lysosomal instability and SHOX-induced cell death. The mechanisms through which SHOX induces oxidative stress remain to be elucidated.
C. Transcriptional targets
To shed light on the transcriptional targets of SHOX, gene expression profiling studies of U2OS cells expressing SHOX or not (induced or not induced U2OS-SHOX stable cell line) were conducted. The most significantly up-regulated gene in SHOX-expressing cells was NPBB, which encodes brain natriuretic peptide (BNP) (MIM: 600295) (17-fold increase 24 hours after SHOX induction). SHOX activated the NPBB promoter in a dual luciferase reporter gene assay by binding specific SHOX-responsive elements within the regulatory region of the NPBB gene. Chromatin immunoprecipitation (ChIP) assay confirmed the direct binding of SHOX to the NPBB regulatory region. Furthermore, SHOX and BNP were coexpressed in the hypertrophic zone of human growth plate chondrocytes. Together, these results provided evidence that BNP is a downstream target of SHOX and may play an important role as a mediator of SHOX cellular functions in the growth plate (103) (discussed in section IV E).
After the initial discovery of NPBB as a cellular target of SHOX, other studies have sought to characterize the pathways and networks through which SHOX regulates bone growth. Using a similar approach to that for the discovery of NPBB—gene expression profiling of SHOX-expressing cell lines—Decker et al (28) identified FGFR3 as a direct SHOX target gene. Numerous studies have demonstrated that FGF/FGF receptor (FGFR) signaling pathways play a critical role in regulating bone development, controlling the function of basically all skeletal cells, including chondrocytes, osteoblasts, and osteoclasts. They exert their activity closely interacting with other signaling pathways involved in the control of skeletal development and homeostasis, including Indian hedgehog, BMPs, CNP, PTHrP, wingless-related integration site proteins, SOXs, and RUNX2 pathways. These studies have been recently reviewed and therefore will not be covered here (92). The finding that SHOX takes part in the regulation of FGFR3 transcription places SHOX at the center of this network of factors controlling bone growth.
It is known that gain-of-function mutations in FGFR3 cause distinct skeletal syndromes including achondroplasia, hypochondroplasia, and thanatophoric dysplasia—all of which are marked by rhizomelic shortening of the limbs (92, 104) due to dysregulated endochondral ossification. Furthermore, Kant et al (105) have recently reported on a novel mutation in FGFR3 causing proportionate short stature. On the contrary, heterozygous (106) and homozygous (107) inactivating FGFR3 mutations cause tall stature associated with skeletal and nonskeletal defects. In agreement with these results, transgenic mice overexpressing activated Fgfr3 mutants in the growth plate present severe dwarfism with decreased chondrocyte proliferation, disorganized chondrocyte columns, and narrowed hypertrophic zone (77, 92, 108–110). Conversely, Fgfr3 knockout mice have been shown to display long bone elongation that correlates with increased chondrocyte proliferation and an elongated growth plate hypertrophic zone (111, 112). Taken together, these results indicate that FGFR3 signaling negatively regulates bone growth by decreasing chondrocyte proliferation, accelerating the onset of hypertrophic differentiation, and decreasing the height of the hypertrophic zone in the postnatal growth plate.
Luciferase reporter assays, ChIP sequencing, and ChIP and electromobility band shift experiments together provided evidence that FGFR3 represents a direct target of SHOX. In agreement with this, several SHOX consensus sites were identified within the FGFR3 promoter region. Using limb bud-derived chicken micromass cultures as a model, Decker et al (28) demonstrated, by quantitative real-time-PCR and in situ hybridization, that retrovirus-mediated SHOX gene transfer down-regulates FGFR3 expression. The fact that SHOX represses FGFR3 promoter activity may explain the almost mutually exclusive expression patterns of Fgfr3 and Shox in embryonic chicken limbs.
Further involvement of SHOX in bone development was supported by the discovery that SHOX interacts with the SOX trio (SOX9, SOX5, and SOX6) transcription factors (113). SOX9, together with SOX5 and SOX6, is a master regulator of chondrocyte differentiation. SOX9 is expressed in resting, proliferative, and prehypertrophic chondrocytes, but not in hypertrophic chondrocytes (114). In the proliferative zone, Sox9 regulates the transcription of multiple genes, including the activation of Col2a1 and aggrecan (ACAN) and the repression of Col10a1 and Runx2, thereby sustaining chondrocyte survival and preventing chondrocyte hypertrophy. In prehypertrophic chondrocytes, instead, Sox9 is responsible for the activation of Col10a1, thereby initiating the onset of hypertrophy (82). Mutations of SOX9 have been associated with campomelic dysplasia, a severe skeletal dysplasia characterized by congenital bowing and angulation of long bones and other skeletal and extraskeletal defects (115).
By interacting with the SOX trio, SHOX regulates the expression of ACAN, which encodes aggregan, a major component of the cartilage extracellular matrix. ACAN plays an important role in normal growth plate function, as exemplified by the fact that homozygous mutations in ACAN are responsible of a severe skeletal dysplasia, spondyloepimetaphyseal dysplasia aggrecan type (116), whereas heterozygous mutations cause a milder skeletal dysplasia, spondyloepimetaphyseal dysplasia, Kimberley type, or short stature without evident radiographic signs of dysplasia (117).
SHOX was shown to bind SOX6 through its homeodomain. Different SHOX missense mutations, which had been described in individuals with LWD or ISS, failed to interact with the SOX trio. Immunohistochemistry of human fetal growth plates demonstrated that SHOX is coexpressed with SOX5, SOX6, and SOX9 (113). More recently, SHOX2 has been shown to dimerize with SHOX and, similarly to SHOX, to activate expression of NPBB and ACAN (118). Because SHOX and SHOX2 are homologous (80%) proteins that share the same homeodomain, the two proteins may cooperate, by forming heterodimers, in modulating expression of a similar subset of genes in limb development (eg, NPBB) and in other tissues and organs.
More recently, transgenic mice, in which SHOX expression was under the control of a murine Col2a1 promoter and enhancer region resulting in SHOX expression in chondrocytes, were used to study SHOX cellular activities (119). No major skeletal anomalies were seen in these transgenic mice; however, statistically significant up-regulation of several cartilage and bone markers during embryonic phase E12.5 to E14.5 was observed by gene expression profiling in transgenic vs wild-type limb RNA. Up-regulated genes included connective tissue growth factor (Ctgf), periostin, asporin, EGF-containing fibulin-like extracellular matrix protein 1, and matrilin 4, all genes known to be involved in limb development, extracellular matrix, or skeletal pathways. To confirm these results, U2OS and normal human dermal fibroblast cell lines were used that expressed either the wild-type or the transcriptionally defective Y141D mutant SHOX. Wild-type SHOX, but not the Y141D mutant, significantly up-regulated the Ctgf/CTGF target gene.
Further evidence for Ctgf as a direct target of SHOX was derived from ChIP sequencing data using chicken micromass cultures in which SHOX was introduced retrovirally. With this approach, several SHOX-binding sites in the Ctgf upstream region were identified. Accordingly, in silico analysis showed that the human CTGF 5′-regulatory region contains more than 40 potential consensus sequences for SHOX. Luciferase gene reporter and electromobility shift assays demonstrated that SHOX specifically binds to these sequences. Finally, SHOX and CTGF coexpression was detected in the hypertrophic zone of the growth plate. Together, these results strongly suggest that SHOX regulates CTGF expression in the developing limbs (119).
D. Upstream regulators
Clustered genes from the Hox family of transcription factors (Hox A/D 9–13) have been shown to perform pivotal roles during limb development and axial skeletal patterning (120, 121). For instance, ablation of Hox9 and Hox10 gene clusters in mice leads to shortened stylopodal elements (humerus and femur) (122, 123), and loss of Hox11 results in truncated zeugopodial elements (radius/ulna and fibula/tibia) (124), whereas deletion of Hox13 leads to reduced formation of the autopod (metacarpals/metatarses) (125).
HOXA9 has been identified as the first upstream regulator of SHOX expression (126). By use of luciferase assays, ChIP, and electromobility shift assay, a HOXA9 binding site, consisting of two 31 nucleotide-long AT-rich sequences, was identified within the SHOX promoter 2. Virus-induced Hoxa9 overexpression in a chicken micromass model was associated with down-regulation of Shox. Because Hoxa9 and Shox were expressed in the same regions of developing limb buds, a regulatory relationship has been proposed between Hoxa9 and Shox during limb development (126).
Further evidence of interactions between the Hox and SHOX families derived from elegant studies in mice in which the effects of Shox2 dosage variations were examined in the context of different HoxA/D cluster deletion background (38). Shox2 was found to be coexpressed in the proximal limb with Hoxd9 and Hoxa11 during embryonic limb development. Shox2 overexpression could partly compensate for Hox gene loss. It was shown that both Shox2 and Hox genes functionally interact in regulating cartilage maturation by modulating the expression levels of Runx2 in the stylopodal and zeugopodal elements of vertebrate limbs (38). Shox2c/− animals do not display changes in the expression levels of Hox genes, indicating that Shox2 does not regulate Hox gene transcription (34, 38, 127). Conversely, Hox11 genes seem to be required for Shox2 expression in the proliferating chondrocytes of the zeugopodal elements (38, 128), although this observation is still under debate (38). Interestingly, Hox genes regulate Shox2 expression in the perichondrium because their deletion results in the complete loss of Shox2 in this structure (38). Together, these results provide the first evidence of a mutual interaction between SHOX and HOX genes.
In chicken embryos, Shox is expressed in the medial, proximal portion of limb buds and promotes chondrogenesis. Graft experiments with soaked beads in chicken embryos indicated that Shox expression is negatively regulated by Bmp4, Fgf4, and Fgf8 distally and RA proximally (27). How these signaling pathways contribute to the regulation of SHOX expression during chondrogenesis remains to be elucidated. SHOX overexpression in chicken has had no detectable effect on the proximal-distal pattern of skeletal elements, but it increases the length of the skeletal elements, and occasionally the timing of ossification is altered.
E. Possible roles of SHOX in bone development
1. SHOX as a regulator of chondrocyte hypertrophy
As will be discussed in section V, SHOX deficiency is implicated in short stature syndromes characterized by skeletal defects as well as in nonsyndromic ISS. SHOX expression in the growth plate implies a role of SHOX in this structure. However, the exact role of SHOX in the growth plate is only beginning to be elucidated. The results described above in osteosarcoma cell lines and primary cultures indicate that ectopic expression of SHOX induces cell cycle arrest and apoptosis, suggesting that the protein may also control proliferation of chondrocytes in the growth plate and promote their maturation. Multiple signaling pathways have been described to control chondrocyte maturation in the growth plate (4, 72, 73, 77, 82). In this section, based on the results described above and current knowledge of endochondral ossification, we propose a model of SHOX involvement in some of the signaling pathways that control proliferation and maturation of growth plate chondrocytes during bone elongation (see also Table 1 and Figure 3B for cellular factors interconnected with SHOX).
2. Influence of SHOX on FGFR3 signaling
FGFR3 is a negative regulator of chondrocyte proliferation and differentiation (92). In the growth plate, FGFR3 is expressed in proliferating chondrocytes but is down-regulated in the hypertrophic zone—a pattern complementary to that of SHOX and supporting the concept that SHOX represses FGFR3 transcription (Figure 4A). A number of pathways have been found to act downstream of FGFR3 activation (92, 129), including, but not limited to, the Janus kinase-signal transducer and activator of transcription (JAK-STAT) and MAPK pathways, which inhibit chondrocyte proliferation and differentiation (4, 92, 130). SHOX, by repressing FGFR3 transcription, may therefore impact on these pathways; eg, SHOX-mediated FGFR3 down-regulation may result in inactivation of the JAK-STAT and MAPK pathways, allowing normal chondrocyte proliferation and maturation (Figure 4B). Clarification on how SHOX-mediated down-regulation of FGFR3 influences FGFR3 signaling and other regulative pathways involved in bone formation is an important area for future research.
Figure 4.
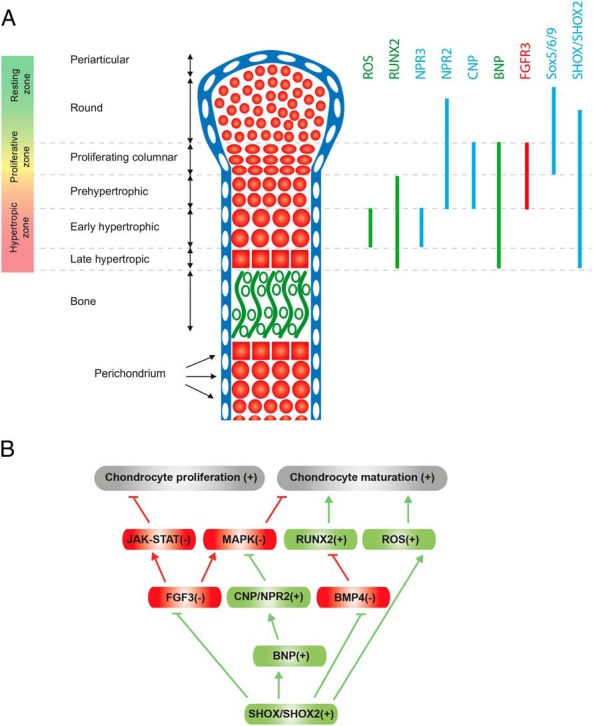
SHOX as a regulator of endochondral ossification. A, Expression pattern of cellular factors involved in growth plate regulation interacting with SHOX. A schematic representation of the mouse long bone growth plate at E15.5-E16.5 is displayed. The growth plate is subdivided into different zones that contain chondrocytes at different stages of maturation. Chondrocytes at the end of their differentiation process undergo cell death and are replaced by bone. The expression pattern of FGFR3, BNP, NPR2, NPR3, and RUNX2 is illustrated according to Kozhemyakina et al (82). SHOX is not expressed in mice, but the mouse genome contains the closely related SHOX2 gene. Here we speculate that SHOX2 and BNP in mouse growth plate have similar expression patterns to those found for SHOX and BNP in human growth plate specimens (98, 103). Expression patterns of SHOX/Shox2 target genes are depicted in green (up-regulated) or in red (down-regulated). B, Tentative model illustrating the involvement of SHOX in pathways regulating chondrocyte proliferation and maturation in the growth plate. SHOX-mediated down-regulation of FGFR3 may repress FGFR3 signaling, whereas up-regulation of the NPPB gene may stimulate the CNP/NPR2 pathway. This results in the repression of JAK-STAT and MAPK signaling pathways, which negatively regulate chondrocyte proliferation and maturation, respectively. Note that FGFR3 and CNP/NPR2 signaling pathways converge in the regulation of the levels of activated MAPK, which blocks the initiation of chondrocyte hypertrophy (FGFR3 signaling being an activator and CNP/NPR2 signaling being an inhibitor of the MAPK pathway). It has been shown that through the repression of BMP4 signaling, SHOX2 may regulate the levels of RUNX2, a master regulator of chondrocyte hypertrophy. Although a similar role of SHOX in activating RUNX2 remains to be demonstrated, this is hypothesized in the model, given the high homology and functional redundancy between the two homeodomain proteins. Depicted in green are pathways promoting chondrocyte hypertrophy, whereas in red are those having a negative impact. Plus and minus signs indicate the possible effects in response to SHOX/SHOX2 expression.
As discussed above, Fgfr3 and Shox have almost mutually exclusive expression patterns in embryonic chicken limbs (28). These results provide an intriguing hypothesis for the rhizomelic short stature seen in FGFR3-mutated achondroplasia patients and its potential interrelationship with SHOX. The presence of SHOX in the mesomelic bone segments, by blocking FGFR3 mutant expression, may allow normal development of these bones, whereas the lack of SHOX in the rhizomelic portion, by allowing FGFR3 mutant expression, may account for the rhizomelic phenotype (28).
3. Influence of SHOX on CNP/Npr2 signaling
In addition to FGFR3, the activity of SHOX may be mediated, at least in part, by its cellular target gene natriuretic peptide B (NPPB). NPPB encodes BNP, a member of the natriuretic peptide family that also includes the related atrial natriuretic peptide (ANP) and CNP. Three receptors mediate the activity of these peptides: natriuretic peptide receptor (NPR) 1, NPR2, and NPR3 (also called guanylyl cyclase A, B, and C, respectively). NPR1 recognizes ANP and BNP, whereas NPR2 is specific for CNP. NPR3 is a decoy/clearance receptor that regulates the levels of natriuretic peptides available for interaction with NPR1 and NPR2. ANP and BNP, which function as cardiac hormones, are produced primarily by the atrium and ventricle of the heart, respectively (131). Together with their receptor NPR1, ANP and BNP are involved mainly in cardiovascular homeostasis by regulating blood pressure and body fluid volume. They are used as serum markers of disease severity in heart failure, myocardial infarction, cardiac hypertrophy, and hypertension (131–133). Importantly, ANP and BNP are used clinically in the treatment of heart failure (132).
CNP is produced mainly in the brain, where it is thought to act as a neuropeptide. However, studies in genetically engineered mice indicate that CNP and its receptor Npr2 are also expressed in growth plate, where they are implicated primarily in regulating endochondral bone growth (82) (Figure 4A). CNP-deficient mice exhibit severe dwarfism, with a 50–80% reduction in the length of endochondral bones such as the femur, tibia, and vertebrae. Histological studies in these mice show decreased growth plate width due to smaller proliferative and hypertrophic zones (93). A similar phenotype was observed in Npr2 (CNP receptor)-deficient mice (134). Conversely, CNP transgenic mice and Npr3 (clearance receptor)-deficient mice exhibit prominent bone elongation with extended growth plate proliferative and hypertrophic zones (135). In humans, loss-of-function mutations in the NPR2 gene cause acromesomelic dysplasia type Maroteaux, characterized by severe dwarfism (136, 137). By contrast, an NPR2 gain-of-function mutation has been identified in three individuals with skeletal overgrowth (138, 139). Thus, the CNP/NPR2 signaling pathway regulates bone growth in both humans and mice. CNP increases chondrocyte hypertrophy by opposing FGF signaling via MAPK pathway repression (82, 108, 140, 141). Consistent with the above findings, targeted expression of CNP in the growth plate—or systemic administration of synthetic CNP-22—ameliorated both the skeletal defects and the growth deficit in a mouse model of achondroplasia (135), thus providing preclinical proof-of-concept that CNP may be useful for the treatment of achondroplasia in humans.
The role of BNP in endochondral bone ossification is less well-defined than that of CNP. Despite marked skeletal overgrowth in BNP transgenic mice (142), resembling the phenotype of CNP transgenic mice, BNP knockout animals do not exhibit skeletal or growth abnormalities but instead exhibit only cardiovascular defects (143). Therefore, the overgrowth phenotype in BNP transgenic mice has been proposed to represent unphysiologically high levels of BNP that either cross-reacted with NPR2 or saturated the NPR3 clearance receptor, causing reduced clearance of CNP. The resulting increased concentration of growth plate CNP could augment endochondral bone growth, thereby accounting for the overgrowth phenotype in BNP transgenic animals.
However, the discovery that BNP is a cellular target of SHOX and that the two proteins are coexpressed in the growth plate hypertrophic zone (103) argues for an important but as yet uncharacterized physiological role for BNP in growth plate regulation, for example, by offsetting FGF signaling. Recently, NPR2 mutations have been associated with disproportionate short stature (similar to LWD but without Madelung deformity) in patients in whom no mutations were detected in SHOX or its enhancer regions (144–146). Conceivably, SHOX-mediated BNP expression may increase CNP/NPR2 signaling, perhaps by competing with CNP for the NPR3 clearance receptor and thereby increasing CNP half-life and its stimulatory effect upon the growth plate (Figure 4B). Further studies are required to shed light on such a potential physiological role of BNP in bone elongation.
Because of the role of BNP in body fluid and blood pressure homeostasis, the common occurrence of hypertension and varied cardiovascular and renal anomalies in SHOX-deficient girls with TS is noteworthy (147). Hypothetically, these defects may be due to reduced circulating BNP levels as a consequence of SHOX deficiency.
4. Influence of SHOX on Bmp4 signaling and RUNX activity
In addition to the above pathways, SHOX may act in concert with RUNX2 and RUNX3 to trigger chondrocyte hypertrophy. As described above, a SHOX ortholog does not exist in mice, but the mouse genome does contain the SHOX-related gene Shox2, which also plays a central role in skeletal development.
Specifically, conditional deletion of the Shox2 gene in embryonic limb mesenchyme (Prx1–Cre–Shox2, in which Shox2 deletion was dependent on activation of the limb-specific Prx1 promoter) produced animals displaying severely shortened limbs due to the nearly complete absence of humerus and femur (34). A significant delay in chondrocyte maturation—due to a down-regulation of Runx2 (34) or up-regulation of Bmp4 (127)—underlies the observed phenotype. Because BMP4 functions as a repressor of Runx2 gene expression, it was proposed that Shox2 regulates Runx2 expression via Bmp4 (127) (Figure 4B).
Col2a1–Cre-driven conditional Shox2 deletion in chondrocytes also causes significant shortening of humerus and femur (39). However, this rhizomelia is caused by precocious hypertrophic differentiation of stylopodial chondrocytes rather than the delayed maturation observed in Prx1–Cre–Shox2 animals. This apparent inconsistency may have its explanation in the higher levels of Bmp4 found in Col2a1–Cre–Shox2 chondrocytes than in Prx1–Cre–Shox2 chondrocytes (39). If expressed at very high levels, Bmp4 would trigger accelerated chondrocyte hypertrophy, whereas at lower levels it would be sufficient only to promote early chondrogenesis. Together, these results suggest that Shox2 may act as a regulator of chondrogenesis in mouse limb development by repressing the expression of Bmp4 (39). Supportive of this hypothesis is the discovery that decreased Shox2 expression correlates with a concomitant increase in Bmp4 expression during normal mouse endochondral ossification in two different phases: first, during the initial differentiation of the stylopodial cartilage anlage; and second, during final chondrocyte maturation and hypertrophy (39).
Interestingly, experiments in mice demonstrated partial functional redundancy between human SHOX and mouse Shox2 genes because SHOX can rescue both defective sinoatrial node formation (by restabilizing normal pacemaking function) and forelimb stylopodial shortening (26). Furthermore, by sharing the same homeodomain, SHOX and SHOX2 may regulate the same subset of genes (118). The fact that Shox2 conditional knockout animals have severe shortening of the stylopods (humerus and femur), whereas individuals with SHOX deficiency have defects primarily in zeugopods (radius and ulna in the forelimb, and tibia and fibula in the hindlimb), suggests that hSHOX and hSHOX2 may have similar roles in limb development but in different proximodistal segments. This hypothesis is consistent with in situ hybridization studies showing that the two genes exhibit a distinct, partially overlapping expression pattern in human embryos (25).
5. SHOX as regulator of the terminal phase of chondrocyte hypertrophy
At the end of their differentiation, hypertrophic chondrocytes undergo cell death and are replaced by bone cells. The intracellular signaling pathway(s) that governs cell death in growth plate chondrocytes remains largely unknown.
The chondrocyte cell death process itself presents a unique combination of both apoptotic and nonapoptotic morphological changes. Roach et al (148) proposed the term chondroptosis to highlight the unique features of this nonclassical apoptosis. Initially, the process involves enlargement of endoplasmic reticulum and Golgi apparatus—reflecting an increase in protein synthesis—followed by digestion of cellular material within autophagic vacuoles and chondrocyte self-destruction. These authors have proposed that lysosomal proteases are at least as important as caspases in chondroptosis (148).
Four major components characterize the SHOX-induced cell death mechanism in the U2OS osteosarcoma cell line: 1) initial oxidative stress with intracellular accumulation of ROS and RNS; 2) an increase in the number of acidic lysosome vesicles and a strong up-regulation of cathepsin B expression; 3) lysosomal membrane permeabilization with relocation of activated lysosomal cathepsin B into the cytoplasm, where it participates in cell death via MOMP and caspase activation; and 4) substantial release of cathepsin B outside the cells.
Accumulating evidence indicates important roles of ROS, RNS, and cathepsin B in the growth plate. For instance, increased ROS levels in hypertrophic chondrocytes inhibit proliferation and promote hypertrophic differentiation (149) (Figure 4A). Nitric oxide production and elevated extracellular inorganic phosphate levels are also known to be involved in chondrocyte hypertrophy (150–152). Cathepsin B is highly expressed in the growth plate (153, 154), where it is hypothesized to participate in degradation of extracellular matrix components (155, 156). Notably, although U2OS cells do not replicate the complex physiological and cellular interactions of the growth plate, SHOX is able to induce a cell death process in U2OS cells that is reminiscent of that seen physiologically in the growth plate. These findings should propel further investigations to understand how closely SHOX-induced cell death in U2OS cells reflects SHOX-induced events in growth plate chondrocytes. For example, characterization of the ability of SHOX to trigger oxidative stress and up-regulation of cathepsin B in growth plate may provide new insights into the cell death process that occurs during endochondral ossification.
In conclusion, the observations described above suggest that SHOX is one of several critical factors regulating chondrocyte hypertrophy. SHOX positively regulates chondrocyte differentiation through down-regulation of FGFR3 and up-regulation of NPPB, thereby inhibiting FGFR3 signaling and activating CNP/NPR2 signaling, respectively. Furthermore, as suggested by experiments in mouse, SHOX/SHOX2 may also affect chondrocyte maturation by activating RUNX2 through down-regulation of BMP4 signaling. Finally, based upon in vitro data in osteosarcoma cell lines, SHOX may activate the chondrocyte cell death pathway through increased ROS production (Figure 4B).
6. SHOX functions in pattern formation
In human patients with SHOX deficiency, mainly forearms and lower legs are affected, suggesting that SHOX is expressed in some but not all growth plates. Experiments in chicken limb buds indicate that Bmps, Fgfs, and RA signaling are involved in restricting the spatiotemporal expression of shox to the proximal-medial region of the early limb bud; RA inhibits Shox expression proximally, whereas Bmp and Fgf signal distally. However, it remains to be elucidated whether mechanisms similar to those in chick embryo govern SHOX expression during human embryogenesis. Furthermore, in addition to the previously described mechanisms regulating SHOX gene expression (Figure 2), some evidence indicates that HOX genes may play a role in determining SHOX expression pattern (38, 126) .
SHOX functions during embryogenesis may differ from those exerted by the protein in postnatal growth plate. This is suggested by the fact that some of the effects of SHOX deficiency in humans manifest only in middle to late childhood.
V. Clinical Implications of SHOX Deficiency
After identification of the SHOX gene as a cause of short stature (19), SHOX mutation screening studies linked mutations or deletions in one copy of the SHOX gene (as well as in its extragenic enhancer regulatory regions) with the short stature phenotype and the skeletal deformities found in patients with LWD (54, 157). Homozygous loss of the SHOX gene was shown to cause Langer dysplasia (LD) (66, 158, 159), a rare syndrome characterized by more severe growth retardation and some skeletal abnormalities. SHOX overdose in contrast usually leads to long limbs and tall stature (160).
With a combination of multiplex ligation-dependent probe amplification (MLPA) analysis and sequencing, SHOX defects have been found in approximately 10% of children with previously unexplainable short stature in the absence of other symptoms (ISS; Table 2). This makes SHOX deficiency the most frequent monogenic cause of short stature, with an incidence of up to 1:300 in the total population. In adults with SHOX deficiency, the proportion of LWD vs short stature without features of LWD is not well defined. Approximately 80% of detected SHOX mutations are deletions of different size encompassing the entire gene or its extragenic enhancer regions (161, 162). Microdeletions encompassing only single or multiple exons are rare and occur in approximately 5% of cases (data compiled from 134 individuals) (65, 162, 163). Missense or nonsense mutations are also found randomly, distributed throughout the gene but with a higher frequency in exons 3 and 4, which encompass the homeobox region encoding the DNA-binding homeodomain (164) (Figure 5). Duplications of the SHOX gene have also been reported in patients with short stature (11, 165–169). These duplications seem to reduce SHOX gene expression by disrupting gene organization, particularly the proper distance between the promoter and the enhancer regions. Microduplications at the SHOX locus have also recently been suggested as low penetrance risk factors for autism spectrum disorder, and the SHOX isoforms 7–1, 7–2, and 7–3 (Figure 1) (21) with a restricted expression in embryonic and fetal brain have been suggested to play a role in this (170). As of May 2016, 229 unique DNA sequence variants of SHOX have been identified in short stature patients. An updated and complete list of these allelic variants of SHOX can be found in the SHOX database at http://www.shox.uni-hd.de (171).
Table 2.
SHOX Gene Mutations in Idiopathic Short Stature
| First Author, Year (Ref.) | Nationality | Methodology | SHOX Defect |
||
|---|---|---|---|---|---|
| Deletion | Point Mutation | Overall Frequency, % | |||
| Rao, 1997 (19) | German | SSCP | 0/91 | 1/91 | 1.1 |
| Binder, 2000 (205) | German | MS (2) | 3/68 | Not analyzed | 2.1 |
| Rappold, 2002 (58) | German/Japanese | SSCP + FISH | 3/150 | 3/750 | 2.4 |
| Binder, 2003 (184) | German | MS (2) | 3–11/140 | Not analyzed | 2.5 |
| Stuppia, 2003 (209) | Italian | S + FISH | 3/56 | 4/46 | 12.5 |
| Huber, 2006 (174) | French | S + MS* (20) + SNP (49) | 8/84 | 4/84 | 14.3 |
| Rappold, 2007 (180) | Different nations | SSCP + MS (3) + (FISH) | 22/1534 | 8/1553 | 2.2 |
| Jorge, 2007 (179) | Brazilian | S + MS + FISH | 0/63 | 2/36 | 3.2 |
| Funari, 2010 (211) | Brazilian | MLPA + FISH | 4/36 | Not analyzed | 11.1 |
| Benito-Sanz, 2012 (50) | Spanish | MLPA (only for 47 kb) | 11/576 | Not analyzed | 1.9 |
| Hirschfeldova, 2012 (207) | Czech | MLPA | 4/51 | 2/51 | 11.7 |
| Rosilio, 2012 (186) | French | MLPA + S | 49/290 | Not analyzed | 16.9 |
| Sandoval, 2014 (166) | Colombian | MLPA | 5/62 | Not analyzed | 8.1 |
| van Duyvenvoorde, 2014 (167) | Dutch | CNV | 4/149 | Not analyzed | 2.7 |
| Poggi, 2015 (210) | Chilean | MLPA + S | 4/18 | 0/18 | 22.2 |
Abbreviations: CNV, copy number variation (SNP arrays); FISH, fluorescence in situ hybridization (detects deletions >20 kb using cosmid probes); MS*, study of many microsatellite markers (can give good indication of whether a deletion exists or not, but requires parental DNA); MS, study of only a few microsatellite markers (can give indication of whether a deletion exists or not, but requires parental DNA); S, sequencing; SNP, single nucleotide polymorphism analysis (can detect deletions including enhancer deletions; parental DNA is needed); SSCP, single-strand conformation polymorphism (detects 70% of all intragenic mutations). Several of these technologies are no longer in use, eg, SSCP, SNP, MS.
Figure 5.

Summary of 230 exonic SHOX mutations. Data were extracted by the SHOX database (www.shox.uni-hd.de). Exon 2 encloses part of the 5′-UTR, and exons 6a and 6b enclose part of the 3′-UTR; exon 1 only encloses 5′ untranslated sequences and is usually not screened in a diagnostic analysis. The homeobox resides in exon 3 and exon 4. The most frequent recurrent mutations are pArg147 in exon 3 (seven times) and pArg195 in exon 5 (10 times). Stops occurring in exons 2 to 6a are frameshift mutations. No stops were identified in exon 6b. Blue indicates unique mutations, and red indicates total mutations in the respective exon/region. Ex, exon; HB, homeobox region.
SHOX mutations cause short stature with a high phenotypic heterogeneity (161, 172). However, no correlation between the severity of the phenotype and the underlying SHOX mutation has been found so far. Identical SHOX mutations can produce either LWD or ISS, probably depending on the genetic background of the individual (173). In some families, individuals carrying the same mutation can even be asymptomatic with normal height (174) (W. Blum, personal communication). The size of a deletion is also not related to severity of the clinical phenotype; however, if large deletions extend beyond the pseudoautosomal region, a contiguous gene syndrome can result in males, with variable combinations of short stature, chondrodysplasia punctata, intellectual disability, ichthyosis, Kallman syndrome, and ocular albinism (175).
Haploinsufficiency implies that one allele of a gene is mutated, whereas the other functional allele, while intact, does not produce enough of the protein to bring about a wild-type condition, leading to an abnormal or diseased state. According to this concept, SHOX gene function is dosage-dependent. In patients with one mutated copy of SHOX, there could be variations in the expression levels of the other remaining functional SHOX allele that would lead to different degrees of SHOX deficiency and thus result in a more or less severe clinical phenotype. Alternatively, variants in other growth-regulating genes may affect SHOX gene functions, either as direct or indirect regulators of SHOX or by exerting additive effects. Below we provide a brief overview of SHOX-related disorders.
A. Léri-Weill dyschondrosteosis
Léri-Weill dyschondrosteosis (MIM: 127300), also called Léri-Weill syndrome, was first described by André Léri and Jean Weill and is characterized by mesomelic short stature and Madelung deformity (176). Mesomelic short stature is a type of disproportionate short stature, due to symmetric shortening of the forearms and lower legs. Madelung deformity of the wrist and forearm denotes a bilateral shortening and bowing of the radius, distal dislocation of the ulna, and wedged carpal bones (177) (Figure 6A). The primary lesion of Madelung deformity appears to be a premature fusion of the distal radial epiphysis, which possibly results in an aberrant cell death process in the growth plate. This skeletal anomaly is sometimes associated with pain and often with a limited wrist movement. Different operative procedures have been attempted to decrease pain and restore the wrist function. Short stature in LWD is variable; the height of affected adults ranges from 135 cm to normal height. The reason for this variability is not known.
Figure 6.

Skeletal defects associated with SHOX deficiency. A, Madelung deformity in a patient with LWD. The 19-year-old female with a 46,XX karyotype harbors a paternally inherited heterozygous microdeletion involving the SHOX coding region and the 3′ enhancer region. She shows severe short stature (−3.7 SD) and full pubertal development with regular menses. B, Hypoplasia of the ulna and fibula and severe shortening of the radius and tibia in an individual with Langer mesomelic dysplasia. The 19-month-old girl has a 45,X[191]/46,X,r(X)(p22.3q24)[9] karyotype. The ring X chromosome missing SHOX is formed as a de novo event in the X chromosome of paternal origin, whereas the structurally normal X chromosome harboring a microdeletion involving the SHOX enhancer(s) at the 3′ region is derived from the mother with subtle but SHOX-haploinsufficiency compatible skeletal features. The upper, middle, and lower images represent the roentgenograms of the right arm, the left arm, and the lower legs, respectively. C, Short metacarpal in a patient with TS (lower panel) compared to normal metacarpal (upper panel). Shown in the upper panel is an apparently normal hand roentgenogram of a 14-year and 7-month-old female with 45,X[15]/46,X,idic(X)(p11.2)[15] TS. She has been placed on GH treatment since 8 years and 9 months of age and on sex steroid supplementation therapy since 13 years and 8 months of age. Shown in the lower panel is the hand roentgenogram of a 13-year-old female with 45,X TS, showing a short fourth metacarpal associated with premature fusion of the growth plate. D, Radial bowing with decreased carpal angle. Forearm roentgenograms in an 11-year and 6-month-old girl (proband) with apparent ISS and her parents. The proband has a 46,XX karyotype and a paternally derived microdeletion affecting the SHOX 3′ enhancer region. She exhibits mild mesomelic short stature (−2.3 SD) and Tanner 3 breast development. Radial bowing, epiphyseal hypoplasia of the medial side of the distal radius, and decreased carpal angle are observed. The father with the same microdeletion shows mildly decreased carpal angle as the sole recognizable abnormality. His height remains within the normal range (−1.9 SD). This indicates that SHOX haploinsufficiency can permit an apparently normal phenotype as well as an ISS phenotype. The mother is free from discernible genetic and clinical abnormality.
SHOX deficiency has been described as the cause of LWD (54, 157); SHOX mutations have been found in approximately 70–90% of patients with LWD, suggesting that either unknown genes or unknown regulatory mechanisms may contribute to this disorder (54, 55, 65, 67, 157, 173, 174, 178–182).
Approximately 80% of patients have complete or partial gene or enhancer deletions, with the remaining 20% having missense mutations (55–58, 64, 181, 183, 184). Deletions of the enhancer elements are quite common, occurring in roughly 15- 40% of individuals with LWD (see Table 3 and next paragraph) (31, 65, 162, 174, 185, 186). This represents the highest known rate of enhancer element deletions in any disease. The high frequency of crossovers within PAR1 of the sex chromosomes may explain this peculiarity (187).
Table 3.
SHOX Enhancer Deletions in LWD and ISS
| First Author, Year (Ref.) | No. of Patients | Nationality | Methodology | Frequencies | ||
|---|---|---|---|---|---|---|
| Benito-Sanz, 2005 (185) | 80 LWD | French, Spanish, British | SNP analysis | 12/80 | 15% | LWD |
| Microsatellite | ||||||
| Benito-Sanz, 2006 (65) | 26 LWD | Spanish | SNP analysis | 10/26 | 38% | LWD |
| Microsatellite, MLPA | ||||||
| Huber, 2006 (174) | 56 LWD | French | SNP analysis | 9/140 | 6.4% | LWD/ISS |
| 84 ISS | Microsatellite | |||||
| Sabherwal, 2007 (31) | 122 LWD | Various | FISH with SHOX adjacent cosmid G0411; then SNP + microsatellite analysis | 4/122 | 3.3% | LWD |
| Chen, 2009 (162) | 58 LWD | Various | Microsatellite, sequencing FISH | 29/58 | 50% | LWD |
| 735 ISS | 31/735 | 4.2% | ISS | |||
| Rosilio, 2012 (186) | 178 LWD | Mainly French | MLPA, FISH, sequencing | 31% | LWD | |
| 290 ISS | 59% | ISS | ||||
| 69 DSS | ||||||
| Benito-Sanz, 2012 (50) | 124 LWD | Mainly Spanish | MLPA–47 kb del | 19/124 | 15.3% | LWD |
| 576 ISS | 11/576 | 1.9% | ISS | |||
| Bunyan, 2013 (213) | 377 LWD/ISS | English | MLPA–47 kb del | 17/377 | 12% | LWD |
| 4.5% | LWD/ISS | |||||
| Donze, 2015 (212) | 233 LWD/ISS | Dutch | MLPA, Sequencing | 44/233 | 18% | LWD/ISS |
Abbreviation: DSS, disproportionate short stature.
LWD typically develops in middle to late childhood, with a 4-fold higher prevalence in females than in males, and higher estrogen levels have been proposed as a mechanism for the more severe symptoms in girls vs boys (160, 161). It has been suggested that gonadal estrogens exert a maturational effect on skeletal tissues that are susceptible to unbalanced premature growth plate fusion and skeletal maturation because of SHOX haploinsufficiency, facilitating the development of skeletal lesions in a female-dominant and pubertal tempo-influenced fashion.
It is still under debate whether patients with typical features of SHOX deficiency but without any apparent mutations in the SHOX gene have yet uncharacterized mutations in SHOX regulatory regions further apart from the gene or in another yet unknown gene involved in the etiology of LWD. Recently 173 individuals, with suspected LWD (but no Madelung deformity) without evident defects in SHOX, were screened for mutations in the NPR2 gene, which encodes an important regulator of bone development. Alterations in NPR2 affecting receptor activity were found in 3% of these patients; however, the archetypal sign of this syndrome, Madelung deformity, was not present (144). It is intriguing to think that SHOX may be indirectly affecting the activity of this receptor via up-regulation of the NPPB gene (encoding the BNP).
B. Langer mesomelic dysplasia
Leonard Langer Jr. first described what is now termed Langer mesomelic dysplasia (LD) (MIM: 249700) (188). It is a rare disorder characterized by more extreme short stature and skeletal dysplasia than LWD, with height ranging from −5.5 to −8.9 SDS (189). Several couples in which both members are affected with LWD have had offspring with LD (66, 190). In particular, individuals with LD present an aplasia or marked hypoplasia of the ulna and/or fibula and severe shortening of the radius and tibia (Figure 6B). Other features include hypoplasia of the mandible and micrognathia; typically Madelung deformity is not part of LD. Homozygous loss of SHOX has been shown to underlie LD (66, 159, 191).
C. Turner syndrome
TS is a genetic disease caused by complete or partial absence of one copy of the X chromosome (termed monosomy of the X chromosome; 45, X/partial monosomy of the X). Also called Ullrich-Turner syndrome, the disease was first described by Otto Ullrich and Henry Turner (192, 193). It affects about one in 2500 women. Almost all individuals with TS exhibit short stature (final average adult height in Caucasians, 142–147 cm), gonadal dysgenesis (dysfunctional ovaries), and resulting amenorrhea (absence of menstrual cycle) and infertility. Skeletal defects, which appear less frequently, include short fourth metacarpals (Figure 6C), cubitus valgus (forearm angled away from the body), micrognathia, Madelung deformity, high-arched palate, webbed neck, broad chest, and low-set ears (194–196).
Women with TS may also present congenital heart defects, kidney problems, lymphedema, high blood pressure, hypothyroidism, diabetes, vision problems, hearing impairment, and autoimmune diseases (147). Finally, a specific pattern of cognitive deficits is often observed, with particular difficulties in visuospatial, mathematical, and memory areas (197). SHOX deficiency is thought to cause the skeletal abnormalities associated with TS (25, 161, 198). The expression of SHOX in the elbow and knee (and the lack thereof) may explain the cubitus valgus and the bowing and shortening of forearms and lower legs in some TS as well as LWD patients. The Madelung deformity and the shortened metacarpals seen in TS and LWD could result from a loss of SHOX expression in the distal ulna and radius. The prevalence of Madelung deformity remains only 7.5% in TS, and this would be explained by gonadal estrogen production usually being compromised in TS (198). A lack of SHOX expression in the first and second pharyngeal arches is likely to contribute to TS traits such as micrognathia, high-arched palate, and sensorineural deafness (25).
TS is also associated with low bone mineral density (BMD) in cortical rather than trabecular bones (199). In fact, the cortical (but not the trabecular) BMD of the distal forearm is significantly lower in adult TS patients compared to age-matched 46,XX females with premature ovarian insufficiency (200). This ovarian estrogen-independent selective reduction in cortical BMD would primarily be ascribed to SHOX deficiency. Indeed, altered bone geometry and microarchitecture have been found in both TS and SHOX deficiency (199). In addition, although SHOX expression is primarily identified in the growth plate and is absent from osteoblasts and osteoclasts, SHOX is known to interact with multiple factor(s) relevant for skeletogenesis, as described in this paper. Thus, it is likely that SHOX deficiency disturbs the complex molecular network and the cell-cell interactions involved in the cortical bone formation, leading to low cortical BMD.
D. Sex chromosome aneuploidies
Adult heights in patients with sex chromosome aneuploidies do not show a simple correlation with the SHOX gene dosage (201). Although severe short stature in patients with 45,X and mild to moderate tall stature in patients with 47,XXX or 47,XXY are grossly consistent with SHOX gene dosage (15), the adult heights in patients with four or five sex chromosomes (eg, 48,XXXX, 48,XXXY, 48,XXYY, 49,XXXXX, and 49,XXXXY) are usually not increased. In this regard, it has been reported that the distribution of the mean adult heights in apparently nonmosaic patients with sex chromosome aberrations can be explained by four factors: 1) the dosage effect of SHOX; 2) the dosage effect of the putative Y-specific growth gene GCY; 3) the sex difference in gonadal steroids; and 4) the degree of global nonspecific developmental defects including growth failure caused by chromosome imbalance (202). Of these factors, the concept of chromosome imbalance (quantitative alterations of euchromatic or noninactivated regions) is noteworthy. It has been suggested that chromosome imbalance disturbs developmental homeostasis, resulting in multiple nonspecific features common to various aneuploidies such as growth failure, developmental retardation, gonadal dysfunction, and tissue dysplasia, with the deleterious effects being more severe in deletions than in corresponding duplications (203, 204). Furthermore, it has been assumed that the degree of chromosome imbalance is grossly similar between loss or gain of inactivated X chromosomes harboring several noninactivated regions and that of normal Y chromosomes (15, 202). Thus, the marked adult height deficiency in patients with 45,X is explained by the combined effects of SHOX haploinsufficiency and severe growth disadvantage resulting from a sex chromosome deletion, and the mild to moderate adult height gain in patients with 47,XXX or 47,XXY (and 47,XYY as well) is explained by the combined effects of SHOX and/or GCY overdosage and relatively mild growth disadvantage caused by a sex chromosome duplication. In patients with four or five sex chromosomes, such a situation may drastically impair developmental homeostasis because of marked chromosome imbalance, resulting in reduced adult heights despite increased SHOX dosage and/or GCY dosage. Indeed, it is possible that the effects of excessive SHOX and/or GCY dosage cannot be fully reflected in the adult height in such situations. Although this notion remains speculative, it would provide a reasonable explanation for the intriguing findings of the adult heights in patients with sex chromosome aneuploidies.
E. Idiopathic short stature
Idiopathic short stature (MIM: 300582) is a heterogeneous diagnostic category in which the underlying cause of short stature has not been revealed by standard diagnostic procedures. Patients are shorter than 2 SD below the mean for age and gender (ie, below the third percentile) without presenting any apparent clinical signs of systemic, endocrine (eg, thyroid or GH deficiency), nutritional, or chromosomal abnormalities. Approximately 3% of the general population fall below the third percentile in height and are therefore considered short. Among individuals with the initial diagnosis of ISS, approximately 10% harbor SHOX mutations (Table 2). The results vary considerably from cohort to cohort, and the prevalence of SHOX mutations has increased with more sensitive genetic tests that can detect small deletions (eg, MLPA vs fluorescence in situ hybridization [FISH]) and with the examination of SHOX enhancer regions that were omitted in early SHOX mutation screening (18, 50, 58, 161, 166, 167, 174, 179, 180, 184, 186, 205–207, 209–211).
F. SHOX enhancer deletions in LWD and ISS
Remote regulatory elements are thought to play a critical role in how genes are switched on and off during development. Among patients with LWD, LD, or ISS who have an intact SHOX coding sequence, some were found to carry deletions of varying size downstream of the SHOX gene, which suggested that cis-acting enhancer elements regulating SHOX transcription may have been lost in these patients (185, 189). Subsequent studies identified other deletions downstream of the SHOX coding sequence in individuals with LWS or ISS, and these also were hypothesized to delete or disrupt putative enhancer elements (31, 162, 174, 186, 212, 213) (Table 3).
Further insight into the existence of enhancer elements controlling SHOX transcription came from a study including five families with individuals suffering from LWD (49). The affected individuals did not present mutations within SHOX, but instead had deletions downstream of its coding region. Analysis of the 3′ end of these SHOX deletions revealed an overlapping region of approximately 30 kb. Comparative genomic analysis showed that this interval comprised several CNEs that could act as enhancer elements of SHOX transcription. Consistent with this finding, luciferase reporter assays showed that one of these CNEs—cloned at the 3′ end of the luciferase gene—enhanced the SHOX promoter activity in the U2OS osteosarcoma cell line (49).
Further studies of LWD patients with intact SHOX identified other deletions 100–300 kb downstream of the SHOX gene and, more importantly, provided the first evidence of enhancer activity in chicken limb buds (31). Similarly to the study of Fukami et al (49), comparative genomic analysis of this region revealed several putative SHOX long-range regulatory elements that were highly conserved in all vertebrates carrying the SHOX gene (31). When cloned in front of a gene reporter system consisting of a β-globin promoter driving the expression of the GFP-encoding gene, three of these CNEs increased gene expression in limb buds of chicken embryos, indicating that they may function as enhancers of SHOX regulation (31). These three CNEs also exhibited enhancer activity in zebrafish embryos in the pectoral fin and in other tissues including ear, brain, skin, and heart (33). Using zebrafish as a model, smaller, more deeply conserved subsequences that were still endowed with enhancer activity were delineated within these CNEs (33). Interestingly, 4C-seq interaction and H3K27ac ChIP-seq profiles have recently revealed that the cis-regulatory domain of SHOX may extend 1 Mb surrounding SHOX, suggesting that further unknown cis-regulatory elements are located up- and downstream of SHOX (214–216).
Comparative genomic analysis also proved instrumental for the identification of putative enhancer elements upstream of SHOX by revealing three CNEs 250 kb upstream of the SHOX gene with enhancer properties in the developing chicken limb. However, deletion screening of the SHOX upstream region in 60 LWS patients with intact SHOX coding and downstream regions did not pinpoint novel deletions encompassing these putative enhancer elements. These results suggest that deletions upstream of the SHOX gene occur at a lower rate than those affecting the downstream region, most likely due to structural genomic differences between these two regions (30). Nevertheless, one individual with ISS has been reported to have a deletion comprising two SHOX upstream enhancers, CNE-5 and CNE-3 (29), and recently upstream copy number variations were found in three of 501 patients with ISS (214). Altogether, these studies indicate that deletions up- or downstream of the SHOX gene may result in the loss of SHOX enhancer elements, thereby leading to SHOX deficiency due to reduced SHOX expression.
G. Treatment of SHOX deficiency
Before the link between SHOX deficiency and short stature was firmly established, GH was already successfully used for the treatment of short stature in TS patients. This prompted clinical researchers to evaluate whether patients with SHOX deficiency would also benefit from GH therapy. To this end, Eli Lilly sponsored a randomized phase III clinical trial that included three treatment arms (n = 25 per arm): two groups of patients with short stature and SHOX deficiency, who were randomized to receive no treatment or treatment with GH (0.05 mg/kg/d) for 2 years; and one observational comparator group of age-matched girls with TS who were treated with the same GH regimen. The trial showed that GH was effective in children with SHOX deficiency, with 41% of patients reaching a height within the normal range within 2 years, compared to 4% of untreated children and 28% of patients with TS (217). The gain of height in SHOX-deficient children who were treated with GH was 3.5 cm during the first year and 1.9 cm in the second year (217). Based on this study, treatment with GH of SHOX deficiency has been approved by the U.S. Food and Drug Administration and the European Medicines Agency. GH treatment in patients with SHOX deficiency had no systematic effect on the skeletal anomalies in this disorder (218). Continuation of GH treatment to final height yielded, on average, height gains of more than 1.3 SDS, which corresponds to 8 to 9 cm. GH therapy was especially efficacious regarding growth rate and final outcome when it was started early in young children (219). It has also been noted that the response to GH treatment is greater in patients with SHOX enhancer deletions compared with SHOX coding deletions (212).
In addition to GH treatment, GnRH analog (GnRHa) therapy may also be recommended in patients with SHOX deficiency. It is expected that GnRHa therapy serves to prevent or mitigate the development of skeletal features by suppressing gonadal estrogen production. Indeed, it has been suggested and discussed that gonadal estrogens exert a maturational effect on skeletal tissues that are susceptible to unbalanced premature growth plate fusion because of SHOX deficiency, facilitating the development of skeletal lesions in a female-dominant and pubertal tempo-dependent fashion (160, 198, 220). This notion would explain, in terms of gonadal estrogen deficiency, why severe skeletal lesions remain rare in TS patients, despite the presence of SHOX deficiency.
Furthermore, the combination therapy of GH and GnRHa may be most effective in promoting statural growth in patients with SHOX deficiency (221, 222), as well as in those with other types of growth deficiencies (223, 224). In particular, the combination therapy would be worth attempting in girls maturing early or in individuals with early signs of premature fusion of the growth plates. However, clinical experience of combined GH and GnRHa therapy remains poor in SHOX deficiency, and further studies are required to have a definitive conclusion regarding the clinical effects of the combination therapy.
It would also be worth considering whether aromatase inhibitors (AIs) can be a therapeutic option for SHOX deficiency. Theoretically, because AIs hinder the conversion of androgens into estrogens (225), they could suppress estrogen-dependent skeletal lesions and the resulting growth deficiency. However, the therapy with AIs results in an accumulation of androgens and an elevation of gonadotropins (226). Thus, although clinical features of SHOX deficiency are usually more severe in affected girls than in affected boys, AIs cannot be applied to affected girls because of the risk of virilization and ovarian stimulation (226). Furthermore, although the AI therapy could be performed in a small fraction of affected boys with relatively severe phenotype, the long-term efficacy and safety of the AI therapy still remain uncertain in previous studies for boys with ISS (225–228). Thus, the AI therapy for SHOX deficiency is regarded as experimental and is not recommended at present.
H. Clinical indicators of SHOX deficiency
Treatment with GH is effective for ameliorating the growth deficit and skeletal anomalies found in children with SHOX deficiency. However, among children with ISS it may be difficult before puberty to distinguish those who have constitutional delay of growth and adolescence and are destined eventually to reach normal height without intervention. Because a similar late attainment of normal height is unlikely in children with SHOX deficiency (229), it is especially important to identify those short children harboring SHOX mutations so that they can benefit from timely GH treatment.
As reported above, SHOX deficiency results in a wide spectrum of short stature phenotypes, including LWD, LD, or short stature without any typical features in ISS individuals (Figure 6). Birth length is usually only mildly reduced in children with SHOX deficiency, but growth failure is already observed in early childhood (161). Despite there being little or no correlation between genotype and phenotype in individuals with SHOX deficiency, a disproportionate mesomelic short stature (shortening of the forearms and lower legs) characterizes a large fraction of this population. Mesomelic skeletal disproportion usually appears first in school-aged children and increases in frequency and severity with age (181, 230). Therefore, detailed anthropometric analysis of body proportion provides an important selection criterion to allow more cost-effective use of genetic testing. This analysis normally includes measurement of standing height, arm span, and sitting height and the calculation of subischial leg length as the difference between standing and sitting height. When compared to age-related reference standards, children with SHOX deficiency often exhibit a lower than expected arm span and subischial leg lengths in relation to their standing and sitting heights. Mainly based on this observation, several scoring systems have been developed to assist pediatric endocrinologists in their decision to request SHOX molecular analysis before a final diagnosis. Binder et al (184) proposed a limb:trunk ratio (leg length + arm span)/sitting height as a good predictor of SHOX deficiency. Abnormal body proportions are also the criteria for another scoring system that predicts SHOX mutations in children with ISS by considering the ratio of sitting height to standing height for age and sex (179).
Rappold et al (180) developed a more comprehensive scoring system that includes body disproportion (ratios of arm span to standing height and of sitting height to standing height) and other indices of SHOX deficiency such as body mass index, muscular hypertrophy, cubitus valgus, short forearm, bowing of forearm, and dislocation of ulna (at elbow or wrist). The score items and their weights were derived by multivariate analysis in 1608 individuals with sporadic or familiar short stature, 68 (4.2%) of whom had SHOX deficiency (Table 4). At a cutoff score of 7 (of a total score of 24), the positive prediction rate for identifying a SHOX gene point mutation or deletion was 19%. Downstream or upstream SHOX enhancers were not known at the time, and patients were therefore not tested for such deletions. Including the testing for SHOX enhancers would theoretically have increased the positive prediction value. Validation of this score in an independent large cohort of short children confirmed its usefulness (186).
Table 4.
Scoring System Based on Clinical Criteria
| Score Item | Criteria | Score Points |
|---|---|---|
| Dislocation of ulna (at elbow) | Yes | 5 |
| Body mass index | >50th percentile | 4 |
| Short forearm | Yes | 3 |
| Bowing of forearm | Yes | 3 |
| Muscular hypertrophy | Yes | 3 |
| Arm span/height ratio | <96% | 2 |
| Sitting height/height ratio | >55% | 2 |
| Cubitus valgus | Yes | 2 |
| Total | 24 |
Scoring system for identifying patients that qualify for SHOX testing based on clinical criteria.
Scrutinizing left hand radiographs, which are a diagnostic standard in children with short stature for bone age assessment, may further reveal hints of SHOX deficiency (218). It is important to recognize metaphyseal lucency and epiphyseal hypoplasia at the medial side of the distal radius as early skeletal signs of SHOX deficiency (Figure 7), as well as short fourth metacarpals and carpal sign (ie, decreased carpal angle ≤117°) (208, 231). In particular, metaphyseal lucency appears to be fairly specific to SHOX deficiency in childhood, although it becomes obscure with skeletal maturation. Carpal sign is also characteristic of SHOX deficiency, especially at later ages. Although these features are not invariably identified in patients with SHOX deficiency, they can be good indicators for SHOX deficiency. Thus, when such findings are observed on hand and wrist roentgenograms, forearm radiographs should be obtained to examine radial curvature and/or shortening indicative of SHOX deficiency.
Figure 7.

Radiological indications for SHOX deficiency. Hand roentgenograms obtained at 8 years and 2 months of age (A and B) and at 16 years of age (C) in patient 1 and those obtained at 11 years and 9 months of age (D–F) in patient 2. Patient 1 has a 46,XY karyotype and a de novo microdeletion encompassing the SHOX coding and enhancer regions. Patient 2 has a 46,XX karyotype and a paternally derived microdeletion involving the SHOX-downstream enhancer region. Both patients show metaphyseal lucency of the medial side of the distal radius (arrows), epiphyseal hypoplasia of the medial side of the distal radius (arrowheads), and decreased carpal angle.
In addition, familial members of a proband with SHOX deficiency should be studied irrespective of clinical phenotype. Indeed, familial studies have identified SHOX deficiency in individuals, especially in males, with apparent ISS or low-normal height (Figure 6D).
Selecting patients for genetic SHOX testing requires therefore a four-step procedure: 1) patient history including family history; 2) auxological/anthropometric assessments; 3) examination for dysmorphic signs including family members (eg, Madelung deformity); and 4) radiological examinations (180).
VI. Conclusions
The studies summarized here have helped to elucidate the role of SHOX during bone development, the mechanisms regulating its activity, and the etiopathogenesis of SHOX deficiency phenotypes in ISS, LWD, LD, and TS. However, our understanding of the SHOX role in bone development, of SHOX-related pathways, and of mechanisms of regulating SHOX remains incomplete. Such SHOX-related studies have been hampered by the absence of orthologous SHOX genes in mouse and rat, impeding the establishment of rodent knockout models. Consequently, SHOX-expressing cell lines have been useful for early characterizations of SHOX and may continue to be valuable tools for the further investigation of SHOX cellular functions. However, the physiological relevance of SHOX studies in monolayer cellular systems remains questionable, given the complexity of the growth plate and the spatiotemporal expression of SHOX during the different phases of bone development. Because mouse Shox2 and human SHOX are homologous genes sharing a common homeodomain, and because Shox2 may have undertaken some of the functions of Shox, it is plausible that studies using mouse Shox2 knockout models or embryonic limb bud micromass cultures will contribute to elucidation of the developmental roles of human SHOX and SHOX2. Other animal models, such as the chick embryo or zebrafish, may also be useful for deciphering the distinct cellular functions of these growth regulatory proteins. Further characterization of SHOX-related pathways is crucial not only for elucidating SHOX functions and their link to disease, but also to pave the way for novel therapeutic strategies targeted against SHOX-related disorders.
Acknowledgments
We thank Katja Beiser-Schneider, Werner Blum, Gordon Cutler, Maki Fukami, Caroline Hadley, and Marcel Karperien for important comments on the manuscript and Jutta Jung for helping with the art work. We also thankfully acknowledge the work of our clinical colleagues who supported this work by providing patient material and to the patients and their families. This article is dedicated to Rüdiger Blaschke and Werner Blum and all the members of the Rappold lab who have contributed to these studies in the last 20 years.
Disclosure Summary: A.M. and T.O. have nothing to declare. G.A.R. received lecture fees from Eli Lilly, Pfizer, Serono and Ipsen. G.A.R. is a holder of EU 0946721 and US 7252974 patents.
Footnotes
- ACAN
- aggrecan
- AI
- aromatase inhibitor
- ANP
- atrial natriuretic peptide
- BMD
- bone mineral density
- BMP
- bone morphogenetic protein
- BNP
- brain natriuretic peptide
- ChIP
- chromatin immunoprecipitation
- CKII
- casein kinase II
- CNE
- conserved noncoding element
- CNP
- C-type natriuretic peptide
- CS
- Carnegie stage
- Ctgf
- connective tissue growth factor
- E
- embryonic day
- FGF
- fibroblast growth factor
- FGFR
- fibroblast growth factor receptor
- FISH
- fluorescence in situ hybridization
- GFP
- green fluorescent protein
- GnRHa
- GnRH analog
- ISS
- idiopathic short stature
- JAK-STAT
- Janus kinase-signal transducer and activator of transcription
- LD
- Langer dysplasia
- LWD
- Léri-Weill dyschondrosteosis
- MLPA
- multiplex ligation-dependent probe amplification
- MOMP
- mitochondrial outer membrane polarization
- NPPB
- natriuretic peptide B
- NPR
- natriuretic peptide receptor
- PAR1
- pseudoautosomal region 1
- RA
- retinoic acid
- RNS
- reactive nitrogen species
- ROS
- reactive oxygen species
- RUNX2
- runt-related transcription factor 2
- SHOX
- short stature homeobox-containing gene on chromosome X
- SNP
- single nucleotide polymorphism
- SOX9
- SRY-box 9
- SRY
- sex-determining region Y
- TS
- Turner syndrome
- UTR
- untranslated region.
Reference
- 1. Ranke MB. The KIGS aetiology classification system. In: Ranke MB, Gunnarson R, eds. Progress in Growth Hormone Therapy - 5 Years of KIGS. Mannheim, Germany: J&J Verlag; 1994. [Google Scholar]
- 2. Durand C, Rappold GA. Height matters-from monogenic disorders to normal variation. Nat Rev Endocrinol. 2013;9:171–177. [DOI] [PubMed] [Google Scholar]
- 3. Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. 2016;174:R145–R173. [DOI] [PubMed] [Google Scholar]
- 4. Baron J, Sävendahl L, De Luca F, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015;11:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukami M, Naiki Y, Muroya K, et al. Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J Hum Genet. 2015;60:553–556. [DOI] [PubMed] [Google Scholar]
- 6. Wagner T, Wirth J, Meyer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79:1111–1120. [DOI] [PubMed] [Google Scholar]
- 7. Horton WA. Molecular genetic basis of the human chondrodysplasias. Endocrinol Metab Clin North Am. 1996;25:683–697. [DOI] [PubMed] [Google Scholar]
- 8. Prinos P, Costa T, Sommer A, Kilpatrick MW, Tsipouras P. A common FGFR3 gene mutation in hypochondroplasia. Hum Mol Genet. 1995;4:2097–2101. [DOI] [PubMed] [Google Scholar]
- 9. Stoilov I, Kilpatrick MW, Tsipouras P. A common FGFR3 gene mutation is present in achondroplasia but not in hypochondroplasia. Am J Med Genet. 1995;55:127–133. [DOI] [PubMed] [Google Scholar]
- 10. Kant SG, Wit JM, Breuning MH. Genetic analysis of short stature. Horm Res. 2003;60:157–165. [DOI] [PubMed] [Google Scholar]
- 11. Wit JM, van Duyvenvoorde HA, van Klinken JB, et al. Copy number variants in short children born small for gestational age. Horm Res Paediatr. 2014;82:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ogata T, Goodfellow P, Petit C, Aya M, Matsuo N. Short stature in a girl with a terminal Xp deletion distal to DXYS15: localisation of a growth gene(s) in the pseudoautosomal region. J Med Genet. 1992;29:455–459. [PMC free article] [PubMed] [Google Scholar]
- 13. Davis RM. Localisation of male determining factors in man: a thorough review of structural anomalies of the Y chromosome. J Med Genet. 1981;18:161–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vollrath D, Foote S, Hilton A, et al. The human Y chromosome: a 43-interval map based on naturally occurring deletions. Science. 1992;258:52–59. [DOI] [PubMed] [Google Scholar]
- 15. Ogata T, Matsuo N. Sex chromosome aberrations and stature: deduction of the principal factors involved in the determination of adult height. Hum Genet. 1993;91:551–562. [DOI] [PubMed] [Google Scholar]
- 16. May CA, Shone AC, Kalaydjieva L, Sajantila A, Jeffreys AJ. Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat Genet. 2002;31:272–275. [DOI] [PubMed] [Google Scholar]
- 17. Lien S, Szyda J, Schechinger B, Rappold G, Arnheim N. Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am J Hum Genet. 2000;66:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rao E, Weiss B, Fukami M, et al. FISH-deletion mapping defines a 270-kb short stature critical interval in the pseudoautosomal region PAR1 on human sex chromosomes. Hum Genet. 1997;100:236–239. [DOI] [PubMed] [Google Scholar]
- 19. Rao E, Weiss B, Fukami M, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54–63. [DOI] [PubMed] [Google Scholar]
- 20. Ellison JW, Wardak Z, Young MF, Gehron Robey P, Laig-Webster M, Chiong W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet. 1997;6:1341–1347. [DOI] [PubMed] [Google Scholar]
- 21. Durand C, Roeth R, Dweep H, et al. Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS One. 2011;6:e18115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gehring WJ, Affolter M, Bürglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526. [DOI] [PubMed] [Google Scholar]
- 23. Boncinelli E. Homeobox genes and disease. Curr Opin Genet Dev. 1997;7:331–337. [DOI] [PubMed] [Google Scholar]
- 24. Blaschke RJ, Monaghan AP, Schiller S, et al. SHOT, a SHOX-related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc Natl Acad Sci USA. 1998;95:2406–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clement-Jones M, Schiller S, Rao E, et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet. 2000;9:695–702. [DOI] [PubMed] [Google Scholar]
- 26. Liu H, Chen CH, Espinoza-Lewis RA, et al. Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J Biol Chem. 2011;286:17029–17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tiecke E, Bangs F, Blaschke R, Farrell ER, Rappold G, Tickle C. Expression of the short stature homeobox gene Shox is restricted by proximal and distal signals in chick limb buds and affects the length of skeletal elements. Dev Biol. 2006;298:585–596. [DOI] [PubMed] [Google Scholar]
- 28. Decker E, Durand C, Bender S, et al. FGFR3 is a target of the homeobox transcription factor SHOX in limb development. Hum Mol Genet. 2011;20:1524–1535. [DOI] [PubMed] [Google Scholar]
- 29. Benito-Sanz S, Aza-Carmona M, Rodríguez-Estevez A, et al. Identification of the first PAR1 deletion encompassing upstream SHOX enhancers in a family with idiopathic short stature. Eur J Hum Genet. 2012;20:125–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Durand C, Bangs F, Signolet J, Decker E, Tickle C, Rappold G. Enhancer elements upstream of the SHOX gene are active in the developing limb. Eur J Hum Genet. 2010;18:527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sabherwal N, Bangs F, Röth R, et al. Long-range conserved non-coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Hum Mol Genet. 2007;16:210–222. [DOI] [PubMed] [Google Scholar]
- 32. Sawada R, Kamei H, Hakuno F, Takahashi S, Shimizu T. In vivo loss of function study reveals the short stature homeobox-containing (shox) gene plays indispensable roles in early embryonic growth and bone formation in zebrafish. Dev Dyn. 2015;244:146–156. [DOI] [PubMed] [Google Scholar]
- 33. Kenyon EJ, McEwen GK, Callaway H, Elgar G. Functional analysis of conserved non-coding regions around the short stature hox gene (shox) in whole zebrafish embryos. PLoS One. 2011;6:e21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cobb J, Dierich A, Huss-Garcia Y, Duboule D. A mouse model for human short-stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long-bone development. Proc Natl Acad Sci USA. 2006;103:4511–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blaschke RJ, Hahurij ND, Kuijper S, et al. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007;115:1830–1838. [DOI] [PubMed] [Google Scholar]
- 36. Espinoza-Lewis RA, Yu L, He F, et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2–5. Dev Biol. 2009;327:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu L, Gu S, Alappat S, et al. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development. 2005;132:4397–4406. [DOI] [PubMed] [Google Scholar]
- 38. Neufeld SJ, Wang F, Cobb J. Genetic interactions between Shox2 and Hox genes during the regional growth and development of the mouse limb. Genetics. 2014;198:1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bobick BE, Cobb J. Shox2 regulates progression through chondrogenesis in the mouse proximal limb. J Cell Sci. 2012;125:6071–6083. [DOI] [PubMed] [Google Scholar]
- 40. Vickerman L, Neufeld S, Cobb J. Shox2 function couples neural, muscular and skeletal development in the proximal forelimb. Dev Biol. 2011;350:323–336. [DOI] [PubMed] [Google Scholar]
- 41. Gu S, Wei N, Yu L, Fei J, Chen Y. Shox2-deficiency leads to dysplasia and ankylosis of the temporomandibular joint in mice. Mech Dev. 2008;125:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosin JM, McAllister BB, Dyck RH, Percival CJ, Kurrasch DM, Cobb J. Mice lacking the transcription factor SHOX2 display impaired cerebellar development and deficits in motor coordination. Dev Biol. 2015;399:54–67. [DOI] [PubMed] [Google Scholar]
- 43. Scott A, Hasegawa H, Sakurai K, Yaron A, Cobb J, Wang F. Transcription factor short stature homeobox 2 is required for proper development of tropomyosin-related kinase B-expressing mechanosensory neurons. J Neurosci. 2011;31:6741–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee KY, Yamamoto Y, Boucher J, et al. Shox2 is a molecular determinant of depot-specific adipocyte function. Proc Natl Acad Sci USA. 2013;110:11409–11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blaschke RJ, Töpfer C, Marchini A, Steinbeisser H, Janssen JW, Rappold GA. Transcriptional and translational regulation of the Leri-Weill and Turner syndrome homeobox gene SHOX. J Biol Chem. 2003;278:47820–47826. [DOI] [PubMed] [Google Scholar]
- 46. van Heyningen V, Bickmore W. Regulation from a distance: long-range control of gene expression in development and disease. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pennacchio LA, Rubin EM. Genomic strategies to identify mammalian regulatory sequences. Nat Rev Genet. 2001;2:100–109. [DOI] [PubMed] [Google Scholar]
- 48. Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fukami M, Kato F, Tajima T, Yokoya S, Ogata T. Transactivation function of an approximately 800-bp evolutionarily conserved sequence at the SHOX 3′ region: implication for the downstream enhancer. Am J Hum Genet. 2006;78:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benito-Sanz S, Royo JL, Barroso E, et al. Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J Med Genet. 2012;49:442–450. [DOI] [PubMed] [Google Scholar]
- 51. Rao E, Blaschke RJ, Marchini A, Niesler B, Burnett M, Rappold GA. The Leri-Weill and Turner syndrome homeobox gene SHOX encodes a cell-type specific transcriptional activator. Hum Mol Genet. 2001;10:3083–3091. [DOI] [PubMed] [Google Scholar]
- 52. Wolberger C. Homeodomain interactions. Curr Opin Struct Biol. 1996;6:62–68. [DOI] [PubMed] [Google Scholar]
- 53. Gehring WJ, Qian YQ, Billeter M, et al. Homeodomain-DNA recognition. Cell. 1994;78:211–223. [DOI] [PubMed] [Google Scholar]
- 54. Belin V, Cusin V, Viot G, et al. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998;19:67–69. [DOI] [PubMed] [Google Scholar]
- 55. Cormier-Daire V, Huber C, Munnich A. Allelic and nonallelic heterogeneity in dyschondrosteosis (Leri-Weill syndrome). Am J Med Genet. 2001;106:272–274. [DOI] [PubMed] [Google Scholar]
- 56. Flanagan SF, Munns CF, Hayes M, et al. Prevalence of mutations in the short stature homeobox containing gene (SHOX) in Madelung deformity of childhood. J Med Genet. 2002;39:758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grigelioniene G, Schoumans J, Neumeyer L, et al. Analysis of short stature homeobox-containing gene (SHOX) and auxological phenotype in dyschondrosteosis and isolated Madelung deformity. Hum Genet. 2001;109:551–558. [DOI] [PubMed] [Google Scholar]
- 58. Rappold GA, Fukami M, Niesler B, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab. 2002;87:1402–1406. [DOI] [PubMed] [Google Scholar]
- 59. Wilson D, Sheng G, Lecuit T, Dostatni N, Desplan C. Cooperative dimerization of paired class homeo domains on DNA. Genes Dev. 1993;7:2120–2134. [DOI] [PubMed] [Google Scholar]
- 60. Wilson DS, Desplan C. Homeodomain proteins. Cooperating to be different. Curr Biol. 1995;5:32–34. [DOI] [PubMed] [Google Scholar]
- 61. Schneider KU, Marchini A, Sabherwal N, et al. Alteration of DNA binding, dimerization, and nuclear translocation of SHOX homeodomain mutations identified in idiopathic short stature and Leri-Weill dyschondrosteosis. Hum Mutat. 2005;26:44–52. [DOI] [PubMed] [Google Scholar]
- 62. Sabherwal N, Blaschke RJ, Marchini A, et al. A novel point mutation A170P in the SHOX gene defines impaired nuclear translocation as a molecular cause for Leri-Weill dyschondrosteosis and Langer dysplasia. J Med Genet. 2004;41:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marchini A, Daeffler L, Marttila T, et al. Phosphorylation on Ser106 modulates the cellular functions of the SHOX homeodomain protein. J Mol Biol. 2006;355:590–603. [DOI] [PubMed] [Google Scholar]
- 64. Huber C, Cusin V, Le Merrer M, et al. SHOX point mutations in dyschondrosteosis. J Med Genet. 2001;38:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Benito-Sanz S, del Blanco DG, Aza-Carmona M, et al. PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Léri-Weill dyschondrosteosis (LWD) probands. Hum Mutat. 2006;27:1062. [DOI] [PubMed] [Google Scholar]
- 66. Shears DJ, Guillen-Navarro E, Sempere-Miralles M, Domingo-Jimenez R, Scambler PJ, Winter RM. Pseudodominant inheritance of Langer mesomelic dysplasia caused by a SHOX homeobox missense mutation. Am J Med Genet. 2002;110:153–157. [DOI] [PubMed] [Google Scholar]
- 67. Binder G, Renz A, Martinez A, et al. SHOX haploinsufficiency and Leri-Weill dyschondrosteosis: prevalence and growth failure in relation to mutation, sex, and degree of wrist deformity. J Clin Endocrinol Metab. 2004;89:4403–4408. [DOI] [PubMed] [Google Scholar]
- 68. Sabherwal N, Schneider KU, Blaschke RJ, Marchini A, Rappold G. Impairment of SHOX nuclear localization as a cause for Léri-Weill syndrome. J Cell Sci. 2004;117:3041–3048. [DOI] [PubMed] [Google Scholar]
- 69. Furukawa T, Kozak CA, Cepko CL. rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc Natl Acad Sci USA. 1997;94:3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Semina EV, Reiter R, Leysens NJ, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. [DOI] [PubMed] [Google Scholar]
- 71. Semina EV, Reiter RS, Murray JC. A new human homeobox gene OGI2X is a member of the most conserved homeobox gene family and is expressed during heart development in mouse. Hum Mol Genet. 1998;7:415–422. [DOI] [PubMed] [Google Scholar]
- 72. Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol. 2013;5:a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. [DOI] [PubMed] [Google Scholar]
- 74. Akiyama H. Control of chondrogenesis by the transcription factor Sox9. Mod Rheumatol. 2008;18:213–219. [DOI] [PubMed] [Google Scholar]
- 75. Inada M, Wang Y, Byrne MH, et al. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci USA. 2004;101:17192–17197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lui JC, Nilsson O, Chan Y, et al. Synthesizing genome-wide association studies and expression microarray reveals novel genes that act in the human growth plate to modulate height. Hum Mol Genet. 2012;21:5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lui JC, Nilsson O, Baron J. Recent research on the growth plate: recent insights into the regulation of the growth plate. J Mol Endocrinol. 2014;53:T1–T9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guo MH, Shen Y, Walvoord EC, et al. Whole exome sequencing to identify genetic causes of short stature. Horm Res Paediatr. 2014;82:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99:3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lango Allen H, Estrada K, Lettre G, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kozhemyakina E, Lassar AB, Zelzer E. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development. 2015;142:817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res. 2005;64:157–165. [DOI] [PubMed] [Google Scholar]
- 84. Sederquist B, Fernandez-Vojvodich P, Zaman F, Sävendahl L. Recent research on the growth plate: impact of inflammatory cytokines on longitudinal bone growth. J Mol Endocrinol. 2014;53:T35–T44. [DOI] [PubMed] [Google Scholar]
- 85. Vanderschueren D, Laurent MR, Claessens F, et al. Sex steroid actions in male bone. Endocr Rev. 2014;35:906–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. De Luca F, Uyeda JA, Mericq V, et al. Retinoic acid is a potent regulator of growth plate chondrogenesis. Endocrinology. 2000;141:346–353. [DOI] [PubMed] [Google Scholar]
- 87. Maes C. Signaling pathways effecting crosstalk between cartilage and adjacent tissues: seminars in cell and developmental biology: the biology and pathology of cartilage [published online May 12, 2016]. Semin Cell Dev Biol. doi: 10.1016/j.semcdb.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 88. De Luca F, Barnes KM, Uyeda JA, et al. Regulation of growth plate chondrogenesis by bone morphogenetic protein-2. Endocrinology. 2001;142:430–436. [DOI] [PubMed] [Google Scholar]
- 89. Pogue R, Lyons K. BMP signaling in the cartilage growth plate. Curr Top Dev Biol. 2006;76:1–48. [DOI] [PubMed] [Google Scholar]
- 90. Andrade AC, Nilsson O, Barnes KM, Baron J. Wnt gene expression in the post-natal growth plate: regulation with chondrocyte differentiation. Bone. 2007;40:1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kuss P, Kraft K, Stumm J, et al. Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Dev Biol. 2014;385:83–93. [DOI] [PubMed] [Google Scholar]
- 92. Xie Y, Zhou S, Chen H, Du X, Chen L. Recent research on the growth plate: advances in fibroblast growth factor signaling in growth plate development and disorders. J Mol Endocrinol. 2014;53:T11–T34. [DOI] [PubMed] [Google Scholar]
- 93. Chusho H, Tamura N, Ogawa Y, et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci USA. 2001;98:4016–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mericq V, Uyeda JA, Barnes KM, De Luca F, Baron J. Regulation of fetal rat bone growth by C-type natriuretic peptide and cGMP. Pediatr Res. 2000;47:189–193. [DOI] [PubMed] [Google Scholar]
- 95. Pejchalova K, Krejci P, Wilcox WR. C-natriuretic peptide: an important regulator of cartilage. Mol Genet Metab. 2007;92:210–215. [DOI] [PubMed] [Google Scholar]
- 96. Fernandez-Vojvodich P, Palmblad K, Karimian E, Andersson U, Sävendahl L. Pro-inflammatory cytokines produced by growth plate chondrocytes may act locally to modulate longitudinal bone growth. Horm Res Paediatr. 2012;77:180–187. [DOI] [PubMed] [Google Scholar]
- 97. Jochmann K, Bachvarova V, Vortkamp A. Heparan sulfate as a regulator of endochondral ossification and osteochondroma development. Matrix Biol. 2014;34:55–63. [DOI] [PubMed] [Google Scholar]
- 98. Marchini A, Marttila T, Winter A, et al. The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J Biol Chem. 2004;279:37103–37114. [DOI] [PubMed] [Google Scholar]
- 99. Munns CJ, Haase HR, Crowther LM, et al. Expression of SHOX in human fetal and childhood growth plate. J Clin Endocrinol Metab. 2004;89:4130–4135. [DOI] [PubMed] [Google Scholar]
- 100. Munns CF, Glass IA, LaBrom R, et al. Histopathological analysis of Leri-Weill dyschondrosteosis: disordered growth plate. Hand Surg. 2001;6:13–23. [DOI] [PubMed] [Google Scholar]
- 101. Hristov G, Marttila T, Durand C, Niesler B, Rappold GA, Marchini A. SHOX triggers the lysosomal pathway of apoptosis via oxidative stress. Hum Mol Genet. 2014;23:1619–1630. [DOI] [PubMed] [Google Scholar]
- 102. Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012;1824:22–33. [DOI] [PubMed] [Google Scholar]
- 103. Marchini A, Häcker B, Marttila T, et al. BNP is a transcriptional target of the short stature homeobox gene SHOX. Hum Mol Genet. 2007;16:3081–3087. [DOI] [PubMed] [Google Scholar]
- 104. Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev. 2000;21:23–39. [DOI] [PubMed] [Google Scholar]
- 105. Kant SG, Cervenkova I, Balek L, et al. A novel variant of FGFR3 causes proportionate short stature. Eur J Endocrinol. 2015;172:763–770. [DOI] [PubMed] [Google Scholar]
- 106. Toydemir RM, Brassington AE, Bayrak-Toydemir P, et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet. 2006;79:935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Makrythanasis P, Temtamy S, Aglan MS, Otaify GA, Hamamy H, Antonarakis SE. A novel homozygous mutation in FGFR3 causes tall stature, severe lateral tibial deviation, scoliosis, hearing impairment, camptodactyly, and arachnodactyly. Hum Mutat. 2014;35:959–963. [DOI] [PubMed] [Google Scholar]
- 108. Chen L, Adar R, Yang X, et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Naski MC, Colvin JS, Coffin JD, Ornitz DM. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–4988. [DOI] [PubMed] [Google Scholar]
- 110. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. [DOI] [PubMed] [Google Scholar]
- 111. Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. [DOI] [PubMed] [Google Scholar]
- 112. Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. [DOI] [PubMed] [Google Scholar]
- 113. Aza-Carmona M, Shears DJ, Yuste-Checa P, et al. SHOX interacts with the chondrogenic transcription factors SOX5 and SOX6 to activate the aggrecan enhancer. Hum Mol Genet. 2011;20:1547–1559. [DOI] [PubMed] [Google Scholar]
- 114. Hattori T, Müller C, Gebhard S, et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137:901–911. [DOI] [PubMed] [Google Scholar]
- 115. Mattos EP, Sanseverino MT, Magalhães JA, et al. Clinical and molecular characterization of a Brazilian cohort of campomelic dysplasia patients, and identification of seven new SOX9 mutations. Genet Mol Biol. 2015;38:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tompson SW, Merriman B, Funari VA, et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet. 2009;84:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nilsson O, Guo MH, Dunbar N, et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab. 2014;99:E1510–E1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Aza-Carmona M, Barca-Tierno V, Hisado-Oliva A, et al. NPPB and ACAN, two novel SHOX2 transcription targets implicated in skeletal development. PLoS One. 2014;9:e83104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Beiser KU, Glaser A, Kleinschmidt K, et al. Identification of novel SHOX target genes in the developing limb using a transgenic mouse model. PLoS One. 2014;9:e98543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cohn MJ, Patel K, Krumlauf R, Wilkinson DG, Clarke JD, Tickle C. Hox9 genes and vertebrate limb specification. Nature. 1997;387:97–101. [DOI] [PubMed] [Google Scholar]
- 121. Zakany J, Duboule D. The role of Hox genes during vertebrate limb development. Curr Opin Genet Dev. 2007;17:359–366. [DOI] [PubMed] [Google Scholar]
- 122. Fromental-Ramain C, Warot X, Lakkaraju S, et al. Specific and redundant functions of the paralogous Hoxa-9 and Hoxd-9 genes in forelimb and axial skeleton patterning. Development. 1996;122:461–472. [DOI] [PubMed] [Google Scholar]
- 123. Wellik DM, Capecchi MR. Hox10 and Hox11 genes are required to globally pattern the mammalian skeleton. Science. 2003;301:363–367. [DOI] [PubMed] [Google Scholar]
- 124. Davis AP, Witte DP, Hsieh-Li HM, Potter SS, Capecchi MR. Absence of radius and ulna in mice lacking hoxa-11 and hoxd-11. Nature. 1995;375:791–795. [DOI] [PubMed] [Google Scholar]
- 125. Fromental-Ramain C, Warot X, Messadecq N, LeMeur M, Dollé P, Chambon P. Hoxa-13 and Hoxd-13 play a crucial role in the patterning of the limb autopod. Development. 1996;122:2997–3011. [DOI] [PubMed] [Google Scholar]
- 126. Durand C, Decker E, Roeth R, Schneider KU, Rappold G. The homeobox transcription factor HOXA9 is a regulator of SHOX in U2OS cells and chicken micromass cultures. PLoS One. 2012;7:e45369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yu L, Liu H, Yan M, et al. Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev Biol. 2007;306:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Gross S, Krause Y, Wuelling M, Vortkamp A. Hoxa11 and Hoxd11 regulate chondrocyte differentiation upstream of Runx2 and Shox2 in mice. PLoS One. 2012;7:e43553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat. 2012;33:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D, Basilico C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nishikimi T, Kuwahara K, Nakao K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J Cardiol. 2011;57:131–140. [DOI] [PubMed] [Google Scholar]
- 132. Volpe M. Natriuretic peptides and cardio-renal disease. Int J Cardiol. 2014;176:630–639. [DOI] [PubMed] [Google Scholar]
- 133. Suzuki S, Yoshimura M, Nakayama M, et al. Plasma level of B-type natriuretic peptide as a prognostic marker after acute myocardial infarction: a long-term follow-up analysis. Circulation. 2004;110:1387–1391. [DOI] [PubMed] [Google Scholar]
- 134. Tamura N, Doolittle LK, Hammer RE, Shelton JM, Richardson JA, Garbers DL. Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc Natl Acad Sci USA. 2004;101:17300–17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yasoda A, Komatsu Y, Chusho H, et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10:80–86. [DOI] [PubMed] [Google Scholar]
- 136. Bartels CF, Bükülmez H, Padayatti P, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet. 2004;75:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Hachiya R, Ohashi Y, Kamei Y, et al. Intact kinase homology domain of natriuretic peptide receptor-B is essential for skeletal development. J Clin Endocrinol Metab. 2007;92:4009–4014. [DOI] [PubMed] [Google Scholar]
- 138. Moncla A, Missirian C, Cacciagli P, et al. A cluster of translocation breakpoints in 2q37 is associated with overexpression of NPPC in patients with a similar overgrowth phenotype. Hum Mutat. 2007;28:1183–1188. [DOI] [PubMed] [Google Scholar]
- 139. Bocciardi R, Giorda R, Buttgereit J, et al. Overexpression of the C-type natriuretic peptide (CNP) is associated with overgrowth and bone anomalies in an individual with balanced t(2;7) translocation. Hum Mutat. 2007;28:724–731. [DOI] [PubMed] [Google Scholar]
- 140. Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35–44. [DOI] [PubMed] [Google Scholar]
- 141. Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol. 2003;161:1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Suda M, Ogawa Y, Tanaka K, et al. Skeletal overgrowth in transgenic mice that overexpress brain natriuretic peptide. Proc Natl Acad Sci USA. 1998;95:2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tamura N, Ogawa Y, Chusho H, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci USA. 2000;97:4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2015;100:E1133–E1142. [DOI] [PubMed] [Google Scholar]
- 145. Vasques GA, Amano N, Docko AJ, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab. 2013;98:E1636–E1644. [DOI] [PubMed] [Google Scholar]
- 146. Wang SR, Jacobsen CM, Carmichael H, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat. 2015;36:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sybert VP, McCauley E. Turner's syndrome. N Engl J Med. 2004;351:1227–1238. [DOI] [PubMed] [Google Scholar]
- 148. Roach HI, Aigner T, Kouri JB. Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis. 2004;9:265–277. [DOI] [PubMed] [Google Scholar]
- 149. Morita K, Miyamoto T, Fujita N, et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J Exp Med. 2007;204:1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Teixeira CC, Mansfield K, Hertkorn C, Ischiropoulos H, Shapiro IM. Phosphate-induced chondrocyte apoptosis is linked to nitric oxide generation. Am J Physiol Cell Physiol. 2001;281:C833–C839. [DOI] [PubMed] [Google Scholar]
- 151. Rajpurohit R, Mansfield K, Ohyama K, Ewert D, Shapiro IM. Chondrocyte death is linked to development of a mitochondrial membrane permeability transition in the growth plate. J Cell Physiol. 1999;179:287–296. [DOI] [PubMed] [Google Scholar]
- 152. Teixeira CC, Costas AP, Nemelivsky Y. Apoptosis of growth plate chondrocytes occurs through a mitochondrial pathway. Angle Orthodont. 2007;77:129–134. [DOI] [PubMed] [Google Scholar]
- 153. Söderström M, Salminen H, Glumoff V, Kirschke H, Aro H, Vuorio E. Cathepsin expression during skeletal development. Biochim Biophys Acta. 1999;1446:35–46. [DOI] [PubMed] [Google Scholar]
- 154. Ohsawa Y, Nitatori T, Higuchi S, Kominami E, Uchiyama Y. Lysosomal cysteine and aspartic proteinases, acid phosphatase, and an endogenous cysteine proteinase inhibitor, cystatin-β, in rat osteoclasts. J Histochem Cytochem. 1993;41:1075–1083. [DOI] [PubMed] [Google Scholar]
- 155. Goto T, Kiyoshima T, Moroi R, et al. Localization of cathepsins B, D, and L in the rat osteoclast by immuno-light and -electron microscopy. Histochemistry. 1994;101:33–40. [DOI] [PubMed] [Google Scholar]
- 156. Hill PA, Buttle DJ, Jones SJ, et al. Inhibition of bone resorption by selective inactivators of cysteine proteinases. J Cell Biochem. 1994;56:118–130. [DOI] [PubMed] [Google Scholar]
- 157. Shears DJ, Vassal HJ, Goodman FR, et al. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet. 1998;19:70–73. [DOI] [PubMed] [Google Scholar]
- 158. Zinn AR, Ross JL. Critical regions for Turner syndrome phenotypes on the X chromosome. In: Saenger PH, Pasquino AM, eds. Optimizing Health Care for Turner Patients in the 21st Century. Proceedings of the 5th International Symposium on Turner Syndrome, Naples, Italy. Amsterdam, The Netherlands: Elsevier Science; 2000:19–28. [Google Scholar]
- 159. Robertson SP, Shears DJ, Oei P, et al. Homozygous deletion of SHOX in a mentally retarded male with Langer mesomelic dysplasia. J Med Genet. 2000;37:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ogata T, Matsuo N, Nishimura G. SHOX haploinsufficiency and overdosage: impact of gonadal function status. J Med Genet. 2001;38:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011;75:81–89. [DOI] [PubMed] [Google Scholar]
- 162. Chen J, Wildhardt G, Zhong Z, et al. Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J Med Genet. 2009;46:834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Fukami M, Dateki S, Kato F, et al. Identification and characterization of cryptic SHOX intragenic deletions in three Japanese patients with Léri-Weill dyschondrosteosis. J Hum Genet. 2008;53:454–459. [DOI] [PubMed] [Google Scholar]
- 164. Marchini A, Rappold G, Schneider KU. SHOX at a glance: from gene to protein. Arch Physiol Biochem. 2007;113:116–123. [DOI] [PubMed] [Google Scholar]
- 165. Benito-Sanz S, Barroso E, Heine-Suñer D, et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab. 2011;96:E404–E412. [DOI] [PubMed] [Google Scholar]
- 166. Sandoval GT, Jaimes GC, Barrios MC, Cespedes C, Velasco HM. SHOX gene and conserved noncoding element deletions/duplications in Colombian patients with idiopathic short stature. Mol Genet Genomic Med. 2014;2:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. van Duyvenvoorde HA, Lui JC, Kant SG, et al. Copy number variants in patients with short stature. Eur J Hum Genet. 2014;22:602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Gervasini C, Grati FR, Lalatta F, et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet Med. 2010;12:634–640. [DOI] [PubMed] [Google Scholar]
- 169. Bunyan DJ, Baffico M, Capone L, et al. Duplications upstream and downstream of SHOX identified as novel causes of Leri-Weill dyschondrosteosis or idiopathic short stature. Am J Med Genet A. 2016;170:949–957. [DOI] [PubMed] [Google Scholar]
- 170. Tropeano M, Howley D, Gazzellone MJ, et al. Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions [published online April 12, 2016]. J Med Genet. doi: 10.1136/jmedgenet-2015-103621. [DOI] [PubMed] [Google Scholar]
- 171. Niesler B, Röth R, Wilke S, Fujimura F, Fischer C, Rappold G. The novel human SHOX allelic variant database. Hum Mutat. 2007;28:933–938. [DOI] [PubMed] [Google Scholar]
- 172. Blaschke RJ, Rappold G. The pseudoautosomal regions, SHOX and disease. Curr Opin Genet Dev. 2006;16:233–239. [DOI] [PubMed] [Google Scholar]
- 173. Blaschke RJ, Rappold GA. SHOX: growth, Léri-Weill and Turner syndromes. Trends Endocrinol Metab. 2000;11:227–230. [DOI] [PubMed] [Google Scholar]
- 174. Huber C, Rosilio M, Munnich A, Cormier-Daire V. High incidence of SHOX anomalies in individuals with short stature. J Med Genet. 2006;43:735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ballabio A, Bardoni B, Carrozzo R, et al. Contiguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proc Natl Acad Sci USA. 1989;86:10001–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Léri A, Weill J. Une affection congenitale et symetrique du developpement osseux: la dyschondrosteose. Bull Mem Soc Med Hosp Paris. 1929;35:1491–1494. [Google Scholar]
- 177. Seki A, Jinno T, Suzuki E, Takayama S, Ogata T, Fukami M. Skeletal deformity associated with SHOX deficiency. Clin Pediatr Endocrinol. 2014;23:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Grigelioniene G, Eklöf O, Ivarsson SA, et al. Mutations in short stature homeobox containing gene (SHOX) in dyschondrosteosis but not in hypochondroplasia. Hum Genet. 2000;107:145–149. [DOI] [PubMed] [Google Scholar]
- 179. Jorge AA, Souza SC, Nishi MY, et al. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf). 2007;66:130–135. [DOI] [PubMed] [Google Scholar]
- 180. Rappold G, Blum WF, Shavrikova EP, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007;44:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ross JL, Scott C, Jr, Marttila P, et al. Phenotypes associated with SHOX deficiency. J Clin Endocrinol Metab. 2001;86:5674–5680. [DOI] [PubMed] [Google Scholar]
- 182. Schiller S, Spranger S, Schechinger B, et al. Phenotypic variation and genetic heterogeneity in Léri-Weill syndrome. Eur J Hum Genet. 2000;8:54–62. [DOI] [PubMed] [Google Scholar]
- 183. Falcinelli C, Iughetti L, Percesepe A, et al. SHOX point mutations and deletions in Leri-Weill dyschondrosteosis. J Med Genet. 2002;39:E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Binder G, Ranke MB, Martin DD. Auxology is a valuable instrument for the clinical diagnosis of SHOX haploinsufficiency in school-age children with unexplained short stature. J Clin Endocrinol Metab. 2003;88:4891–4896. [DOI] [PubMed] [Google Scholar]
- 185. Benito-Sanz S, Thomas NS, Huber C, et al. A novel class of pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am J Hum Genet. 2005;77:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Rosilio M, Huber-Lequesne C, Sapin H, Carel JC, Blum WF, Cormier-Daire V. Genotypes and phenotypes of children with SHOX deficiency in France. J Clin Endocrinol Metab. 2012;97:E1257–E1265. [DOI] [PubMed] [Google Scholar]
- 187. Kant SG, van der Kamp HJ, Kriek M, et al. The jumping SHOX gene–crossover in the pseudoautosomal region resulting in unusual inheritance of Leri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2011;96:E356–E359. [DOI] [PubMed] [Google Scholar]
- 188. Langer LO., Jr Mesomelic dwarfism of the hypoplastic ulna, fibula, mandible type. Radiology. 1967;89:654–660. [DOI] [PubMed] [Google Scholar]
- 189. Fukami M, Okuyama T, Yamamori S, Nishimura G, Ogata T. Microdeletion in the SHOX 3′ region associated with skeletal phenotypes of Langer mesomelic dysplasia in a 45,X/46,X,r(X) infant and Leri-Weill dyschondrosteosis in her 46,XX mother: implication for the SHOX enhancer. Am J Med Genet A. 2005;137:72–76. [DOI] [PubMed] [Google Scholar]
- 190. Thomas NS, Maloney V, Bass P, Mulik V, Wellesley D, Castle B. SHOX mutations in a family and a fetus with Langer mesomelic dwarfism. Am J Med Genet A. 2004;128A:179–184. [DOI] [PubMed] [Google Scholar]
- 191. Zinn AR, Wei F, Zhang L, et al. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet. 2002;110:158–163. [DOI] [PubMed] [Google Scholar]
- 192. Turner HH. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endrocrinology. 1938;23:566–574. [PubMed] [Google Scholar]
- 193. Ullrich O. Über typische Kombinationsbilder multipler Abartungen. Eur J Pediatr. 1930;49:271–276. [Google Scholar]
- 194. Saenger P. Turner's syndrome. N Engl J Med. 1996;335:1749–1754. [DOI] [PubMed] [Google Scholar]
- 195. Ranke MB, Saenger P. Turner's syndrome. Lancet. 2001;358:309–314. [DOI] [PubMed] [Google Scholar]
- 196. Saenger P, Wikland KA, Conway GS, et al. Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrinol Metab. 2001;86:3061–3069. [DOI] [PubMed] [Google Scholar]
- 197. Rovet JF. The psychoeducational characteristics of children with Turner syndrome. J Learn Disabil. 1993;26:333–341. [DOI] [PubMed] [Google Scholar]
- 198. Kosho T, Muroya K, Nagai T, et al. Skeletal features and growth patterns in 14 patients with haploinsufficiency of SHOX: implications for the development of Turner syndrome. J Clin Endocrinol Metab. 1999;84:4613–4621. [DOI] [PubMed] [Google Scholar]
- 199. Faienza MF, Ventura A, Colucci S, Cavallo L, Grano M, Brunetti G. Bone fragility in Turner syndrome: mechanisms and prevention strategies. Front Endocrinol (Lausanne). 2016;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Bakalov VK, Axelrod L, Baron J, et al. Selective reduction in cortical bone mineral density in turner syndrome independent of ovarian hormone deficiency. J Clin Endocrinol Metab. 2003;88:5717–5722. [DOI] [PubMed] [Google Scholar]
- 201. Ottesen AM, Aksglaede L, Garn I, et al. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. Am J Med Genet A. 2010;152A:1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Ogata T, Matsuo N. Turner syndrome and female sex chromosome aberrations: deduction of the principal factors involved in the development of clinical features. Hum Genet. 1995;95:607–629. [DOI] [PubMed] [Google Scholar]
- 203. Gilbert EF, Opitz JM. Developmental and other pathologic changes in syndromes caused by chromosome abnormalities. Perspect Pediatr Pathol. 1982;7:1–63. [PubMed] [Google Scholar]
- 204. Epstein CJ. The consequences of chromosome imbalance: principles, mechanisms, and models. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 205. Binder G, Schwarze CP, Ranke MB. Identification of short stature caused by SHOX defects and therapeutic effect of recombinant human growth hormone. J Clin Endocrinol Metab. 2000;85:245–249. [DOI] [PubMed] [Google Scholar]
- 206. Calabrese G, Fischetto R, Stuppia L, et al. X/Y translocation in a family with Leri-Weill dyschondrosteosis. Hum Genet. 1999;105:367–368. [DOI] [PubMed] [Google Scholar]
- 207. Hirschfeldova K, Solc R, Baxova A, et al. SHOX gene defects and selected dysmorphic signs in patients of idiopathic short stature and Léri-Weill dyschondrosteosis. Gene. 2012;491:123–127. [DOI] [PubMed] [Google Scholar]
- 208. Tauber M, Lounis N, Coulet J, Baunin C, Cahuzac JP, Rochiccioli P. Wrist anomalies in Turner syndrome compared with Leri-Weill dyschondrosteosis: a new feature in Turner syndrome. Eur J Pediatr. 2004;163:475–481. [DOI] [PubMed] [Google Scholar]
- 209. Stuppia L, Calabrese G, Gatta V, et al. SHOX mutations detected by FISH and direct sequencing in patients with short stature. J Med Genet. 2003;40:E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Poggi H, Vera A, Avalos C, et al. A deletion of more than 800 kb is the most recurrent mutation in Chilean patients with SHOX gene defects. Horm Res Paediatr. 2015;84:254–257. [DOI] [PubMed] [Google Scholar]
- 211. Funari MF, Jorge AA, Souza SC, et al. Usefulness of MLPA in the detection of SHOX deletions. Eur J Med Genet. 2010;53:234–238. [DOI] [PubMed] [Google Scholar]
- 212. Donze SH, Meijer CR, Kant SG, et al. The growth response to GH treatment is greater in patients with SHOX enhancer deletions compared to SHOX defects. Eur J Endocrinol. 2015;173:611–621. [DOI] [PubMed] [Google Scholar]
- 213. Bunyan DJ, Baker KR, Harvey JF, Thomas NS. Diagnostic screening identifies a wide range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. Am J Med Genet A. 2013;161A:1329–1338. [DOI] [PubMed] [Google Scholar]
- 214. Verdin H, Fernández-Miñán A, Benito-Sanz S, et al. Profiling of conserved non-coding elements upstream of SHOX and functional characterisation of the SHOX cis-regulatory landscape. Sci Rep. 2015;5:17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bunyan DJ, Taylor EJ, Maloney VK, Blyth M. Homozygosity for a novel deletion downstream of the SHOX gene provides evidence for an additional long range regulatory region with a mild phenotypic effect. Am J Med Genet A. 2014;164A:2764–2768. [DOI] [PubMed] [Google Scholar]
- 216. Tsuchiya T, Shibata M, Numabe H, et al. Compound heterozygous deletions in pseudoautosomal region 1 in an infant with mild manifestations of Langer mesomelic dysplasia. Am J Med Genet A. 2014;164A:505–510. [DOI] [PubMed] [Google Scholar]
- 217. Blum WF, Crowe BJ, Quigley CA, et al. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92:219–228. [DOI] [PubMed] [Google Scholar]
- 218. Child CJ, Kalifa G, Jones C, et al. Radiological features in patients with short stature homeobox-containing (SHOX) gene deficiency and Turner syndrome before and after 2 years of GH treatment. Horm Res Paediatr. 2015;84:14–25. [DOI] [PubMed] [Google Scholar]
- 219. Blum WF, Ross JL, Zimmermann AG, et al. GH treatment to final height produces similar height gains in patients with SHOX deficiency and Turner syndrome: results of a multicenter trial. J Clin Endocrinol Metab. 2013;98:E1383–E1392. [DOI] [PubMed] [Google Scholar]
- 220. Ogata T. SHOX haploinsufficiency: lessons from clinical studies. Curr Opin Endocrinol Diabetes Obes. 2002;9:13–20. [Google Scholar]
- 221. Ogata T, Onigata K, Hotsubo T, Matsuo N, Rappold G. Growth hormone and gonadotropin-releasing hormone analog therapy in haploinsufficiency of SHOX. Endocr J. 2001;48:317–322. [DOI] [PubMed] [Google Scholar]
- 222. Scalco RC, Melo SS, Pugliese-Pires PN, et al. Effectiveness of the combined recombinant human growth hormone and gonadotropin-releasing hormone analog therapy in pubertal patients with short stature due to SHOX deficiency. J Clin Endocrinol Metab. 2010;95:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. van Gool SA, Kamp GA, Visser-van Balen H, et al. Final height outcome after three years of growth hormone and gonadotropin-releasing hormone agonist treatment in short adolescents with relatively early puberty. J Clin Endocrinol Metab. 2007;92:1402–1408. [DOI] [PubMed] [Google Scholar]
- 224. Lem AJ, van der Kaay DC, de Ridder MA, et al. Adult height in short children born SGA treated with growth hormone and gonadotropin releasing hormone analog: results of a randomized, dose-response GH trial. J Clin Endocrinol Metab. 2012;97:4096–4105. [DOI] [PubMed] [Google Scholar]
- 225. Wit JM, Hero M, Nunez SB. Aromatase inhibitors in pediatrics. Nat Rev Endocrinol. 2012;8:135–147. [DOI] [PubMed] [Google Scholar]
- 226. Ranke MB. Treatment of children and adolescents with idiopathic short stature. Nat Rev Endocrinol. 2013;9:325–334. [DOI] [PubMed] [Google Scholar]
- 227. Dunkel L. Treatment of idiopathic short stature: effects of gonadotropin-releasing hormone analogs, aromatase inhibitors and anabolic steroids. Horm Res Paediatr. 2011;76(suppl 3):27–29. [DOI] [PubMed] [Google Scholar]
- 228. Geffner ME. Aromatase inhibitors to augment height: continued caution and study required. J Clin Res Pediatr Endocrinol. 2009;1:256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Rappold GA, Durand C, Decker E, Marchini A, Schneider KU. New roles of SHOX as regulator of target genes. Pediatr Endocrinol Rev. 2012;9(suppl 2):733–738. [PubMed] [Google Scholar]
- 230. Fukami M, Nishi Y, Hasegawa Y, et al. Statural growth in 31 Japanese patients with SHOX haploinsufficiency: support for a disadvantageous effect of gonadal estrogens. Endocr J. 2004;51:197–200. [DOI] [PubMed] [Google Scholar]
- 231. Kosowicz J. The carpal sign in gonadal dysgenesis. J Clin Endocrinol Metab. 1962;22:949–952. [DOI] [PubMed] [Google Scholar]