

Abstract
The aim of this study was to explore the signaling and neuroprotective effect of transactivator of transcription (TAT) protein transduction of the apoptosis repressor with CARD (ARC) in in vitro and in vivo models of cerebral ischemia in mice. In mice, transient focal cerebral ischemia reduced endogenous ARC protein in neurons in the ischemic striatum at early reperfusion time points, and in primary neuronal cultures, RNA interference resulted in greater neuronal susceptibility to oxygen glucose deprivation (OGD). TAT.ARC protein delivery led to a dose-dependent better survival after OGD. Infarct sizes 72 h after 60 min middle cerebral artery occlusion (MCAo) were on average 30 ± 8% (mean ± SD; p = 0.005; T2-weighted MRI) smaller in TAT.ARC-treated mice (1 μg intraventricularly during MCAo) compared with controls. TAT.ARC-treated mice showed better performance in the pole test compared with TAT.β-Gal-treated controls. Importantly, post-stroke treatment (3 h after MCAo) was still effective in affording reduced lesion volume by 20 ± 7% (mean ± SD; p < 0.05) and better functional outcome compared with controls. Delayed treatment in mice subjected to 30 min MCAo led to sustained neuroprotection and functional behavior benefits for at least 28 d. Functionally, TAT.ARC treatment inhibited DAXX–ASK1–JNK signaling in the ischemic brain. ARC interacts with DAXX in a CARD-dependent manner to block DAXX trafficking and ASK1–JNK activation. Our work identifies for the first time ARC–DAXX binding to block ASK1–JNK activation as an ARC-specific endogenous mechanism that interferes with neuronal cell death and ischemic brain injury. Delayed delivery of TAT.ARC may present a promising target for stroke therapy.
SIGNIFICANCE STATEMENT Up to now, the only successful pharmacological target of human ischemic stroke is thrombolysis. Neuroprotective pharmacological strategies are needed to accompany therapies aiming to achieve reperfusion. We describe that apoptosis repressor with CARD (ARC) interacts and inhibits DAXX and proximal signals of cell death. In a murine stroke model mimicking human malignant infarction in the territory of the middle cerebral artery, TAT.ARC salvages brain tissue when given during occlusion or 3 h delayed with sustained functional benefits (28 d). This is a promising novel therapeutic approach because it appears to be effective in a model producing severe injury by interfering with an array of proximal signals and effectors of the ischemic cascade, upstream of JNK, caspases, and BIM and BAX activation.
Keywords: behavioral outcome, brain ischemia, endogenous neuroprotection, middle cerebral artery occlusion, penumbra, TAT protein transduction
Introduction
Stroke is a leading cause of death and disability worldwide (Saver et al., 2009). For ischemic stroke, the only therapeutic options that have proved to result in a better outcome are intravenous thrombolysis, thrombectomy, hemicraniectomy, and stroke unit care (Fisher and Saver, 2015; Moretti et al., 2015). Cerebral ischemia initiates a cascade of pathological pathways that cause cell death in the ischemic core within minutes after the onset of ischemia. Therefore, the aim of neuroprotective strategies is to interrupt this cascade and stabilize metabolically compromised but still viable brain cells in the ischemic penumbra. This concept involves inhibition of pathological molecular events that eventually lead to the influx of calcium, oxidative stress, and neuronal death (Broughton et al., 2009). Despite encouraging results from animal experiments, clinical trials with neuroprotective therapies have been unsuccessful until now. New therapeutic strategies are needed.
Therapeutic approaches targeting neuronal cell death in stroke must grapple with a complex challenge posed by multiple signaling pathways, heterogenous mechanisms of cell death in different subregions of the ischemic territory, and a limited time window. Although damaged neurons often die from necrosis in the core of the infarct, cerebral ischemia triggers two central pathways of apoptosis: the extrinsic or death receptor pathway and the intrinsic or mitochondrial death pathway. Both result in the activation of caspases that lead to the proteolytic destruction of the cell (Namura et al., 1998; Broughton et al., 2009). Other upstream modulators of apoptosis are mitogen-activated protein kinases (MAPKs), JNK, ASK1, and DAXX (Wang et al., 1996; Kiriakidou et al., 1997; Chang et al., 1998; Herdegen et al., 1998; Ko et al., 2001; Irving and Bamford, 2002; Borsello et al., 2003; Kuan et al., 2003; Putcha et al., 2003; Kuida and Boucher, 2004; Nishina et al., 2004; Okuno et al., 2004; Gao et al., 2005; Salomoni and Khelifi, 2006; Murata et al., 2012).
Apoptosis repressor with CARD (ARC) is a highly potent and multifunctional inhibitor of apoptosis that is predominantly expressed in postmitotic cells, such as cardiomyocytes, skeletal muscle cells, and neurons (Koseki et al., 1998). ARC was originally described as an inhibitor of the death receptor pathway because it blocks apoptosis induced by a variety of death receptors (CD95/FAS, TNFR1, and TRAMP/DR3) and their adaptors (FADD and TRADD; Koseki et al., 1998). We recently showed that ARC is able to block apoptosis induced by activators of the mitochondrial death pathway such as ischemia/reperfusion injury in the heart and doxorubicin-induced cardiotoxicity (Donath et al., 2006; An et al., 2009). ARC inhibits both death receptor and mitochondrial apoptotic death pathways through nonhomotypic death-fold interactions. The death receptor pathway is disrupted by interactions between ARC and FAS, FADD, and procaspase-8 (Nam et al., 2004). The mitochondrial death pathway is inhibited by ARC binding BAX (An et al., 2013). Furthermore, treatment with exogenous ARC protein abrogates TNF-mediated liver damage through the direct interaction of ARC with JNK1/2 and thus inhibits JNK-mediated TNFα expression (An et al., 2012). Additionally, ARC protein transduction interferes with the RIP pathway by inhibiting JNK1/2 activation and counteracts acetaminophen overdose-induced hepatocellular necrosis (An et al., 2013).
Given ARCs multiple interactions in both apoptotic death pathways, its interference with necrosis/programmed necrotic cell death, and its comparably high protein expression in neurons, we hypothesized that it could be acting like a toggle switch between death and survival of neurons and thereby constitute a novel therapeutic target. This led us to evaluate the application of exogenous ARC protein in mice as a novel stroke therapy.
Materials and Methods
All experiments were conducted in an ISO9001-certified quality management environment. Reporting of the study complies with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (Kilkenny et al., 2010).
Primary neuronal cultures.
Primary neuronal cultures were derived from C57BL/6_N mice at embryonic day E16 as described previously (Harms et al., 2004) and cultured with Neurobasal medium and B27 supplement (Invitrogen; Harms et al., 2007).
Oxygen glucose deprivation.
Ischemic-like stress was induced in neuronal cultures at day in vitro (DIV) 10 by combined oxygen glucose deprivation (OGD) for 150 min after treatment with transactivator of transcription (TAT) proteins or if transduced with lentiviral particles for loss of function.
Lactate dehydrogenase assay to assess cellular injury.
Neuronal injury after OGD was assessed by measuring lactate dehydrogenase (LDH) in culture medium as described previously (Harms et al., 2004). Briefly, LDH was measured in culture medium in a kinetic photometric assay (at 340 nm) at 24 h after the injury paradigm. Fifty microliters of culture media were pipetted into 96-well plates and mixed with 200 μl of β-nicotinamide adenine dinucleotide solution (0.15 mg/ml in 1× LDH buffer). Measurement was started rapidly after addition of the reaction substrate pyruvate (50 μl of 22.7 mm pyruvate solution). Optical density was measured at 340 nm using a microplate reader, by 10 counts with 30 s intervals, followed by calculation of results using an LDH standard (Greiner; DiaSys). The maximum release of LDH was achieved by 20 min cell lysis with Triton X-100 as described recently, and data were normalized to these measurements (Datwyler et al., 2011; Yildirim et al., 2014).
Propidium iodide incorporation.
Cell viability was assessed after staining naive cell cultures with propidium iodide (PI) to distinguish between living and dead cells (0.001 mg/ml for 5 min with subsequent rinsing), and five images per well were taken using an inverted IX81 microscope (Olympus) as described previously (Datwyler et al., 2011) with duplicates per condition. Viable neurons not incorporating PI (PI−) were counted in transmission images and quantified as ratios versus all neurons as described previously (Harms et al., 2007).
Lentiviral particle generation and transduction.
Third-generation lentiviral particles were used to express microRNA embedded small-hairpin RNAs (shRNAs) targeting ARC. They were produced and titered using enhanced green fluorescent protein (EGFP) expression and woodchuck posttranscriptional regulatory element as described previously (Datwyler et al., 2011; Reich et al., 2011). Loss-of-function experiments were performed using lentiviral transfer vectors driven by the synapsin promoter. Briefly, microRNA 155 embedded control and ARC target-specific shRNAs (miR-shRNA) were designed using the software algorithm of Block-iT RNAi designer (Invitrogen) and blasted against the mouse genome. Three different target regions in the 5′UTR (TTGGCCCTTTCTGCACTGTCA), the open reading frame (ORF; AGCTATGACCCTTCATGCCCA), and the 3′UTR (AGTCTTGGCGCTCACAGTCTT) of Nol3/ARC (National Center for Biotechnology Information nucleotide accession number NM_030152.4) were used to design oligonucleotides for oligo annealing and ligation into pcDNA6.2-GW/EmGFP-miR (Invitrogen) with the following sequences: NM_030152.4 (Nol3, ARC)_5′UTR_8, TGCTGTGACAGTGCAGAAAGGGCCAAGTTTTGGCCACTGACTGACTTGGCCCTCTGCACTGTCA; NM_030152.4_ORF_479, TGCTGTGGGCATGAAGGGTCATAGCTGTTTTGGCCACTGACTGACAGCTATGACTTCATGCCCA; and NM_030152.4_3′UTR_1271, TGCTGAAGACTGTGAGCGCCAAGACTGTTTTGGCCACTGACTGACAGTCTTGGCTCACAGTCTT.
The nontargeting negative control miR-shRNA (scrambled) was TGCTGAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT.
These miR-shRNAs with an EGFP reporter were subcloned into a modified derivate of Addgene plasmid 27232 driven by a neuron-specific RNA polymerase II-dependent synapsin promoter as described previously (Datwyler et al., 2011). Neuronal cultures were transduced on DIV 3. After 96 h, transduction efficiencies (>95% of neurons) and multiplicity of infection (∼5) were determined and calculated from serial dilutions in neuronal cultures using EGFP fluorescence as a reporter. Three different ARC miR-shRNAs were tested for knockdown efficacy; the best candidate was used for additional analysis (miR-shRNA_ARC_8_5′UTR) and compared with control miR-shRNA.
Immunoblotting and immunoprecipitation.
Brains were cut in three sagittal slices from median to lateral and collected for each hemisphere. Protein extracts were prepared and subjected to either SDS page and immunoblot analysis or immunoprecipitations as previously described (Hauck et al., 2007, 2008; An et al., 2012, 2013). For immunoprecipitation, protein extracts were mixed with antibodies (1–5 μg/ml) for 2 h on a rotating wheel. This was followed by the addition of 50 μl of protein A or G Plus-Sepharose beads (Roche) or 30 μl of agarose conjugated JNK1 (sc-1648 AC)/JNK2 (sc-827 AC) or phospho-SAPK/JNK (Thr183/Tyr185) beads for 1 additional hour at 4°C. Immunoprecipitates were washed four times with RIPA buffer [for activated Bax, we used CHAPS buffer as described (Gustafsson et al., 2004)] and boiled in 50 μl of SDS sample buffer. Samples were resolved over 12 or 15% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. The membrane was probed with primary antibodies and then incubated with the secondary antibody (1:2500 dilution). Immunocomplexes were detected using the enhanced chemiluminescence system (GE Healthcare). Anti-ARC (1:2000 dilution) was purchased from ProSci. Anti-Actin (1:2000 dilution) was obtained from Calbiochem. Anti-caspase-8 antibody (1:1000 dilution) was obtained from Alexis Biochemicals. phospho-ASK1 (S967, 1:500 dilution), phospho-ASK1 (T845, 1:500 dilution), anti-caspase-3 antibody (1:1000 dilution), anti-caspase-9 antibody (1:1000 dilution), anti-phospho-c-Jun (P-c-Jun; 1:1000 dilution), anti-c-Jun (c-Jun, 1:1000 dilution), anti-Daxx (1:500), anti-JNK2 (1:1000 dilution), anti-JNK3 (1:1000 dilution), anti-phospho-JNK (Thr183/Tyr185; P-JNK, 1:1000 dilution), phospho-SAPK/JNK (Thr183/Tyr185; 81E11; Sepharose Bead Conjugate), anti-phospho-p44/42 MAPK (P-p44/42; 1:2000 dilution), and anti-p44/42 MAPK (p44/42, 1:1000 dilution) were purchased from Cell Signaling Technology. Anti-Bax (1:1000 dilution), anti-HA tag (1:1000 dilution), anti-JNK2, agarose conjugated JNK1 (sc-1648 AC), JNK2 (sc-827 AC), and anti-JNK (JNK, 1:1000 dilution) were purchased from Santa Cruz Biotechnology. Anti-Bax6A7 (1:1000 dilution) and anti-JNK1 (1:1000 dilution) were obtained from BD Pharmingen. Anti-His tag (1:1000 dilution) and anti-phospho-JNK (Thr183/Tyr185; P-JNK, 1:1000 dilution) were purchased from Invitrogen. Anti-Bim (1:1000 dilution) was obtained from Stressgen. Anti-ASK1 (1:1000 dilution) and anti-HA-Tag (1:4000 dilution) were purchased from Abcam. Anti-JNK, HRP conjugates was obtained from Abcam. Protein A Sepharose beads for rabbit antibody and Protein G beads for mouse IgG1 were obtained from Roche. HRP-conjugated secondary antibody specific for mouse IgG and specific for rabbit IgG was from GE Healthcare (1:2500 dilution). All other chemicals were purchased from standard commercial sources.
Densitometry of immunoblots.
Western blot quantification were performed after smart background subtraction (rolling ball algorithm) using NIH ImageJ, and each band was normalized to the total gray intensities of a given blot and expressed as the ratio to the housekeeping. Each red spot represents immunoblots of individual animals/time points. Adjustment of densitometric measures to the lesion was done on magnetic resonance imaging (MRI)-based T2 lesion volume per slice, and a dilution factor with non-T2-enhanced “healthy” brain tissue was used to adjust for immunoblot signals derived from a specific stroke subregion.
TAT fusion proteins.
For the generation of recombinant proteins, pTAT.HA and pTAT.β-Gal vectors were obtained from S. Dowdy (Howard Hughes Medical Institute, La Jolla, CA). We produced TAT recombinant proteins as published previously (Hauck et al., 2007, 2008; An et al., 2012, 2013). A scheme of all TAT protein constructs is shown in Figure 6D. Protein purity was evaluated by SDS-PAGE and Coomassie blue staining. Concentrations of the purified proteins were determined by the Bradford method and adjusted to 1 μg/μl with bovine albumin as a standard (Pierce, Thermo Fisher Scientific).
Figure 6.
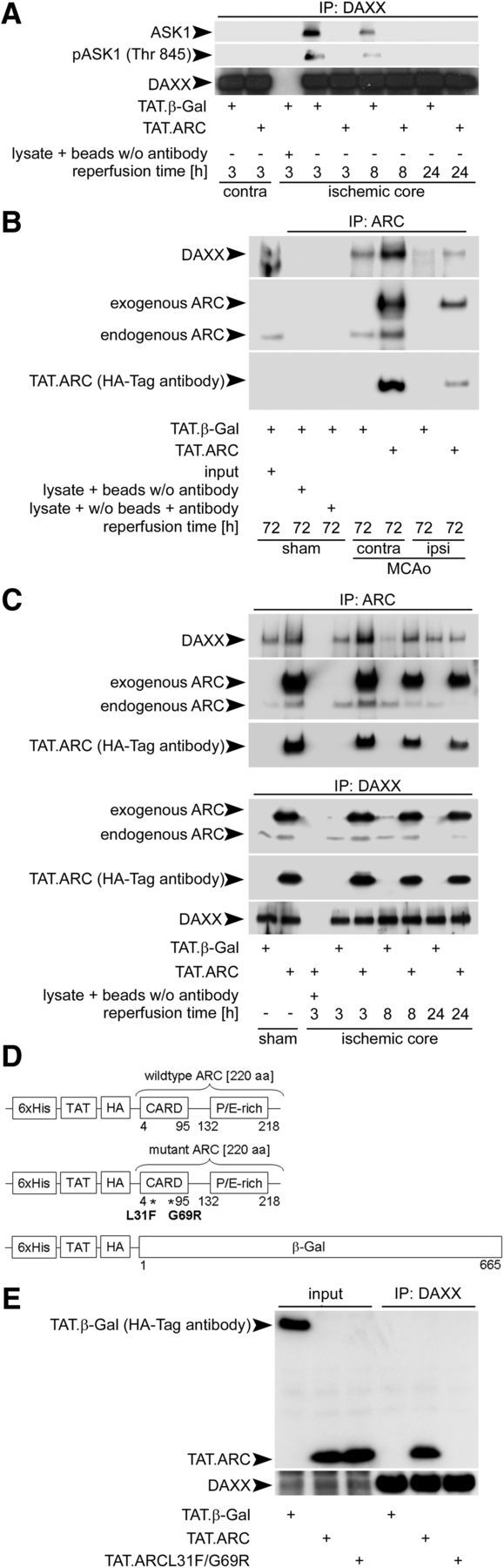
ARC interacts with DAXX and prevents DAXX–ASK1 binding. A, Exogenous ARC prevents DAXX–ASK1 binding. Brain extracts from the middle slice of the reperfusion or corresponding contralateral slice were immunoprecipitated using anti-DAXX antibody. Immunoprecipitates were immunoblotted for DAXX and ASK1, respectively. Brain lysates of the middle slices plus beads without anti-DAXX antibody served as internal loading controls. B, ARC binds DAXX in the cerebral cortex under physiological and ischemic conditions 72 h after reperfusion. Brain homogenates of the middle slices were immunoprecipitated with anti-ARC antibody and immunoblotted with antibodies against DAXX, ARC, and HA. Brain lysate plus beads without anti-ARC or lysate plus anti-ARC antibody without beads served as controls. C, Endogenous and exogenous ARC interacts with endogenous DAXX in vivo. Immunoprecipitation of brain lysates of the middle slice after ischemia or sham operation were performed with anti-ARC or anti-DAXX as primary antibodies. Brain lysates plus beads without anti-ARC or anti-DAXX antibody served as controls. D, Domain structures of TAT proteins with their tags and mutations. His, Histidin tag; HA, hemagglutinin tag; P/E-rich, proline/glutamate rich domain; β-Gal, β-galactosidase; L, leucine; F, phenylalanine; G, glycine; R, arginine. E, ARC interacts via its CARD domain with DAXX. Brains were transduced with TAT.β-Gal, TAT.ARC, or TAT.ARC.mutant (L31F; G69R) and immunoprecipitated with anti-DAXX antibody. Immunoblots were probed as indicated and derived from two mice and replicated in triplicate.
Middle cerebral artery occlusion and TAT protein delivery.
Filamentous middle cerebral artery occlusion was performed for 60 min with indicated reperfusion time points according to the protocol published as a Standard Operating Procedure (SOP) protocol by Dirnagl et al. (doi:10.1038/npre.2010.3492.2). In addition, 30 min middle cerebral artery occlusion (MCAo) was performed with a 28 d observation period to corroborate sustainability of the neuroprotective and neurorestorative effects (Fig. 8). All animal experiments were approved by the Berlin governmental authorities (Landesamt für Gesundtheit und Soziales, G0385/08 and G197/12). Male C57BL/6NCrl mice were derived from Charles River at the age of 8 weeks and were used in the experiments at the age of 10–12 weeks. All animals had access to food and water ad libitum and were kept under a 12 h light/dark cycle. No specific exclusion criteria were set. One mouse was excluded because it showed no lesion in the MRI or a functional deficit in the first setting of in vivo experiments (see Fig. 2). Table 1 lists the survival and time of death events in detail.
Figure 8.
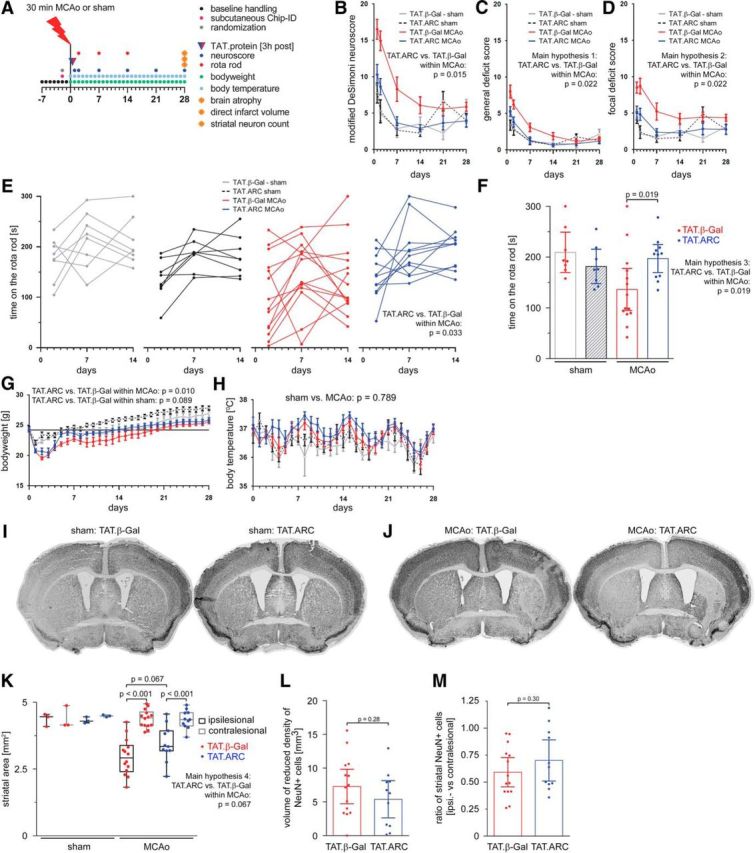
Delayed administration of TAT.ARC protein attenuates focal ischemic brain injury and fosters recovery in the long term. A, Experimental setup of MCAo, 3 h delayed intrathecal application of 5 μg of TAT proteins, and reperfusion time points for distinct analyses. Composite modified DeSimoni neuroscore (B) with general (C) and focal deficit (D) scores and rotarod data (E, F). Rotarod data after 14 d is shown as the mean time on the rod with CIs. G, Body weight was measured before surgery (day 0) and after non-invasive transponder-mediated body temperature measurements (H). Data in B–D and G, H are presented as mean ± SEM and in F, L, and M as mean ± CI. I, Multiple image alignment of neuronal staining using anti-NeuN–DAB of sham mice and 28 d after MCAo (J). Striatal area in coronal section a3 (K) in both ipsilateral and contralateral hemispheres and residual infarct area/scar as defined by decreased neuronal density (L) and ratios of ipsilesional versus contralesional neurons counted in TAT.β-Gal or TAT.ARC mice (M) were illustrated by scatter dot plots and box and whiskers (minimum to maximum) for K and mean + CI (L, M).
Figure 2.
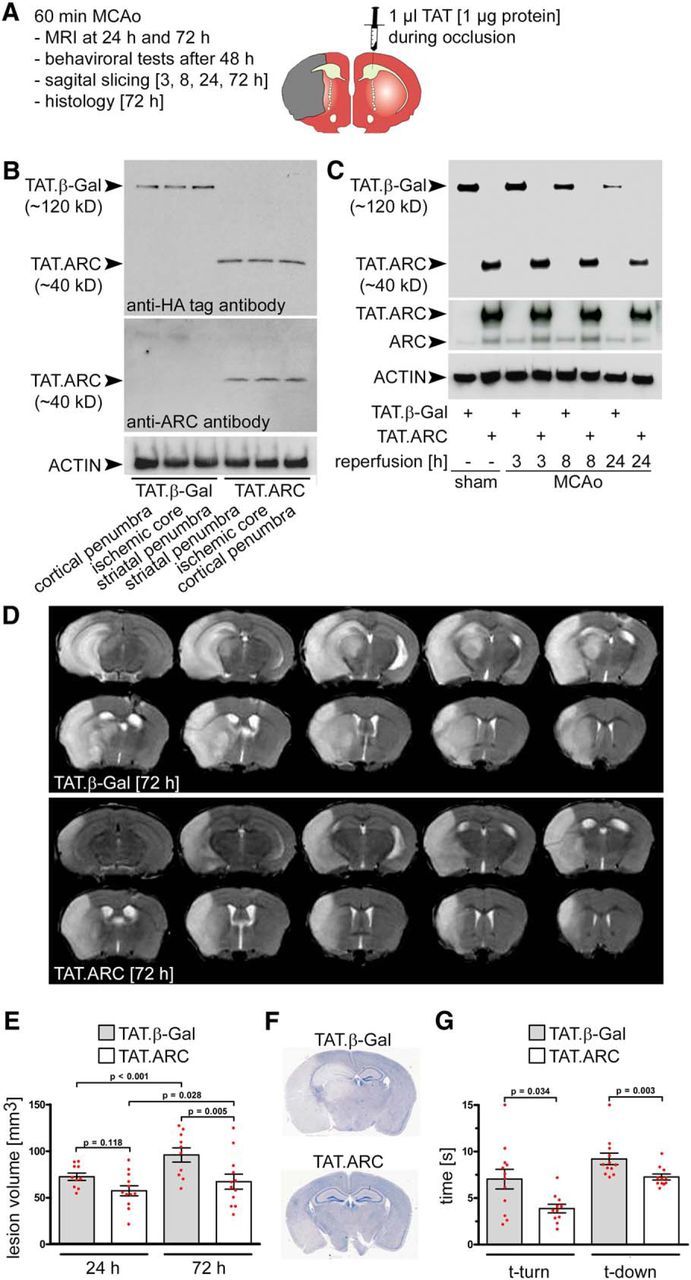
Administration of TAT.ARC protein attenuates focal ischemic brain injury. A, Experimental setup of MCAo, intrathecal application of TAT proteins, and reperfusion time points for distinct analyses. B, Mice were subjected to 60 min MCAo, and TAT proteins were administered intrathecally in the contralateral hemisphere during occlusion. Exogenous protein transduction with TAT.β-Gal or TAT.ARC was detected by immunoblots with the indicated antibodies and shows homogenous distribution of TAT proteins throughout the brain after 72 h (brain lysates were derived from 2-mm-thick sagittal slices derived from subregions of the ischemic and surrounding tissue of the ipsilateral hemisphere). C, Stability of TAT.β-Gal or TAT.ARC proteins at indicated time points after reperfusion derived from sham mice (3 h after delivery) or from the striatal ischemic core after 60 min of MCAo. D, Representative MRI scan 72 h after 60 min MCAo showing significantly attenuated brain injury in the TAT.ARC compared with the TAT.β-Gal-treated mouse. E, Quantification of lesion volume 24 and 72 h after 60 min MCAo. Data are presented as mean and scattered dot plots ± SEM. T2 stroke volumes were analyzed by two-way RM ANOVA with F(1,43) = 5.8 and p = 0.03 for TAT treatment, F(1,43) = 27.5 and p < 0.001 for reperfusion time point, and F(1,43) = 4.5 with p = 0.046 for TAT × reperfusion time point interaction. F, Hematoxylin staining after 72 h reperfusion time treated with TAT proteins as indicated. G, Pole test, Data are presented as mean and scattered dot plots ± SEM. Mann–Whitney U rank-sum test for time-to-turn: T = 97.000 n(small) = 11 n(big) = 12 (p = 0.034); and time-to-descend: T = 83.000 n(small) = 11 n(big) = 12 (p = 0.003).
Table 1.
Survival data of 145 animals
| Animals (n) | MCAo length (min) | TAT.application: Time/dose | TAT.β-Gal–sham: n/death event (day) | TAT.β-Gal–MCAo: n/death event (day) | TAT.ARC–sham: n/death event (day) | TAT.ARC–MCAo: n/death event (day) | Number of animals at survival endpoint | Data shown |
|---|---|---|---|---|---|---|---|---|
| 24 | 60 | During MCAo [1 μg] | — | 12/1 (1); | — | 12/0 (n/a) | 23/3 d | Figs. 2, 3, 5, 6 |
| 30 | 60 | 3 h post MCAo [5 μg] | — | 15/0 (n/a) | — | 15/1a (1) | 29/3 d | Fig. 7 |
| 40 | 60 | During MCAo [1 μg] | 5/0 (n/a) | 15/0 (n/a) | 5/0 (n/a) | 15/0 (n/a) | 40/3–24 h | Figs. 4, 6 |
| 5 | n/a | TAT.ARCL31F/G69R | — | — | 5/0 (n/a) | — | 5/24 h | Fig. 6 |
| 46 | 30 | 3 h post MCAo [5 μg] | 8/0 (n/a) | 15/1 (7) | 8/0 (n/a) | 15/4 (1, 7, 8, 10) | 41/28 db | Fig. 8 |
aSurvival was tested with the log-rank Mantel–Cox test with χ2 = 6.081 (df = 3) and p = 0.11 indicative that survival did not differ between groups in the last experiment.
bAnimal died after MCAo before TAT.application.
TAT protein delivery in the contralateral ventricle.
TAT proteins were delivered by stereotaxic inoculation in the lateral ventricle of the contralateral hemisphere with injection over 5 min starting 15 min after MCAo and a total volume of 1 μl with 1 μg of highly purified recombinant TAT-fusion proteins with the following coordinates: 0.7 mm caudal to bregma, 1.3 mm lateral to sagittal suture (right side), and 1.3 mm in depth. The needle was retracted carefully within 5 min. In case of delayed treatment, a total volume of 5 μl was injected with 5 μg of protein of the respective TAT protein.
MRI.
MRI was performed using a 7 Tesla rodent scanner (Bruker Pharmascan 70/16AS) with a 16 cm horizontal bore magnet and a 9 cm (inner diameter) shielded gradient with an H-resonance frequency of 300 MHz and a maximum gradient strength of 300 mT/m. For imaging, a 1-hydrogen proton radio frequency quadratur-volume resonator with an inner diameter of 20 mm was used. Data acquisition and image processing were performed with the Bruker software Paravision 4.0. During the examinations, mice were placed on a heated circulating water blanket to ensure constant body temperature of 37°C. Anesthesia was induced with 2.5% and maintained with 1.0–2.0% isoflurane delivered in a O2/N2O mixture (30/70%) via a facemask under constant ventilation monitoring. A rapid acquisition with relaxation enhancement (RARE) T2-weighted sequence was used. Imaging parameters were as follows: for T2, repetition time, 4200; echo time, 36 ms; RARE factor 8, 4 averages. Twenty axial slices with a slice thickness of 0.5 mm, a field of view of 2.75 × 2.75 cm, and a matrix of 256 × 256 were positioned over the brain excluding the olfactory bulb.
Stroke volume and regional stroke volume in 2 mm sagittal slices used for the biochemical assays were analyzed using Analyze 5.0 and NIH ImageJ software. For stroke volumetry, hyperintense areas of ischemic tissue in T2-weighted images were assigned with a region of interest tool. This enabled a threshold-based segmentation that was performed by connecting all pixels within a specified threshold range around the selected seed pixel and resulted in a 3D object map of the whole stroke region. The total volume of the whole object map was calculated automatically.
To calculate the regional ischemic lesion volumes that would correspond to slices prepared for immunoblots, axial MR images were divided with NIH ImageJ into three equal slices from medial to lateral. The volumes of hyperintense areas in each brain section were calculated by Analyze 5.0 as described above. Brain parenchyma were cropped for presentation purposes.
Pole test.
The pole test was performed to analyze sensory motor function. The test was performed in a blinded manner with treatment allocation concealed throughout. Mice were habituated to the procedure the day before testing. They were placed head upward near the top of a vertical rough-surfaced pole (1 cm diameter, 50 cm height) and then allowed to descend five times during one experimental session. The total time needed to turn completely head downward (“time-to-turn”) and the time it took the mouse to reach the floor with all four paws (“time-to-come-down”) were recorded. Criteria for inclusion of mice for evaluation in the pole test were a non-interrupted “smooth” run to measure the time-to-descend and an immediate initiation to turn with no additional interruption to come down the pole. Because of this criteria, we had to exclude five animals in the TAT.β-Gal-treated group and three animals in the TAT.ARC-treated group in the delayed application series (see Fig. 7).
Figure 7.
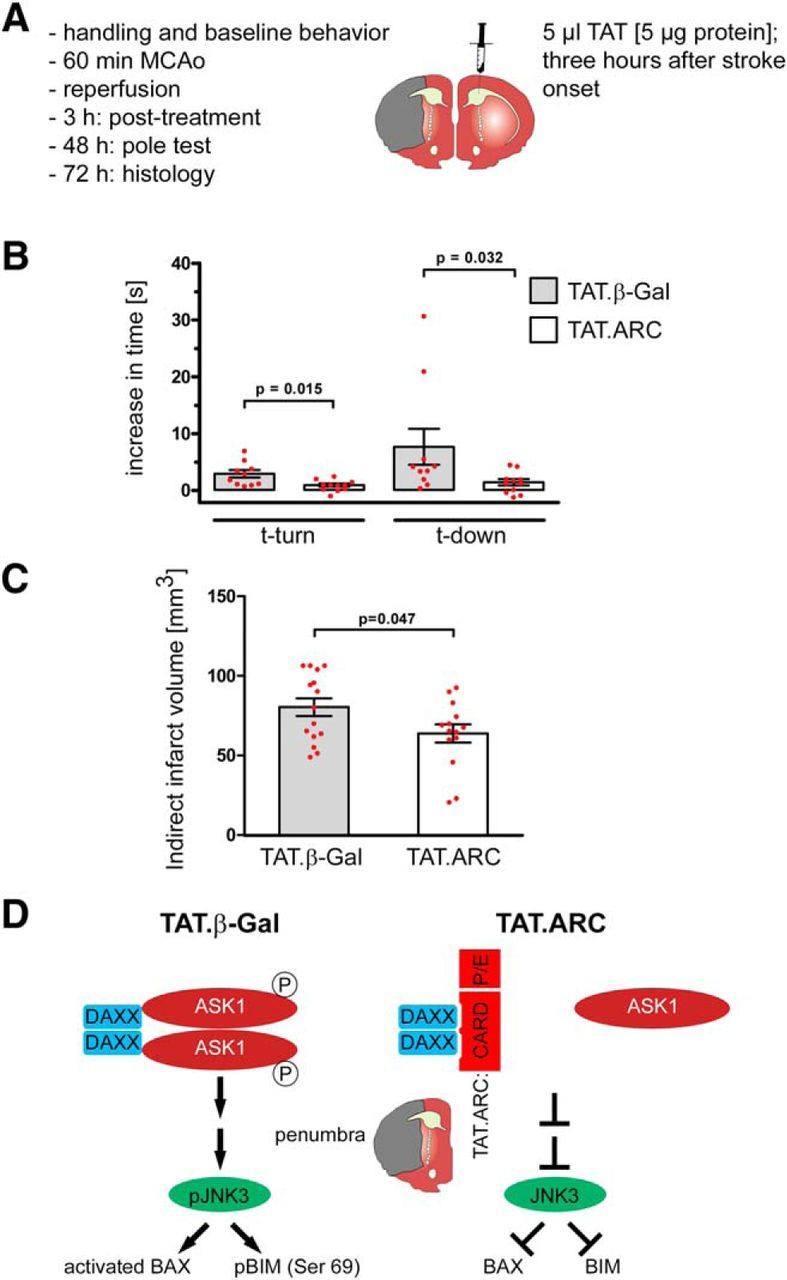
Delayed administration of TAT.ARC protein attenuates focal ischemic brain injury. A, Experimental setup of MCAo, intrathecal application of TAT proteins, and reperfusion time points for distinct analyses. B, Pole test, Data are presented as the increase of time to turn and time to descend after MCAo compared with individual performance in mice before ischemia. Data are presented as mean and scattered dot plots ± SEM; n = 10 and n = 11 animals (TAT.β-Gal vs TAT.ARC). For specific exclusion criteria and mortality, see Materials and Methods and Results. C, Indirect infarct size was measured using hematoxylin staining after 72 h as described in Materials and Methods. Data are presented as mean and scattered dot plots ± SEM with n = 15 animals in TAT.β-Gal and n = 14 animals in the TAT.ARC group. D, Proposed model of molecular mechanisms of TAT.ARC treatment targeting the penumbra after brain ischemia.
Modified DeSimoni neuroscores, rotarod test, and non-invasive body temperature measures.
DeSimoni neuroscore was performed at the indicated time points (see Fig. 8) as described previously (De Simoni et al., 2003; Orsini et al., 2012) with some modifications. In brief, general health (Table 2) and specific focal assessments (Table 3) were separately scored, analyzed, and finally added to form a summation score. Summative scores added up to a maximum of 43 points, with more points meaning more deficits.
Table 2.
Modified DeSimoni neuroscore for the assessment of general deficits
| No. | General versus focal deficits | Objective | Assessment/instruction | Score = 0 | Score = 1 | Score = 2 | Score = 3 | Score = 4 | Summative score |
|---|---|---|---|---|---|---|---|---|---|
| 1 | General deficits | Hair | Mouse observed on open bench top (OS). Observation with no interference | Hair neat and clean | Lack of grooming, piloerection and dirt on the fur around nose and eyes | Lack of grooming, piloerection, and dirty coat, extending beyond just nose and eyes | 0–2 | ||
| 2 | General deficits | Ears | Mouse on OS. Observation at the beginning with no interference and then stimulation by clicking one's tongue | Normal. Ears are stretched laterally and behind. They react by straightening up after noise | Stretched laterally but not behind (one or both). They react to noise | Same as 1. They do not react to noise | 0–2 | ||
| 3 | General deficits | Eyes | Mouse on OS. Observation with no interference or stimulation | Open and clear (no discharge) | Open and characterized by milky white mucus | Open and characterized by milky dark mucus | Eyes clotted (one or both sides) | Closed | 0–4 |
| 4 | General deficits | Posture | Place the mouse on the palm of your hand and rock gently to observe stability | The mouse stands in the upright position on four limbs with the back parallel to the palm. During the rocking movement, it uses its limbs to stabilize itself | The mouse stands humpbacked. During the rocking movement, it lowers its body instead of using its limbs to gain stability | The head or part of the trunk lies on the palm | The mouse reclines to one side but may be able to turn to an upright position with some difficulty | No upright position possible | 0–4 |
| 5 | General deficits | Spontaneous Activity | Mouse on OS. Observation with no interference or stimulation | The mouse is alert and explores actively | The mouse seems alert, but it is calm and quiet | The mouse starts and stops exploring repeatedly and slowly. The mouse is listless, moves sluggishly but does not explore | The mouse is lethargic or stuporous and barely moves during the 60 s | No spontaneous movements | 0–4 |
| 0–16 |
Table 3.
Modified DeSimoni neuroscore for the assessment of focal deficits
| No. | General versus focal deficits | Objective | Assessment/instruction | Score = 0 | Score = 1 | Score = 2 | Score = 3 | Score = 4 | Summative score |
|---|---|---|---|---|---|---|---|---|---|
| 6 | Focal deficits | Body symmetry | Mouse on OS, observation of undisturbed resting behaviour and description of the virtual nose–tail line | Normal. a, Body: normal posture, trunk elevated from the bench, with forelimbs and hindlimbs leaning beneath the body. b, Tail: straight | Slight asymmetry. a, Body: leans on one side with forelimbs and hindlimbs leaning beneath the body. b, Tail: slightly bent | Moderate asymmetry. a, Body: leans on one side with forelimbs and hindlimbs stretched out. b, Tail: slightly bent | Clear asymmetry. a, Body leans on one. b, Tail: clearly bent | Complete asymmetry. a, Body. b, Tail | 0–4 |
| 7 | Focal deficits | Gait | Mouse on OS. Observation of undisturbed movements | Normal. Gait is flexible, symmetric, and quick | Stiff, inflexible. The mouse walks humpbacked, slower than normal mice | Limping with asymmetric movements | More severe limping, drifting, falling with obvious deficiency in gait | Does not walk spontaneously. (In this case, stimulation will be performed gently pushing the mouse with a pen. When stimulated, the mouse walks no longer than three steps.) | 0–4 |
| 8 | Focal deficits | Climbing | Mouse is placed in the center of a gripping surface at an angle of 45° to OS | Normal. The mouse climbs quickly | Climbs slowly, limb weakness present | Holds onto slope, does not slip or climb | Slides down slope; difficulty to prevent fall | Slides down slope, unsuccessful effort to prevent fall | 0–4 |
| 9 | Focal deficits | Circling behavior | Mouse on OS. Observation of the mouse walking undisturbed on the OS | Circling behavior absent. The mouse turns equally to left or right | Predominantly one-sided turns. Optional: record to which side the mouse turns | Circles to one side, although not constantly | Circles constantly to one side. This one is now highlighted in yellow | No movements | 0–4 |
| 10 | Focal deficits | Forelimb symmetry | Mouse suspended by the tail. Movements and position of forelimbs are observed | Normal. Both forelimbs are extended towards the bench and move actively | Light asymmetry. Contralateral forelimb does not extend entirely | Marked asymmetry. Contralateral forelimb bends towards the trunk. The body slightly bends on the side ipsilateral to the stroke | Prominent asymmetry. Contralateral forelimb adheres to the trunk | Slight asymmetry, no body/limb movement | 0–4 |
| 11 | Focal deficits | Compulsory circling | Forelimbs on bench, hindlimbs suspended by the tail. This position reveals the presence of the contralateral limb palsy. In this handstand position, limb weakness is displayed by a circling behavior when the animal attempts forward motion | Absent. Normal extension of both forelimbs | Both forelimbs extended but begins to circle predominantly to one side | Circles only to one side and may fall to one side | Pivots to one side sluggishly and does not rotate in a full circle. Mouse will fall to one side | No or rare movements | 0–4 |
| 12 | Focal deficits | Gripping of the forepaw | Mouse is held by the tail on the wire bar cage lid, so that the forepaws touch the grid | Mouse grasps the grid firmly with forepaws and tries to place the hind paws also onto the grid by pulling the hindpaws under the body | Mouse accesses the grid but has less power. A slight pull breaks the grip of the forepaws | Mouse cannot grip with the impaired forepaw | Mouse cannot grip the grid | 0–3 | |
| 0–27 |
Rotarod was assessed as described recently (Hoffmann et al., 2015), and the best run of three replicates at a given time point was used for statistical analysis.
Body temperature was non-invasively assessed at the same time of the day before body weight measurements using subcutaneous transponders (IPTT-300; Bio Medic Data Systems) for unambiguous identification of mice in their home cages as described previously (Kort et al., 1998; Langer and Fietz, 2014).
Coronal sections and mouse brain coordinates for histology.
Mouse brains were cut in coronal sections using a mouse brain atlas (Franklin and Paxinos, 2007) to analyze direct and indirect infarct volumes (a1–a5, area), striatal area, and neuronal densities within the striatum were determined in a3. a1 = interaural 6.6 mm/bregma = 2.80; a2 = interaural 5.34 mm/bregma = 1.54; a3 = interaural 3.94 mm/bregma = 0.14; a4 = interaural 1.86 mm/bregma = −1.94; a5 = interaural −0.08 mm/bregma = −3.88.
Histology.
Immunohistochemistry for Figure 1A was performed in paraffin-embedded 4-μm-thick coronal sections after perfusion with physiological saline and 4% paraformaldehyde (PFA) in deep anesthesia. Rabbit anti-ARC antibody (F100) and mouse anti-NeuN (F100) were incubated overnight after blocking with 10% normal donkey serum in 0.1% Triton X-100. Secondary antibodies were derived from donkey and conjugated to Rhodamine Red X (anti-rabbit) or FITC (anti-mouse). Sections were mounted in Immunomount Gold (Invitrogen) and visualized using confocal microscopy. Immunohistochemistry, hematoxylin staining, and histology were performed as described previously (Reich et al., 2011). Neuronal densities were achieved by anti-NeuN DAB staining as described previously (Hoffmann et al., 2015).
Figure 1.
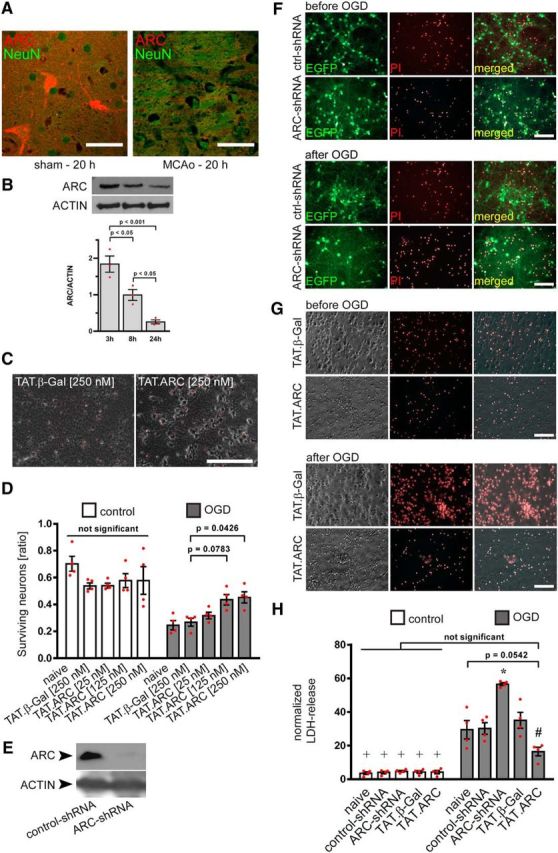
ARC is expressed in neurons and serves as an endogenous and exogenous neuroprotective protein against OGD. A, C57BL/6 mice were subjected to 30 min of MCAo or sham procedure. PFA and paraffin-embedded 4 μm sections were stained for ARC protein (Rhodamine Red X) and neuronal counterstain with NeuN (FITC) after 20 h of reperfusion. Microscopic pictures were taken with a confocal microscope. Scale bar, 50 μm. B, C57BL/6 mice were subjected to 60 min of MCAo, and the striatal ischemic core was isolated at indicated time points. Immunoblots for ARC and ACTIN protein was performed from brain lysates and densitometric analysis [n = 3 animals (red dot plots and mean ± SEM), ordinary ANOVA and Tukey's post hoc test]. C, D, Exogenous ARC protein transduction leads to a dose-dependent increase in neuronal survival after OGD compared with TAT.β-Gal-treated neurons. F[Degrees of freedom for the numerator: DFn, Degrees of freedom for the denominator: DFd](4,30) = 3.858 with p = 0.0121 for TAT treatment × OGD interaction and F[DFn, DFd](4,30) = 64.28 with p < 0.0001 for effects of OGD. Data were derived from four independent experiments and shown as red dot plots, whereas bars were presented as mean ± SEM. Primary neuronal cultures were transduced with lentiviral particles for shRNA delivery (shRNA) either nontargeting (shRNA-control) or interfering with ARC mRNA along with EGFP reporter expression and staining of dead cells with PI (visualized in F). Knockdown of endogenous ARC was demonstrated by immunoblots (E). Primary neuronal cultures were counterstained with PI for visualization of neuronal cell death before and after 150 min OGD (F, G). Normalized LDH release after ARC knockdown or best dose of titration experiments for TAT proteins (D; 250 nm) was measured 24 h after 150 min OGD (H). Data are presented as the mean normalized LDH release of four independent experiments and presented as mean and scatter dot plots ± SEM. F[DFn, DFd](4,30) = 13.84 with p < 0.0001 for miR-shRNA or TAT treatment interaction with OGD. F[DFn, DFd](4,30) = 14.85 with p < 0.0001 for treatment effects and F[DFn, DFd](1,30) = 290.3 with p < 0.0001 for OGD effects. *p < 0.05 versus all other treatment groups within OGD; #p < 0.05 versus all other treatment groups except naive neurons within OGD.
Imaging acquisition and quantification of neuronal densities.
Image collection of NeuN–DAB-stained brain slices from mice 28 d after MCAo were collected as transmission images using a Leica DMI8 microscope equipped with an LED light source, a Fluotar 10×/030 dry objective, and a DFC300G camera and stitched within the Leica LAS X2.0 software. Striatal neuronal areas were measured from coronal section three (a3) directly, and NeuN-positive cell counts were derived from thresholded and segmented images in the public domain program NIH ImageJ. Direct lesion volumes were calculated from five coronal brain sections as described previously (Hoffmann et al., 2015).
Statistical analysis.
Our study was designed as an exploratory analysis of the endogenous neuroprotective mechanisms of ARC as a potential therapeutic target (Kimmelman et al., 2014). The first part of our study (see Figs. 1–7) uses post hoc tests (e.g., Tukey's test) to correct for multiple testing for each figure separately. The second part of our study (see Fig. 8) focuses on sustained effects of TAT.ARC treatment in MCAo mice with 28 d survival, and we used five hypotheses with hierarchical testing. All data are presented as means and scattered dot plots ± SEM. We calculated 95% confidence intervals (CIs) for Figure 8, F, L, and M, and presented all animals with box and whiskers for Figure 8K. Two-way repeated-measures (RM) ANOVA with Bonferroni's post hoc tests for comparison of simple treatment effects within control or OGD was used for Figure 1D. Two-way ANOVA with Tukey's post hoc analysis was performed for miR-shRNA effects on OGD and TAT protein transduction of neuronal cultures (Fig. 1H). Two-way RM ANOVA, followed by Tukey's post hoc analysis, was performed for MRI stroke volume assessment after 24 and 72 h (see Fig. 2C). Two separate mixed linear models were performed for sagittal slice-specific MRI stroke volume assessment for 24 and 72 h Sidak's post hoc tests (see Fig. 3A), including treatment and slice area as covariates. Mann–Whitney U rank-sum test was performed for behavioral analysis and stroke volume analysis (pole test; Figs. 2E, 7B, histology, C). Two-tailed Student's t test was performed for direct stroke volume analysis after 28 d and for testing differences in ratios of counted NeuN-positive cells within the striatum (coronal slice a3; see Fig. 8L,M). The number of experimental units required to detect a standardized effect size >0.25 was calculated by a priori power analysis with the following assumptions: α = 0.05; β = 0.2; mean, SD 20% of the mean for in vivo MCAo experiments.
Figure 3.

Administration of TAT.ARC protein attenuates focal ischemic brain injury in the cortical penumbra but attenuates cell death signaling independent of ischemic subregions. A, In analogy to the biochemical analysis, ischemic hemispheres were divided into 2 mm sagittal slices for regional lesion volume analysis after 60 min MCAo and quantified as described in Materials and Methods. Gray bar graphs indicate TAT.β-Gal-treated mice, and open bar graphs indicate TAT.ARC-treated mice. Data are presented as mean and scattered dot plots ± SEM. T2 stroke volumes were analyzed by two separate mixed linear regression models with Sidak's post hoc analysis. B–G, Immunoblots show blocked formation of cleaved caspase-8, caspase-9, caspase-3, and BAX activation (B, C), reduced JNK activity (D, E), and blunted BIM phosphorylation (F, G) in response to TAT.ARC administration compared with TAT.β-Gal-treated mice. Different ischemic zones are shown as indicated. Data are representative of three independent experiments (B, D, F). Mixed linear regression models were applied for semiquantitative densitometric Western blot analysis of three animals per TAT protein group and presented as red scattered dot plots per animal and mean ± SEM. Quantification was referred to the indicated reference protein and the dilution factor of healthy tissue within a subregion (characterized by T2 intensity in MRI) was used to adjust the Western blot signal to the lesion (for details, see Materials and Methods).
Figure 4.

TAT.ARC-mediated inhibition of DAXX–ASK1–JNK signaling during focal ischemic infarction. A, Immunoblots demonstrate that TAT.ARC attenuates ischemia-induced activation of the DAXX–ASK1–JNK signaling pathway at 24 h after ischemia compared with the corresponding area in TAT.β-Gal-treated mice. The blots are representative of three independent experiments. B, Semiquantitative analysis of immunoblots of three mice are presented as mean and scattered dot plots ± SEM. The right panel was adjusted to lesion volume as measured by T2 sequence in MRI in the core slice after 24 h. DAXX, pASK1, and activated BAX showed significant TAT treatment × time interactions as analyzed by two-way ANOVA, followed by Tukey's post hoc analysis. pJNK1-3 showed significant treatment (p = 0.03) and time (p = 0.0002) effects but no interaction (p = 0.0988). pBim showed no significant treatment × time interaction (p = 0.065). Post hoc tests were significant as indicated by asterisks between TAT β-Gal and TAT.ARC comparisons per time point in the graphs. C, Immunoblots show no effects on MAP44/42 kinase pathway in response to TAT.ARC administration compared with TAT.β-Gal-treated mice. Different reperfusion times from the middle ischemic slice are shown as indicated. Data are representative of three independent experiments.
Figure 5.

TAT.ARC treatment inhibits JNK phosphorylation after focal brain ischemia. A, Whole-brain lysates from damaged or corresponding contralateral zone 72 h after ischemia or sham operation treated with TAT.ARC or TAT.β-Gal were immunoprecipitated with an anti-phospho-JNK antibody and then immunoreacted as indicated. Brain lysate plus Protein A or G: Plus-Sepharose beads without anti-pJNK or lysate plus anti-pJNK antibody without beads served as internal loading controls. B, Higher phospho-JNK signals were not detected in the contralateral hemisphere of mice subjected to focal brain ischemia and treated with TAT.β-Gal or TAT.ARC proteins. Whole-brain lysates from sham mice or contralateral to the ischemic hemisphere were immunoprecipitated with anti-phospho-JNK antibody and detected by Western blots as indicated.
Seven separate linear mixed models were used to analyze total neuroscore values (see Fig. 8B), general neuroscore values (see Fig. 8C), focal deficit values (see Fig. 8D), rotarod values (see Fig. 8E,F), striatal area per hemisphere in coronal slice a3 (see Fig. 8K), body weight (see Fig. 8G), and body temperature (dependent variables; see Fig. 8H) to test differences between TAT.ARC and β-Gal within sham and MCAo mice (random intercept models to account for average differences in outcome between mice and clustered data structure). To analyze the rotarod data, we used the best value of three measures for each time point and each animal. Main hypotheses were related to differences between TAT.ARC- and β-Gal-treated mice with MCAo. Specifically, we tested five hierarchical ordered main hypotheses [hypothesis 1, differences in MCAo group between TAT.ARC- and β-Gal-treated mice in the general neuroscore; hypothesis 2, same as in 1 but focal score; hypothesis 3, same as in 1 but sustainability of neuroprotection by TAT.ARC in MCAo mice compared with β-Gal-treated mice as shown by rotarod values at 14 d; hypothesis 4, morphological correlates presenting less striatal atrophy by TAT treatment (compared to β-Gal-treated mice) within MCAo; and hypothesis 5, smaller direct lesion volume or lower number of NeuN-positive neurons within the striatum] by using a hierarchical test procedure to control the familywise error rate for these subanalyses. For these hierarchical testing, a two-sided significance level of α = 0.05 was considered. All additional hypotheses were tested exploratory. For the linear mixed regression models, we used all available measures over time for all mice (255 measures for 45 mice for the total neuroscore/general neuroscore/focal deficit; 128 measures for 45 mice for rotarod, 62 measures for 31 mice for striatal area in a3, 1173 measures for 46 mice for body temperature/body weight). As independent variables, we included the group (sham or MCAo), the intervention (TAT.ARC or β-GAL), and the interaction between group and intervention in all models. For the models for neuroscore values (total, general, and focal deficit), for the rotarod data, and for body temperature and body weight, we additionally included time (day centered and for the neuroscore models as well as for body temperature and body weight additionally, a squared term for time to account for the curvilinear relation between time and the dependent variable) and the interaction terms group × time and intervention × time. For the two-lesion volume model, we included the information on hemisphere (ipsilateral or contralesional) and the interaction between group × hemisphere and intervention × hemisphere. In case of skewed distribution of outcome, we transformed the values before regression. All reported p values are related to post hoc tests from the mixed models. Differences in survival rates were calculated by the log-rank test (Table 1).
Methods to prevent bias.
Cell counts for survival of neurons that are not incorporating propidium iodide (PI−) were performed after blinding, randomization, and blinding of the allocation to treatment groups. Ten high-power fields derived from duplicate wells per experiment was used to calculate the mean that was further analyzed as an independent data point. A total of four independent cell culture platings were used, and 689 and 780 cells in control conditions and after OGD were analyzed overall.
Survival and the number of 145 animals allocated to the various groups and finally evaluated are described in Table 1. Twelve mice per group (in 24 mice) were used for Figure 2, but one animal died and one animal was excluded before analysis and unblinding to the groups because it had no visible stroke (both from the TAT.β-Gal group). Thirty mice were used for the delayed treatment protocol in Figure 7 (15 per group). One mouse died from the TAT.ARC-treated group. Ten animals were subjected to sham surgery and received TAT.β-Gal or TAT.ARC (each five animals), and 30 animals were transduced with TAT proteins and were subjected to 60 min of MCAo with 3, 8, or 24 h reperfusion time (each 5 animals) for immunoblots in Figures 3B, 4, A and B, 5, and 6, A–C and E. An additional five animals were transduced with TAT.ARC.mutant (see Fig. 6E). No animals died or had no lesion in the time course study. In summary, of the 145 animals, 137 animals were included in the study and reached the endpoint. Survival in the 28 d sustained neuroprotection study (see Fig. 8) is documented in detail in Table 1, and no animals were excluded in this substudy. The experimental outlines are depicted in Figures 2, 7, and 8 and Table 1.
Animals were randomized using the GraphPad calculator tool (http://graphpad.com/quickcalcs/randomize1/) before the experiments by a scientist who was not involved in the surgery, the TAT application, or behavioral or MRI analysis. All details on cage number, group of surgery cohort (day), and mouse identification can be found in the open data resources that are available for this publication on Figshare Repository and that are described under original data deposition. Concealment of treatment allocation was maintained throughout the study. The surgeon was blinded for the grouping to either the control or the TAT.ARC protein. MRI analysis was performed by a computer-aided algorithm by scientists who were not involved in surgery, TAT application, or behavioral analysis, and both the grouping and the treatment allocation were concealed for stroke volume analysis and behavioral tests. The specific inclusion criteria for the pole test were set before the experiment.
Criteria for inclusion of mice for evaluation in the pole test were a non-interrupted smooth run to measure the time-to-descend and an immediate initiation to turn with no additional interruption to come down the pole. Because of these criteria, we had to exclude five animals in the TAT.β-Gal-treated group and three animals in the TAT.ARC-treated group in the delayed application series (see Fig. 7).
Original data deposition.
The following primary datasets, MRI scans, original behavioral datasets, histological analyses, or stitched histology images of NeuN are available as open data on Figshare Repository in raw data format: Figure 1, C and D, cell counts, https://doi.org/10.6084/m9.figshare.3405787 (Donath et al., 2016a); Figure 1 H, normalized LDH data, https://doi.org/10.6084/m9.figshare.3406282 (Donath et al., 2016h); Figures 2 and 3, MRI original data, https://doi.org/10.6084/m9.figshare.3398722 (Donath et al., 2016g); Figure 3A, MRI slice results of T2 lesion after 24 and 72 h, https://doi.org/10.6084/m9.figshare.3 4 2 0 8 8 3 (Donath et al., 2016f); Figure 7C, histology of infarct volumes, https://doi.org/10.6084/m9.figshare.3420868 (Donath et al., 2016e); Figure 8A–H, behavior, body weight, and body temperature, https://doi.org/10.6084/m9.figshare.3420901 (Donath et al., 2016d); Figure 8K--M, striatal volume, chronic lesion volume, and striatal neuronal cell counts, https://doi.org/10.6084/m9.figshare.3398761 (Donath et al., 2016c); and Figure 8, I and J, NeuN-DAB_stainings_in_sham_MCAo_mice_with_28-days_survival, https://doi.org/10.6084/m9.figshare.3413683 (Donath et al., 2016b).
Results
Endogenous and exogenous ARC provides neuroprotection against OGD
We identified an early loss of ARC protein in neurons in the ischemic striatum at 20 h of reperfusion after MCAo in vivo (Fig. 1A). We corroborated loss of ARC protein by Western blot analysis of the ischemic striatal core after 3, 8, and 24 h of 60 min of transient MCAo with a gradual decrease over time (Fig. 1B). Next, we investigated whether increasing ARC protein levels using TAT.ARC protein transduction would provide neuroprotection. We tested neuronal cultures and applied TAT proteins 1 h before OGD (Fig. 1C,D). Exogenous ARC protein provided strong, dose-dependent neuroprotection that was similar to and not significantly different from control cultures (Fig. 1D). To investigate whether endogenous ARC had an effect on neuronal vulnerability, we used RNA interference to gradually reduce ARC protein content in primary neuronal cultures (Fig. 1E,F,H). Lentiviral particles coding for a scrambled microRNA-embedded shRNA (control-shRNA) served as a control. shRNAs were driven by a neuron-specific synapsin promoter. EGFP was used to serve as a reporter for bicistronic expression of miR-shRNA. Although the baseline number of neuronal cells did not vary, neurons with reduced ARC protein levels were more vulnerable and prone to neuronal cell loss after combined OGD (Fig. 1F,H). We aimed to directly compare the effects of gain and loss of function and applied the most effective dose derived from titration experiments (Fig. 1D) and the corresponding TAT.β-Gal control and lentiviral particles for RNA interference in one set and performed four independent experiments with 150 min of OGD (Fig. 1F–H).
TAT.ARC protein transduction protects mice from transient focal cerebral ischemia
We exposed mice to MCAo, applied TAT proteins intracerebroventricularly, and analyzed several time points of reperfusion. The experimental setup is shown in detail in Figure 2A. We tested the distribution of TAT.β-Gal or TAT.ARC in the brain and their protein stability in the ischemic territory over time in Figure 2, B and C. The ipsilateral hemispheres were cut in three 2-mm-thick sagittal slices to demonstrate the spread of the proteins in brain lysates of these subregions after contralateral intrathecal TAT protein administration 72 h after 60 min MCAo (Fig. 2B). Both TAT.β-Gal and TAT.ARC showed lower immunoreactive protein on the same immunoblot membrane over time when we compared their presence in the ischemic core at 3, 8, and 24 h of reperfusion (Fig. 2C). TAT.ARC-treated mice showed infarct sizes that were significantly smaller than those with TAT.β-Gal-treated controls 72 h after 60 min MCAo by MRI measurement (Fig. 2D,E). This was confirmed by histological analysis at 72 h (Fig. 2F). In the cortical subregion of the ischemic territory, MRI had already revealed differences in lesion volume between treatment groups at 24 h (Fig. 3A), indicative of a protective effect of TAT.ARC treatment in the penumbra. This reduction in the cortical penumbra was more pronounced at 72 h and resulted in a significantly smaller entire ischemic territory in TAT.ARC-treated mice (Fig. 3A). This lower lesion volume after MCAo was associated with significantly better behavioral function compared with control animals (Fig. 2G).
In analogy to the biochemical analysis, in which brains were cut in three sagittal slices from medial to lateral and collected for each hemisphere, we cut/separated the brain MRI scans into slices that corresponded to cortical penumbra (lateral), ischemic core (middle), and striatal penumbra (medial) and analyzed lesion volumes for each slice (see Materials and Methods and Fig. 3A). We performed this type of analysis because smaller lesion volumes in the treatment group and differing pathophysiological processes in the core and the penumbra of the ischemic territory may confound the analysis of the mechanisms underlying the neuroprotective effect of ARC.
MRI-based regional subgroup analysis of lesion volumes showed significant effects of TAT protein delivery on the cortical penumbra after 24 and 72 h of reperfusion (Fig. 3A), whereas the ischemic core and medial striatum showed no significant difference between TAT.β-Gal- and TAT.ARC-treated mice at either reperfusion time point (Fig. 3A).
Exogenous ARC prevents cleavage of caspase-8, caspase-9, and caspase-3 and prevents activation of BH3-only proteins BAX/ BIM and JNK pathways
Brain sections were analyzed by immunoblots and showed substantially less activation of caspase-8, caspase-9, and caspase-3 in all subregions of the ischemic brains treated with TAT.ARC proteins compared with the TAT.β-Gal-treated animals (Fig. 3B with semiquantitative analysis non-adjusted and adjusted to the lesion volume in a given slice in C). Although the proapoptotic BH3-only protein BAX was strongly activated in TAT.β-Gal-treated animals after MCAo, only weak BAX activation was detected in the ARC-treated group (Fig. 3B,C). Furthermore, phosphorylation of JNK isoforms was lower in case of exogenous ARC protein delivery, whereas total JNK1-3 protein expression was not different (Fig. 3D,E). In turn, this completely blocked BIM phosphorylation at serine 69, because it depends on JNK activity (Fig. 3F,G). Together, both initiator and effector caspases of the extrinsic and intrinsic apoptotic death pathway, as well as initiator and effector targets of the JNK signaling pathway were attenuated by TAT.ARC protein transduction at 72 h of reperfusion.
TAT.ARC inhibits DAXX–ASK1–MKK–JNK signaling after focal ischemia
We observed less activation of JNK at 72 h of reperfusion and therefore analyzed the time course of upstream DAXX, ASK1, and JNK signaling in ischemic brains (Fig. 4A,B). We subjected five animals per group to 60 min MCAo with TAT.β-Gal or TAT.ARC treatment and indicated time points of reperfusion and analyzed the core ischemic region and corresponding contralateral section with immunoblotting. Lesion size did not differ significantly in this section (Fig. 3A). By so doing, we accounted for the fact that any differences seen in TAT.β-Gal- or TAT.ARC-treated animals in the assays may be a mere reflection of a difference in lesion size. DAXX and ASK1 signaling was attenuated in TAT.ARC-treated mice at 8 and 24 h after MCAo but not at 3 h (Fig. 4A,B). A significant attenuation of activated BAX and slightly decreased pJNK and pBIM proteins were detected at 8 or 24 h (Fig. 4B,C). Notably, phosphorylation of MAP44/42 kinase was not altered at 3, 8, and 24 h of reperfusion in mice treated with TAT.ARC or TAT.β-Gal proteins (Fig. 4C).
TAT.ARC protein does not bind to activated JNK isoforms
To disentangle which JNK isoforms are phosphorylated after focal cerebral ischemia, immunoprecipitation experiments were performed to immobilize phospho-JNK1-3 and then probe with an antibody against all JNK isoforms (i.e., 1–3). JNK phosphorylation was observed predominantly for JNK3, but JNK1 and JNK2 were also activated in the TAT.β-Gal-treated group (Fig. 5A). The exact ratio of activated isoforms cannot be predicted because cross-reactivity of antibodies might be a confounder so that proportions between activated isoforms might be a little different. In contrast to our recent results in the liver (An et al., 2012, 2013), activated endogenous JNK did not bind to exogenous ARC protein in the brain (Fig. 5A,B). Notably, there was no difference between the two groups in the lesion volumes of the middle slice representing the core of the infarct at 24 h (Fig. 3A) as analyzed by MRI.
We aimed to show that JNK phosphorylation is restricted to the ischemic territory and performed immunoprecipitations from brain lysates of the contralateral hemisphere of mice subjected to MCAo and administration of TAT.β-Gal or TAT.ARC (Fig. 5B). In contrast to the ischemic territory, neither ARC binding nor phospho JNK signaling was detected in non-ischemic tissue.
TAT.ARC directly interacts with DAXX and inhibits recruitment to ASK1
We immunoprecipitated DAXX and blotted for ASK1 and phosphorylated ASK1 after treatment with TAT.β-Gal or TAT.ARC protein, respectively. Notably, coimmunoprecipitation was observed as early as 3 and 8 h after reperfusion in TAT.β-Gal-treated animals but was below the threshold of the detection limit in TAT.ARC transduced animals (Fig. 6A). At later time points (24 h), interaction of DAXX and ASK1 was not detectable in either group. DAXX immunoblotting confirmed the efficacy of pull-down throughout all groups. Subsequently, we used immunoprecipitation to test whether or not endogenous and/or exogenous ARC interacts with DAXX. Both exogenous and endogenous ARC protein bound to DAXX and immobilized, as indicated by immunoblotting the precipitates at 72 h after reperfusion in the contralateral hemisphere (Fig. 6B). No endogenous ARC protein was detected in the ischemic hemisphere, and DAXX–ARC binding was lower at this time point. Consequently, we tested the earlier time points (3, 8, and 24 h) and immunoprecipitated both proteins (Fig. 6C). In line with the initiating complex of DAXX with ASK1 after 3 and 8 h of reperfusion (Fig. 6A), an attenuated ARC–DAXX interaction was observed at these time points in the ischemic hemisphere (Fig. 6C). Endogenous ARC protein was diminished to under the threshold of detection at 24 h (Fig. 6C). Furthermore, the interaction of ARC with DAXX was completely abrogated in the mutant form of ARC (L31F, G69R), with two point mutations in the CARD domain of ARC, indicating CARD-dependent ARC–DAXX binding (Fig. 6D,E). Our data thus suggest that ARC interacts with DAXX directly in a CARD-dependent manner and thereby inhibits DAXX–ASK1 binding.
Delayed TAT.ARC administration protects the brain and restores function
To test whether TAT.ARC administration is suitable as a neuroprotectant against brain ischemia, we determined the therapeutic window for delayed treatment. This is important for a clinically relevant setting, in which spontaneous recanalization or thrombolysis might precede a therapeutic intervention and patients have to be transported and diagnosed for ischemic brain injury. We chose a higher dose of 5 μg/mouse and a time point of 3 h after onset of the MCAo (Fig. 7A). We subjected 15 mice per group to 60 min of MCAo. The mice were tested and compared in the pole test for their individual performance for time-to-turn and time-to-descend as functional outcome measures before and 48 h after MCAo (Fig. 7B). Finally, mice were killed, and the infarct volume was determined histologically (Fig. 7C). Eight animals (of 30) were excluded from the analysis of the pole test because they did not fulfill the predefined criteria of a smooth run down the pole (5 of 15 from TAT.β-Gal and 3 animals from TAT.ARC). One animal (of 15) died after surgery before injection of TAT.ARC protein. The animals in the TAT.ARC group performed significantly better in both parameters and had 20% lower infarct volume than controls (Fig. 7B,C).
Delayed TAT.ARC administration provides sustained neuroprotection and functional benefits
To test whether TAT.ARC administration provides sustained better long-term functional recovery and neuroprotection, we subjected 46 mice to either sham (n = 8) or 30 min MCAo (n = 15) surgery and 3 h delayed treatment with TAT.β-Gal or TAT.ARC (Fig. 8).
Within the sham group, overall modified DeSimoni neuroscore values were not significantly different with β-Gal than with TAT.ARC (p = 0.565). Within MCAo mice, neuroscore values were significantly lower with TAT.ARC than with β-Gal (p = 0.015; Fig. 8B). Our first and second hypotheses (general and focal deficit scores) were tested, and, within MCAo mice, both scores were significantly lower with TAT.ARC than with β-Gal (both p = 0.022). Within the sham group, time on the rotarod was not significantly different (p = 0.146) between β-Gal- and TAT.ARC-treated mice. In contrast, within MCAo, time on the rotarod was on average 37 s shorter (p = 0.033) with β-Gal than with TAT-ARC. Mice within MCAo showed sustained neuroprotective effects with longer time on the rotarod after 14 d in the TAT.ARC cohort compared with TAT.β-Gal-treated mice (p = 0.019, main hypothesis 3) with a time difference of on average 61 s longer on the rod [163.3 s (CI, 95–178) vs 197.1 s (CI, 170–225)]. MCAo mice had a steeper drop in body weight (sham vs MCAo, p = 0.002) that recovered in sham mice with TAT.β-Gal after 13 d, in TAT.ARC in sham mice after 5 d, in MCAo mice treated with TAT.ARC after 13 d, and in MCAo mice treated with TAT.β-Gal after 20 d (Fig. 8F). Body temperature was assessed non-invasively. We observed a pattern of fluctuations with a 7 d period but no significant difference between sham versus MCAo mice (p = 0.789).
Our hierarchical hypothesis 4 was an effect of TAT.ARC treatment on histological parameters within MCAo. Striatal area was measured in NeuN–DAB coronal slices as a marker for atrophy in sham (n = 3 animals per TAT.protein) and MCAo (n = 14 for TAT.β-Gal and n = 11 for TAT.ARC) mice. Mice within the MCAo group that were treated with TAT.β-Gal showed on average a steeper drop in striatal area on the ipsilesional hemisphere than TAT.ARC-treated mice (2.9 mm2 in TAT.β-Gal-treated vs 3.4 mm2 in TAT.ARC-treated mice, ipsilateral, p = 0.067) that was not significant. Neuronal densities were analyzed as a surrogate marker for residual stroke volume using coronal sections of five regions from the frontal to the caudal pole of the brain (specific coordinates are described in Materials and Methods and in the open dataset for Figs. 7C and 8K–M).
In summary, mean stroke volume after 28 d was slightly lower with TAT.ARC treatment [7.3 mm3 (CI, 4.7–9.8) vs 5.4 mm3 (CI, 2.6–8.1), p = 0.283; Fig. 8L]. We calculated the ipsilateral number of NeuN–DAB-positive cells in the striatum as a fraction to the number of neurons on the contralesional side and found a slight reduction of the ratios from 0.591 (CI, 0.455–0.728) to 0.701 (CI, 0.51–0.892; p = 0.303; Fig. 8M). We were not able to test our hypothesis 5 because our model of hierarchical testing was underpowered to show a significant effect on striatal atrophy (p = 0.067) by TAT.ARC single post-stroke treatment in mice with 28 d survival after 30 min MCAo. Nevertheless, we cannot conclude that TAT.ARC treatment within MCAo has no effect on the number of neurons within the ipsilesional striatum and stroke volume compared with TAT.β-Gal-treated mice 28 d after MCAo.
Discussion
The results of the present study identify endogenous ARC as a novel neuroprotective factor and hence a potential therapeutic target after cerebral ischemia. Four major findings from cell culture and murine experimental stroke experiments support this conclusion. First, we observed that endogenous ARC protein was significantly downregulated as early as 20 h after 30 min MCAo or at 8 h after 60 min MCAo. This loss of neuronal ARC preceded neuronal apoptosis, which occurred predominantly at 48 h and reached a maximum at 72 h of reperfusion in this model (Katchanov et al., 2001). ARC knockdown in primary neurons by RNA interference resulted in higher susceptibility to cell death in response to OGD compared with control shRNA. Conversely, administration of exogenous ARC protein led to a dose-dependent neuronal survival in response to OGD compared with TAT.β-Gal. Second, there is a significantly lower infarct size in TAT.ARC-treated mice (−30%) compared with TAT.β-Gal-treated controls after 60 min MCAo and 72 h of reperfusion. Lower lesion volume after MCAo in TAT.ARC-treated mice was associated with significantly better behavioral function when compared with control animals. Third, we demonstrated that, beyond inhibition of caspase activation and activation of proapoptotic BH3-only proteins BAX and BIM by ARC, ARC also blocks DAXX–ASK1–JNK-mediated cell death via direct interaction with DAXX. The specificity of these findings was demonstrated by the absence of an effect on the MAP44/42 kinase pathway. Finally, we provided evidence that TAT.ARC could be administered as late as 3 h after the onset of ischemia/reperfusion injury and still provided significant neuroprotective functional benefits for brain protection in a 60 min MCAo model with 3 d and in a 30 min MCAo with 28 d survival. Together, these observations suggest that ARC plays a crucial role during the ischemic stress response in the brain by providing early antiapoptotic neuroprotective signals that promote neuronal survival, minimize tissue injury, and preserve behavioral performance. Previous studies showed that ASK1–JNK signaling plays a crucial role during cell death (Chang et al., 1998). Recently, more attention has been paid to the connection between DAXX and ASK1–JNK signaling (Karunakaran et al., 2007; Ryo et al., 2007). Besides the activation of downstream targets of death signaling, such as caspase-8, caspase-9, caspase-3, or BAX and BIM, activation of key upstream death mediators, such as DAXX, ASK1, and JNK, were detected after MCAo. Importantly, research has shown that, after exogenous ARC administration, activation of both upstream and downstream death mediators is substantially suppressed compared with levels in mice treated with TAT.β-Gal during late reperfusion. We showed that exogenous ARC interferes with DAXX–ASK1–JNK-mediated death signaling and neuronal cell loss after MCAo by directly binding DAXX. ARC–DAXX binding is specific, dependent on the CARD domain of ARC, and independent of neuronal injury. The absence of a CARD and death domain in DAXX suggests other nonhomotypic death-fold interactions between DAXX and ARC. Additional studies will aim to map the ARC binding domain in DAXX.
The Daxx gene encodes a multifunctional protein that resides in many locations, such as the nucleus and cytoplasm, and functions as a transcriptional regulator. Previous studies suggest that DAXX has both proapoptotic and antiapoptotic functions, depending on its subcellular localization and concentration (Niu et al., 2011). DAXX promotes cell death and JNK activation under physiological conditions (Khelifi et al., 2005). DAXX-depleted cells are resistant to cell killing induced by ultraviolet irradiation and oxidative stress after impaired ASK1–JNK activation (Khelifi et al., 2005). DAXX overexpression sensitizes cells not only to FAS-induced apoptosis but also to TGFβ-induced JNK activation (Perlman et al., 2001). Oxidative stress can trigger DAXX induction and induce neuronal cell death, both of which can be inhibited by antioxidant treatment (Niu et al., 2011). Activated, cytoplasmic DAXX binds to and activates ASK1 and triggers activation of MKK4/7–JNK (Yang et al., 1997; Chang et al., 1998; Song and Lee, 2003; Khelifi et al., 2005; Rodriguez-Nieto and Zhivotovsky, 2006; Niu et al., 2011). DAXX was reported to promote ASK1-mediated apoptosis in a kinase- and caspase-independent manner (Charette et al., 2001). By blocking DAXX trafficking, rat hippocampus CA1 neurons were protected against cerebral ischemia/reperfusion injury (Niu et al., 2011). Interfering with DAXX signaling decreased ASK1 activation, suppressed the assembly of the FAS–DAXX–ASK1 signaling module, and subsequently inhibited JNK activation and c-Jun phosphorylation (Niu et al., 2011). Furthermore, knockdown of endogenous DAXX exerted similar neuroprotective effects during ischemia/reperfusion (Niu et al., 2011).
Recent evidence suggests that activation of JNK signaling plays a crucial role in ischemia-induced neuronal death (Kuan et al., 2003; Guan et al., 2005, 2006a,b). Here, we did not show that ARC directly binds JNK1/2 in a tissue-dependent manner and thereby inhibits JNK activation (An et al., 2012). This argues against a direct effect of ARC on JNK signaling in the brain and might explain why substantial JNK activation was detected at 3 h of reperfusion even in the ARC-treated group. In fact, ARC binding mediated the inhibition of DAXX activation, and ASK1–JNK signaling prevents sustained JNK activation, as seen in the TAT.β-Gal group with subsequent caspase activation and neuronal cell death. Consistent with our findings, others show that inhibition of nuclear export of DAXX prevents JNK activation (Jung et al., 2007).
One major therapeutic advantage of ARC-mediated neuroprotection is the fact that it affects many pathways, whereas most antiapoptotic approaches only operate selectively, interfering with either death receptor only or mitochondrial apoptotic signaling only. Others operate only at the postmitochondrial level, as with caspase inhibitors. The multimodal pleiotropic death-repressing interactions of exogenous ARC protein with key initiators (such as DAXX) of both death pathways suggests that TAT.ARC might be a more powerful tool for stroke treatment than are caspase or small-molecule JNK inhibitors, which target only one pathway (see proposed model of molecular mechanisms in Fig. 7D). We observed a robust and significantly lower lesion volumes of 30% compared with controls and a preserved function of behavior when TAT.ARC was applied during occlusion. This effect was not as strong but still present (20% lower compared with controls) if treatment was delayed to 3 h after onset of ischemia, and it still was associated with better behavioral outcome than in controls. This reduction of effect size fits the well known “time is brain” concept for recanalization in humans. Subregions of the ischemic territory as analyzed in Figure 3 model the tissue at risk in the lateral or inner penumbra. This region includes the cortex and medial striatum. The core of the infarct compromised the lateral striatum. The percentage of salvaged tissue cross-sectionally increases from 30 to 41% in the lateral and in the inner penumbra to even 54% because it is amenable to rescue whereas, as expected, the ischemic core shows the modest cross-sectional reduction of 22% if TAT.ARC is applied during occlusion (Fig. 3A).
Finally, we studied the effect of a single, delayed injection of TAT.ARC protein and compared it with TAT.β-Gal-treated mice subjected to sham/MCAo and used hierarchical testing of hypotheses: most important were better outcome (first three hypotheses) and effect on striatal atrophy and neuronal cell death in the striatum (hypotheses 4 and 5) after 28 d. We were able to show sustained effects on better outcome of a single dose of TAT protein applied 3 h after the onset of ischemia (Fig. 8). Independent functional parameters of TAT.ARC-dependent effects on recovery were the general and focal deficits and longer time on the rotarod of TAT.ARC-treated mice compared with TAT.β-Gal-treated controls. Striatal atrophy after stroke was cross-sectionally reduced 28 d after MCAo but not significantly (p = 0.067 for atrophy reduction by TAT.ARC treatment). At this level, hypothesis testing terminated by a priori definition. Our study was most likely underpowered for effects on loss of neurons within the striatum in the long term. We are confident that the effect sizes in our study are robust, because great emphasis was put on internal validity (reduction of bias) and conformity to ARRIVE reporting guidelines and conducted in a laboratory with certified quality management. TAT.ARC protein transduction or ARC-related small molecules could be of therapeutic benefit for preventing and treating ischemic brain injury in humans along with treatment modalities aiming for reperfusion. This might buy time for the brain and preserve its function. In summary, the strength of this study is the identification of a novel endogenous neuroprotective molecular mechanism and the proof of principle that each treatment starting during occlusion or applied after reperfusion with TAT.ARC is neuroprotective after experimental stroke. Our study warrants additional confirmatory preclinical studies for detailed dose–response curves, comorbidity effects, gender studies, and interference with the only available therapy, that is, thrombolysis or endovascular thrombectomy. A limitation of this study is the application route of TAT proteins. TAT.Bcl-x(L), TAT.Hsp70, and TAT.GDNF have been given intravenously after MCAo (Kilic et al., 2003; Doeppner et al., 2009a,b). This needs to be addressed in future studies and was not within the scope of this mechanistic explorative approach (Kimmelman et al., 2014).
Footnotes
This work was supported by the German Research Foundation (SFB TRR43 and Exc 257), the Federal Ministry of Education and Research (01 EO 08 01), and intramural funding by the Faculty of the Charité-Universitätsmedizin Berlin (Gerok position) to the Center for Stroke Research Berlin. We thank Katarzyna Pogodzinski and Nadine Weser for excellent technical assistance. We are grateful to Catherine Aubel for editing assistance. pTAT.HA and pTAT.β-Gal were kindly provided by S. Dowdy (Howard Hughes Medical Institute, La Jolla, CA).
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- An J, Li P, Li J, Dietz R, Donath S. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med (Berl) 2009;87:401–410. doi: 10.1007/s00109-008-0434-z. [DOI] [PubMed] [Google Scholar]
- An J, Harms C, Lättig-Tünnemann G, Sellge G, Mandić AD, Malato Y, Heuser A, Endres M, Trautwein C, Donath S. TAT-apoptosis repressor with caspase recruitment domain protein transduction rescues mice from fulminant liver failure. Hepatology. 2012;56:715–726. doi: 10.1002/hep.25697. [DOI] [PubMed] [Google Scholar]
- An J, Mehrhof F, Harms C, Lättig-Tünnemann G, Lee SL, Endres M, Li M, Sellge G, Mandić AD, Trautwein C, Donath S. ARC is a novel therapeutic approach against acetaminophen-induced hepatocellular necrosis. J Hepatol. 2013;58:297–305. doi: 10.1016/j.jhep.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- Charette SJ, Lambert H, Landry J. A kinase-independent function of Ask1 in caspase-independent cell death. J Biol Chem. 2001;276:36071–36074. doi: 10.1074/jbc.C100340200. [DOI] [PubMed] [Google Scholar]
- Datwyler AL, Lättig-Tünnemann G, Yang W, Paschen W, Lee SL, Dirnagl U, Endres M, Harms C. SUMO2/3 conjugation is an endogenous neuroprotective mechanism. J Cereb Blood Flow Metab. 2011;31:2152–2159. doi: 10.1038/jcbfm.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E, Bergamaschini L. Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab. 2003;23:232–239. doi: 10.1097/01.WCB.0000046146.31247.A1. [DOI] [PubMed] [Google Scholar]
- Doeppner TR, Dietz GP, El Aanbouri M, Gerber J, Witte OW, Bähr M, Weise J. TAT-Bcl-x(L) improves survival of neuronal precursor cells in the lesioned striatum after focal cerebral ischemia. Neurobiol Dis. 2009a;34:87–94. doi: 10.1016/j.nbd.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Doeppner TR, Nagel F, Dietz GP, Weise J, Tönges L, Schwarting S, Bähr M. TAT-Hsp70-mediated neuroprotection and increased survival of neuronal precursor cells after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2009b;29:1187–1196. doi: 10.1038/jcbfm.2009.44. [DOI] [PubMed] [Google Scholar]
- Donath S, Li P, Willenbockel C, Al-Saadi N, Gross V, Willnow T, Bader M, Martin U, Bauersachs J, Wollert KC, Dietz R, von Harsdorf R. Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation. 2006;113:1203–1212. doi: 10.1161/CIRCULATIONAHA.105.576785. [DOI] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: a novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig_1_C_D (cell counts surviving neurons: open data) Figshare Repository. 2016a doi: 10.6084/m9.figshare.3405787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig_8_I_J_NeuN-DAB_staining_in_sham_MCAo_mice_with_28-days_survival. Figshare Repository. 2016b doi: 10.6084/m9.figshare.3413683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig_8_striatal atrophy, chronic lesion volume, striatal neuronal cell counts. Figshare Repository. 2016c doi: 10.6084/m9.figshare.3398761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig_8_behavior_temp_bodyweight. Figshare Repository. 2016d doi: 10.6084/m9.figshare.3420901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig_7_C_histology_indirect_infarct_volumes. Figshare Repository. 2016e doi: 10.6084/m9.figshare.3420868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig_3_A_MRI_slice_results_24h_and_72h. Figshare Repository. 2016f doi: 10.6084/m9.figshare.3420883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig. 2 and Fig. 3 (MRI: open data) Figshare Repository. 2016g doi: 10.6084/m9.figshare.3398722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath S, An J, Lee SLL, Gertz K, Datwyler AL, Harms U, Müller S, Farr TD, Füchtemeier M, Lättig-Tünnemann G, Lips J, Foddis M, Mosch L, Bernard R, Grittner U, Balkaya M, Kronenberg G, Dirnagl U, Endres M, Harms C. Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. In: Fig. 1 H (normalized LDH: open data) Figshare Repository. 2016h doi: 10.6084/m9.figshare.3406282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Saver JL. Future directions of acute ischaemic stroke therapy. Lancet Neurol. 2015;14:758–767. doi: 10.1016/S1474-4422(15)00054-X. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. Amsterdam: Academic; 2007. [Google Scholar]
- Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, Luo Y, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Guan QH, Pei DS, Zhang QG, Hao ZB, Xu TL, Zhang GY. The neuroprotective action of SP600125, a new inhibitor of JNK, on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampal CA1 via nuclear and non-nuclear pathways. Brain Res. 2005;1035:51–59. doi: 10.1016/j.brainres.2004.11.050. [DOI] [PubMed] [Google Scholar]
- Guan QH, Pei DS, Zong YY, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006a;139:609–627. doi: 10.1016/j.neuroscience.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Guan QH, Pei DS, Liu XM, Wang XT, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006b;1092:36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- Gustafsson AB, Tsai JG, Logue SE, Crow MT, Gottlieb RA. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem. 2004;279:21233–21238. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- Harms C, Bösel J, Lautenschlager M, Harms U, Braun JS, Hörtnagl H, Dirnagl U, Kwiatkowski DJ, Fink K, Endres M. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol Cell Neurosci. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hübner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, von Harsdorf R, Endres M, Hörtnagl H. Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci. 2007;27:4562–4571. doi: 10.1523/JNEUROSCI.5110-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck L, Harms C, Grothe D, An J, Gertz K, Kronenberg G, Dietz R, Endres M, von Harsdorf R. Critical role for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to statin signaling in cardiac myocytes. Circ Res. 2007;100:50–60. doi: 10.1161/01.RES.0000254704.92532.b9. [DOI] [PubMed] [Google Scholar]
- Hauck L, Harms C, An J, Rohne J, Gertz K, Dietz R, Endres M, von Harsdorf R. Protein kinase CK2 links extracellular growth factor signaling with the control of p27(Kip1) stability in the heart. Nat Med. 2008;14:315–324. doi: 10.1038/nm1729. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann CJ, Harms U, Rex A, Szulzewsky F, Wolf SA, Grittner U, Lättig-Tünnemann G, Sendtner M, Kettenmann H, Dirnagl U, Endres M, Harms C. Vascular Stat3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation. 2015;131:1772–1782. doi: 10.1161/CIRCULATIONAHA.114.013003. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Jung YS, Kim HY, Lee YJ, Kim E. Subcellular localization of Daxx determines its opposing functions in ischemic cell death. FEBS Lett. 2007;581:843–852. doi: 10.1016/j.febslet.2007.01.055. [DOI] [PubMed] [Google Scholar]
- Karunakaran S, Diwakar L, Saeed U, Agarwal V, Ramakrishnan S, Iyengar S, Ravindranath V. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson's disease: protection by alpha-lipoic acid. FASEB J. 2007;21:2226–2236. doi: 10.1096/fj.06-7580com. [DOI] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelifi AF, D'Alcontres MS, Salomoni P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005;12:724–733. doi: 10.1038/sj.cdd.4401559. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Dietz GP, Bähr M. Intravenous TAT-GDNF is protective after focal cerebral ischemia in mice. Stroke. 2003;34:1304–1310. doi: 10.1161/01.STR.0000066869.45310.50. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman J, Mogil JS, Dirnagl U. Distinguishing between exploratory and confirmatory preclinical research will improve translation. PLoS Biol. 2014;12:e1001863. doi: 10.1371/journal.pbio.1001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriakidou M, Driscoll DA, Lopez-Guisa JM, Strauss JF., 3rd Cloning and expression of primate Daxx cDNAs and mapping of the human gene to chromosome 6p21.3 in the MHC region. DNA Cell Biol. 1997;16:1289–1298. doi: 10.1089/dna.1997.16.1289. [DOI] [PubMed] [Google Scholar]
- Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ, Kim S. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. J Biol Chem. 2001;276:39103–39106. doi: 10.1074/jbc.M105928200. [DOI] [PubMed] [Google Scholar]
- Kort WJ, Hekking-Weijma JM, TenKate MT, Sorm V, VanStrik R. A microchip implant system as a method to determine body temperature of terminally ill rats and mice. Lab Anim. 1998;32:260–269. doi: 10.1258/002367798780559329. [DOI] [PubMed] [Google Scholar]
- Koseki T, Inohara N, Chen S, Núñez G. ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc Natl Acad Sci USA. 1998;95:5156–5160. doi: 10.1073/pnas.95.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci USA. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Boucher DM. Functions of MAP kinases: insights from gene-targeting studies. J Biochem. 2004;135:653–656. doi: 10.1093/jb/mvh078. [DOI] [PubMed] [Google Scholar]
- Langer F, Fietz J. Ways to measure body temperature in the field. J Therm Biol. 2014;42:46–51. doi: 10.1016/j.jtherbio.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Moretti A, Ferrari F, Villa RF. Pharmacological therapy of acute ischaemic stroke: achievements and problems. Pharmacol Ther. 2015;153:79–89. doi: 10.1016/j.pharmthera.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Murata Y, Fujiwara N, Seo JH, Yan F, Liu X, Terasaki Y, Luo Y, Arai K, Ji X, Lo EH. Delayed inhibition of c-Jun N-terminal kinase worsens outcomes after focal cerebral ischemia. J Neurosci. 2012;32:8112–8115. doi: 10.1523/JNEUROSCI.0219-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam YJ, Mani K, Ashton AW, Peng CF, Krishnamurthy B, Hayakawa Y, Lee P, Korsmeyer SJ, Kitsis RN. Inhibition of both the extrinsic and intrinsic death pathways through nonhomotypic death-fold interactions. Mol Cell. 2004;15:901–912. doi: 10.1016/j.molcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H, Wada T, Katada T. Physiological roles of SAPK/JNK signaling pathway. J Biochem. 2004;136:123–126. doi: 10.1093/jb/mvh117. [DOI] [PubMed] [Google Scholar]
- Niu YL, Li C, Zhang GY. Blocking Daxx trafficking attenuates neuronal cell death following ischemia/reperfusion in rat hippocampus CA1 region. Arch Biochem Biophys. 2011;515:89–98. doi: 10.1016/j.abb.2011.07.016. [DOI] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini F, Villa P, Parrella S, Zangari R, Zanier ER, Gesuete R, Stravalaci M, Fumagalli S, Ottria R, Reina JJ, Paladini A, Micotti E, Ribeiro-Viana R, Rojo J, Pavlov VI, Stahl GL, Bernardi A, Gobbi M, De Simoni MG. Targeting mannose-binding lectin confers long-lasting protection with a surprisingly wide therapeutic window in cerebral ischemia. Circulation. 2012;126:1484–1494. doi: 10.1161/CIRCULATIONAHA.112.103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol. 2001;3:708–714. doi: 10.1038/35087019. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM., Jr JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/S0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- Reich A, Spering C, Gertz K, Harms C, Gerhardt E, Kronenberg G, Nave KA, Schwab M, Tauber SC, Drinkut A, Harms K, Beier CP, Voigt A, Göbbels S, Endres M, Schulz JB. Fas/CD95 regulatory protein Faim2 is neuroprotective after transient brain ischemia. J Neurosci. 2011;31:225–233. doi: 10.1523/JNEUROSCI.2188-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Nieto S, Zhivotovsky B. Role of alterations in the apoptotic machinery in sensitivity of cancer cells to treatment. Curr Pharm Des. 2006;12:4411–4425. doi: 10.2174/138161206779010495. [DOI] [PubMed] [Google Scholar]
- Ryo A, Hirai A, Nishi M, Liou YC, Perrem K, Lin SC, Hirano H, Lee SW, Aoki I. A suppressive role of the prolyl isomerase Pin1 in cellular apoptosis mediated by the death-associated protein Daxx. J Biol Chem. 2007;282:36671–36681. doi: 10.1074/jbc.M704145200. [DOI] [PubMed] [Google Scholar]
- Salomoni P, Khelifi AF. Daxx: death or survival protein? Trends Cell Biol. 2006;16:97–104. doi: 10.1016/j.tcb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Saver JL, Gornbein J, Grotta J, Liebeskind D, Lutsep H, Schwamm L, Scott P, Starkman S. Number needed to treat to benefit and to harm for intravenous tissue plasminogen activator therapy in the 3- to 4.5-hour window: joint outcome table analysis of the ECASS 3 trial. Stroke. 2009;40:2433–2437. doi: 10.1161/STROKEAHA.108.543561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Lee YJ. Role of the ASK1-SEK1-JNK1-HIPK1 signal in Daxx trafficking and ASK1 oligomerization. J Biol Chem. 2003;278:47245–47252. doi: 10.1074/jbc.M213201200. [DOI] [PubMed] [Google Scholar]
- Wang XS, Diener K, Jannuzzi D, Trollinger D, Tan TH, Lichenstein H, Zukowski M, Yao Z. Molecular cloning and characterization of a novel protein kinase with a catalytic domain homologous to mitogen-activated protein kinase kinase kinase. J Biol Chem. 1996;271:31607–31611. doi: 10.1074/jbc.271.49.31607. [DOI] [PubMed] [Google Scholar]
- Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–1076. doi: 10.1016/S0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim F, Ji S, Kronenberg G, Barco A, Olivares R, Benito E, Dirnagl U, Gertz K, Endres M, Harms C, Meisel A. Histone acetylation and CREB binding protein are required for neuronal resistance against ischemic injury. PLoS One. 2014;9:e95465. doi: 10.1371/journal.pone.0095465. [DOI] [PMC free article] [PubMed] [Google Scholar]