

Abstract
Cocaine exposure alters brain-derived neurotrophic factor (BDNF) expression in the brain. BDNF signaling through TrkB receptors differentially modulates cocaine self-administration, depending on the brain regions involved. In the present study, we determined how brain-wide inhibition of TrkB signaling affects cocaine intake, the motivation for the drug, and reinstatement of drug taking after extinction. To overcome the inability of TrkB ligands to cross the blood–brain barrier, the TrkB antagonist cyclotraxin-B was fused to the nontoxic transduction domain of the tat protein from human immunodeficiency virus type 1 (tat-cyclotraxin-B). Intravenous injection of tat-cyclotraxin-B dose-dependently reduced cocaine intake, motivation for cocaine (as measured under a progressive ratio schedule of reinforcement), and reinstatement of cocaine taking in rats allowed either short or long access to cocaine self-administration. In contrast, the treatment did not affect operant responding for a highly palatable sweet solution, demonstrating that the effects of tat-cyclotraxin-B are specific for cocaine reinforcement. Cocaine self-administration increased TrkB signaling and activated the downstream Akt pathway in the nucleus accumbens, and had opposite effects in the prefrontal cortex. Pretreatment with tat-cyclotraxin-B normalized protein levels in these two dopamine-innervated brain regions. Cocaine self-administration also increased TrkB signaling in the ventral tegmental area, where the dopaminergic projections originate, but pretreatment with tat-cyclotraxin-B did not alter this effect. Altogether, our data show that systemic administration of a brain-penetrant TrkB antagonist leads to brain region-specific effects and may be a potential pharmacological strategy for the treatment of cocaine addiction.
SIGNIFICANCE STATEMENT Brain-derived neurotrophic factor (BDNF) signaling through TrkB receptors plays a well established role in cocaine reinforcement. However, local manipulation of BDNF signaling yields divergent effects, depending on the brain region, thereby questioning the viability of systemic TrkB targeting for the treatment of cocaine use disorders. Our study provides first-time evidence that systemic administration of a brain-penetrant TrkB antagonist (tat-cyclotraxin-B) reduces several behavioral measures of cocaine dependence, without altering motor performance or reinforcement by a sweet palatable solution. In addition, although cocaine self-administration produced opposite effects on TrkB signaling in the nucleus accumbens and prefrontal cortex, tat-cyclotraxin-B administration normalized these cocaine-induced changes in both brain regions.
Keywords: BDNF, brain-derived neurotrophic factor, cocaine, cyclotraxin-B, dopamine system, TrkB signaling
Introduction
Numerous studies have shown that brain-derived neurotrophic factor (BDNF) signaling through TrkB receptors plays a key role in cocaine dependence (for review, see Corominas et al., 2007; McGinty et al., 2010; McCarthy et al., 2012). An interaction between BDNF and the mesocorticolimbic dopamine system is believed to modulate the behavioral effects of cocaine (Altar et al., 1992; Martin-Iverson et al., 1994; Guillin et al., 2001; Goggi et al., 2003; Corominas et al., 2007; Lobo et al., 2010; McGinty et al., 2010; McCarthy et al., 2012). Accordingly, forced exposure to cocaine increases BDNF expression in the prefrontal cortex (Le Foll et al., 2005; Fumagalli et al., 2007, 2009; Lu et al., 2010) and the nucleus accumbens (Filip et al., 2006; Graham et al., 2007; Huang et al., 2011). Cocaine self-administration increases BDNF expression not only in these cortical (McGinty et al., 2010; Sadri-Vakili et al., 2010; Fumagalli et al., 2013) and ventral striatal (Grimm et al., 2003; Graham et al., 2007; Fumagalli et al., 2013; Li et al., 2013) regions innervated by dopamine neurons, but also in the ventral tegmental area (Grimm et al., 2003; Pu et al., 2006; Graham et al., 2009; Schmidt et al., 2012) where these dopaminergic neurons originate.
Cocaine intake is modulated by BDNF signaling in a brain region-dependent manner. Local injections of BDNF into the nucleus accumbens or the ventral tegmental area enhance cocaine reward, cocaine self-administration, and reinstatement of cocaine seeking (Horger et al., 1999; Lu et al., 2004, 2009; Graham et al., 2007; Bahi et al., 2008), and intra-accumbens infusion of BDNF antiserum and virally mediated TrkB silencing reduce various measures of cocaine dependence (Graham et al., 2007, 2009; Bahi et al., 2008; Li et al., 2013). In contrast, the local injection of BDNF into the medial prefrontal cortex reduces cocaine seeking (Berglind et al., 2007, 2009; Whitfield et al., 2011) and virally mediated BDNF silencing in this brain region increases the motivation to take cocaine (Sadri-Vakili et al., 2010).
The divergent effects of local TrkB signaling manipulation prompted us to investigate the net effect of brain-wide TrkB antagonism on measures of cocaine self-administration that reflect the motivation to work for the drug and on relapse-like behavior associated with compulsive cocaine seeking. However, BDNF and TrkB ligands do not undergo significant transport across the blood–brain barrier. To allow direct delivery to the brain, the nontoxic transduction domain of the tat protein from human immunodeficiency virus type 1 (HIV-1) was fused to the TrkB antagonist cyclotraxin-B (for details, see Cazorla et al., 2010), resulting in tat-cyclotraxin-B (TC). Intravenous administration of TC following a double-injection procedure was previously shown to inhibit the phosphorylation of forebrain TrkB receptors by ∼50% and to reduce anxiety-related behavior without affecting depression-like behavior or motor performance (Cazorla et al., 2010). In the present study, the behavioral effects of TC were tested in rats given limited (1 h/d) or extended (6 h/d) access to cocaine self-administration (for details, see Ahmed and Koob, 1998; Orio et al., 2009; Wee et al., 2012). The stable short access (ShA) cocaine intake is believed to reflect nondependent drug intake, whereas the long access (LgA)-induced escalation of cocaine intake is thought to reflect the transition to compulsive-like drug taking that is observed in drug dependence (for review, see Koob, 2009; Koob and Volkow, 2010).
In addition, we analyzed TrkB signaling in the nucleus accumbens and prefrontal cortex to determine the role of these two dopamine-innervated brain regions in the modulation of cocaine self-administration by TC. Specifically, we measured the effect of cocaine self-administration and TC pretreatment on the total and phosphorylated levels of TrkB, as well as Akt and ERK, two major downstream effectors of TrkB (for review, see Duman and Voleti, 2012). Finally, we examined the effect of cocaine self-administration and TC pretreatment on TrkB levels in the ventral tegmental area.
Materials and Methods
Animals.
Twenty-two male Wistar rats (Charles River Laboratories), weighing 350 ± 5 g at the beginning of the experiment, were housed in groups of two or three in Macrolon type III cages under a 12 h reversed day/night cycle (lights off at 8:00 A.M.). Food and water were available ad libitum except during behavioral testing. All procedures described below were approved by The Scripps Research Institute Animal Care and Use Committee, and were performed in accordance with national and international laws and guidelines for the care and use of laboratory animals.
Surgery.
Rats were implanted with a Micro-Renathane catheter (inner diameter, 0.3 mm; outer diameter, 0.64 mm; catalog #MRE037, Braintree Scientific, Inc.) into the right external jugular vein according to previously reported procedures (Wee et al., 2007). This aseptic surgery procedure was performed under isoflurane anesthesia (2–3%). After surgery, rats were given an analgesic (2.5 mg/kg, s.c.; Flunixin, Sigma-Aldrich) and an antibiotic (0.033 mg, i.v.; Cefazolin, Sagent Pharmaceuticals) treatment for at least 1 week. The catheter was flushed twice daily with heparinized saline (30 USP/ml, Hospira) during the entire experiment.
Self-administration chambers.
Cocaine self-administration was performed in standard operant chambers (28 × 26 × 20 cm; Med Associates Inc.) that were placed in ventilated, light- and sound-attenuating cubicles. The cocaine self-administration chambers were equipped with a swivel system allowing rats to move freely during self-administration sessions, whereas self-administration chambers for the glucose/saccharin solution were equipped with an acrylic drinking cup in the center of the wall between two levers. In both cages, drugs were delivered by a 15 rpm syringe pump (Razel Scientific Instruments). The start of a session was signaled by the presentation of two retractable levers in the self-administration chamber. Pressing the right lever was programmed to deliver cocaine (volume, 0.1 ml in 4 s) or the glucose/saccharin solution (volume, 0.1 ml in 0.5 s), whereas pressing the left lever had no programmed consequences. During drug administration, a stimulus light above the active lever was illuminated for 20 s for the cocaine solution and for 30 s for the glucose/saccharin solution, both indicating a timeout period when additional lever presses did not result in fluid delivery.
Cocaine training.
One week after surgery, rats were trained to self-administer cocaine (0.5 mg/kg/infusion) for 12 d under a fixed ratio 1 (FR1) schedule of reinforcement (one lever press resulted in one drug injection) for 1 h/d. The first day of cocaine training was labeled experimental day 1 (Fig. 1).
Figure 1.
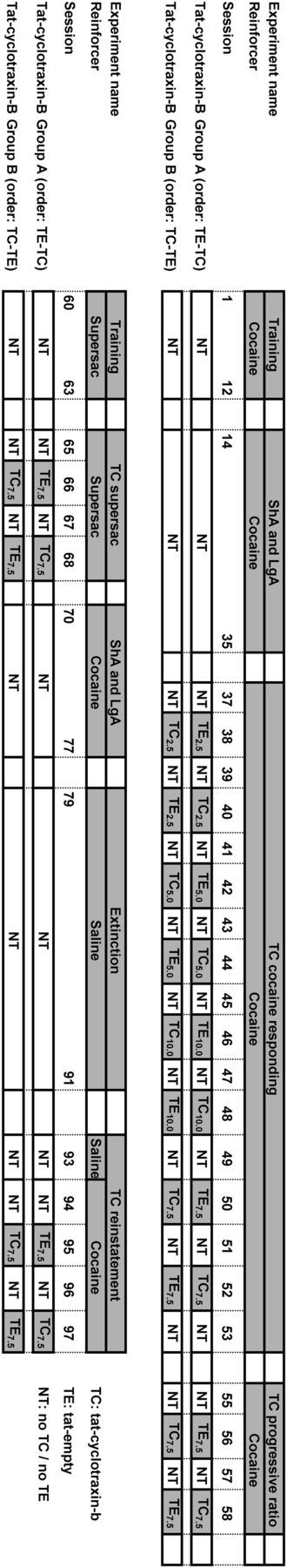
Schematic representation of the cocaine and glucose/saccharin self-administration experiments. The effects of TC and its control peptide (TE) were tested using a within-subjects Latin square design in which test days were separated by a drug-free (NT) day. During these NT days, rats had access to cocaine without drug treatment. Note: the animals in group A received TE followed by TC 2 experimental days later, whereas the animals in group B received TC followed by TE 2 experimental days later.
Escalation of cocaine intake.
After training, rats were divided into two groups matched by the number of infusions during the final training session. One group of rats continued to self-administer cocaine (0.5 mg/kg/infusion) in daily 1 h sessions (ShA), whereas the other group of rats self-administered the same cocaine dose in daily 6 h sessions (LgA) for 22 d (Fig. 1). Twenty-two rats were initially exposed to cocaine self-administration (ShA, n = 11; LgA, n = 11). Over time, three rats had to be excluded because of occlusion of the intravenous catheter (ShA, n = 2; LgA, n = 1). The number of rats included in each experiment is indicated in the figure legends.
Effects of TC on cocaine self-administration in ShA and LgA rats.
After cocaine intake escalation reached a stable level, the effect of intravenous injection of various doses (2.5, 5.0, 7.5, and 10.0 mg/kg/0.5 ml) of the TrkB inhibitor cyclotraxin-B was tested on cocaine self-administration under both ShA and LgA conditions. To allow penetration into the brain, the nontoxic transduction domain of the tat protein from HIV-1 (Gump and Dowdy, 2007) was fused to cyclotraxin-B (for details, see Cazorla et al., 2010), resulting in TC (custom made by NeoPeptide). The ineffective tat peptide lacking the cyclotraxin-B sequence [tat empty (TE)] served as a control (Cazorla et al., 2010). TC and TE were given in a within-subjects Latin-square design, in which 8 test days were separated by drug-free [no tat (NT)] days (Fig. 1). As previously reported, rats received two intravenous injections of TC or TE 60 min apart (Cazorla et al., 2010) and were placed in the self-administration chambers immediately after the second administration.
Effects of TC on progressive ratio responding.
To test the effects of TC on the motivation to work for cocaine, rats were allowed to self-administer cocaine under a progressive ratio (PR) schedule of reinforcement (Hodos, 1961). The number of lever presses required to obtain the next infusion of cocaine exponentially increased according to the following equation: number of responses per infusion = (5 × e(injection number × 0.2)) − 5 (Richardson and Roberts, 1996). When a rat failed to achieve the response requirement within a period of 30 min, the PR session ended and breakpoints were recorded. We chose to test the effect of 7.5 mg/kg TC on PR responding, based on the effect of this dose on FR1 responding in ShA and LgA rats.
Effects of TC on glucose/saccharin self-administration.
To analyze whether TC also altered the self-administration of a nondrug reinforcer, rats were briefly trained (four sessions) to press for the highly palatable reinforcer “supersac” (3% w/v glucose and 0.125% w/v saccharin; Martin-Fardon and Weiss, 2014). Given that the above-mentioned experiments revealed that 7.5 mg/kg TC reduced cocaine self-administration for a period of 90 min (Table 1), the 30 min lasting glucose/saccharin test was performed between 30 and 60 min after the second 7.5 mg/kg TC injection to ensure the best match between behavioral testing and the pharmacokinetic profile of TC.
Table 1.
Effects of TC on absolute number of active and inactive lever presses
| Group | Experiment name | Number of active lever presses | Number of active lever presses during timeout | Number of inactive lever presses |
|---|---|---|---|---|
| A | FR ShA self-administration (0–60 min) | TE: 15.5 ± 2.5 vs TC: 5.3 ± 1.9 | TE: 2.6 ± 0.6 vs TC: 1.5 ± 0.7 | TE: 2.1 ± 0.7 vs TC: 1.3 ± 0.7 |
| F(1,20) = 10.63, p = 0.004 | n.s. | n.s. | ||
| FR LgA self-administration (0–30 min) | TE: 16.0 ± 0.9 vs TC: 8.9 ± 1.6 | TE: 2.2 ± 0.5 vs TC: 1.1 ± 0.5 | TE: 1.4 ± 0.4 vs TC: 1.5 ± 0.6 | |
| F(1,20) = 15.56, p < 0.001 | n.s. | n.s. | ||
| FR LgA self-administration (30–60 min) | TE: 12.5 ± 0.4 vs TC: 7.5 ± 1.5 | TE: 1.2 ± 0.6 vs TC: 0.7 ± 0.3 | TE: 0.1 ± 0.1 vs TC: 0.0 ± 0.0 | |
| F(1,20) = 10.16, p = 0.005 | n.s. | n.s. | ||
| FR LgA self-administration (60–90 min) | TE: 12.3 ± 0.5 vs TC: 8.6 ± 1.6 | TE: 1.5 ± 0.6 vs TC: 0.8 ± 0.7 | TE: 0.0 ± 0.0 vs TC: 0.4 ± 0.4 | |
| F(1,20) = 5.17, p = 0.034 | n.s. | n.s. | ||
| FR LgA self-administration (90–120 min) | TE: 12.0 ± 0.4 vs TC: 10.0 ± 1.3 | TE: 1.0 ± 0.3 vs TC: 0.9 ± 0.5 | TE: 0.1 ± 0.1 vs TC: 0.3 ± 0.3 | |
| n.s. | n.s. | n.s. | ||
| PR self-administration after ShA | TE: 136.7 ± 49.2 vs TC: 25.0 ± 12.4 | TE: 2.7 ± 0.8 vs TC: 2.1 ± 1.0 | TE: 3.3 ± 1.1 vs TC: 3.1 ± 1.1 | |
| F(1,18) = 4.84, p = 0.041 | n.s. | n.s. | ||
| PR self-administration after LgA | TE: 279.4 ± 37.5 vs TC: 150.7 ± 44.6 | TE: 3.1 ± 0.6 vs TC: 1.9 ± 0.9 | TE: 2.9 ± 1.0 vs TC: 3.2 ± 1.0 | |
| F(1,20) = 4.88, p = 0.039 | n.s. | n.s. | ||
| B | Glucose/saccharin self-administration after ShA | TE: 35.9 ± 4.6 vs TC: 35.0 ± 4.6 | TE: 3.4 ± 0.8 vs TC: 2.5 ± 1.1 | TE: 2.7 ± 0.5 vs TC: 1.9 ± 0.5 |
| n.s. | n.s. | n.s. | ||
| Glucose/saccharin self-administration after LgA | TE: 35.9 ± 3.1 vs TC: 35.9 ± 3.4 | TE: 3.5 ± 0.9 vs TC: 2.4 ± 0.9 | TE: 2.2 ± 0.5 vs TC: 2.5 ± 0.7 | |
| n.s. | n.s. | n.s. | ||
| C | Reinstatement of self-administration after ShA | TE: 10.6 ± 2.1 vs TC: 5.2 ± 0.9 | TE: 1.1 ± 0.4 vs TC: 0.2 ± 0.1 | TE: 2.3 ± 0.5 vs TC: 1.4 ± 1.3 |
| F(1,16) = 5.23, p = 0.036 | F(1,16) = 3.94, p = 0.065 | n.s. | ||
| Reinstatement of self-administration after LgA | TE: 14.5 ± 2.1 vs TC: 8.0 ± 1.4 | TE: 4.6 ± 0.7 vs TC: 2.0 ± 0.7 | TE: 2.2 ± 0.5 vs TC: 1.5 ± 1.2 | |
| F(1,18) = 6.42, p = 0.021 | F(1,18) = 6.18, p = 0.023 | n.s. |
Effects of 7.5 mg/kg TC on the number of active and inactive lever presses, as well as active lever pressing during the timeout period. TC reduced the number of active lever presses to obtain cocaine without affecting either inactive lever pressing or the number of active lever presses to obtain a glucose/saccharin solution. Values shown are mean number of lever presses ± SEM following either TE or TC pretreatment. F and p values were obtained using a one-way ANOVA with the factor treatment.
Re-escalation and extinction.
Following self-administration of the glucose/saccharin solution, cocaine intake was reduced by ∼20% (see Results). Rats were re-exposed to cocaine with ShA and LgA until cocaine intake during these re-escalation sessions no longer differed from the cocaine intake observed on experimental day 35 (8 d; Fig. 1). Cocaine was then replaced with saline solution, and extinction sessions of 4 h were performed daily until the rats received no more than three saline infusions per hour.
Effects of TC on reinstatement of cocaine intake.
Following extinction, rats were again given access to cocaine (0.5 mg/kg/infusion) self-administration for four sessions of 1 h duration, during which the reinstatement of cocaine intake was triggered by a single manual intravenous infusion of cocaine. The effect of TC or TE (7.5 mg/kg) pretreatment was tested.
Effects of TC on BDNF system-associated proteins.
A separate cohort of 25 rats was subjected to twelve 1 h sessions of cocaine self-administration followed by twenty-two 6 h sessions, as described above. An additional cocaine self-administration session was conducted, before which rats were pretreated with TC (n = 13) or TE (n = 12), as described above. Another control group of rats (n = 10) underwent surgery, but had no access to cocaine and was pretreated with TE before being killed. These animals were not exposed to saline self-administration because rats do not lever press for this nonrewarding solution. However, rats were handled and their catheters were flushed on a daily basis. All rats were killed by decapitation 60 min after the start of the final self-administration session (i.e., 60 min after the second TC/TE injection). Bilateral punches of nucleus accumbens and ventral tegmental area, and a medial punch of prefrontal cortex were collected from freshly dissected brain sections of 2 mm using a 1.2-mm-diameter needle (Verheij et al., 2008). Western blot analysis was conducted to measure the levels of total and phosphorylated TrkB, Akt, and ERK, as previously described by Giannotti et al. (2014). Briefly, punched brain regions were homogenized by sonication using a cold buffer containing 0.32 m sucrose, 1 mm HEPES solution, 0.1 mm EGTA, and 0.1 mm PMSF, pH 7.4, in the presence of a complete set of protease inhibitors and a phosphatase inhibitor cocktail. Equal amounts of protein were measured under reducing conditions on the criterion TGX precast gels (Bio-Rad Laboratories) and then were electrophoretically transferred onto polyvinylidene difluoride membranes (GE Healthcare). Blots were blocked for 1 h at room temperature with 10% nonfat dry milk in TBS plus 0.1% Tween-20 buffer, incubated with antibodies against the phosphorylated forms of the proteins, and then stripped and reprobed with the antibodies against the corresponding total proteins. The following primary antibodies were used: anti-phospho-TrkB Y706 (1:1000; Santa Cruz Biotechnology); anti-TrkB (1:750; Santa Cruz Biotechnology); anti-phospho-ERK1 T202/Y204 (1:1000; Cell Signaling Technology); anti-total ERK1 (1:5000; Santa Cruz Biotechnology); anti-phospho-Akt S473 (1:1000; Cell Signaling Technology); anti-total Akt (1:1000; Cell Signaling Technology); and anti β-actin (1:10,000; Sigma-Aldrich). Results were standardized using β-actin as the control protein, which was detected by evaluating the band density at 43 kDa. Immunocomplexes were visualized by chemiluminescence using the Chemidoc MP Imaging System (Bio-Rad Laboratories).
Data analysis.
Data are expressed as the mean ± SEM. The effects of TC on cocaine or glucose/saccharin self-administration were analyzed using a two-way or one-way ANOVA with a correction for repeated measures when required. The effects of TC on cocaine-induced protein regulation were analyzed using a one-way ANOVA. Post hoc comparisons were performed by means of a Student's t test or least significant difference test, depending on the number of experimental groups to be compared. Prism 6.0 (GraphPad) was used to analyze all the data.
Results
Systemic injection of a brain-penetrant TrkB antagonist reduces responding and motivation for cocaine self-administration
After training, rats responded 12.3 ± 1.6 times/h for cocaine (Fig. 2A, session 12). LgA, but not ShA, cocaine self-administration resulted in an escalation of the drug intake over sessions [Fig. 2A; access × session interaction (two-way ANOVA for repeated measures): F(22,440) = 25.4, p < 0.001; session effect LgA (one-way ANOVA for repeated measures): F(22,220) = 10.74, p < 0.001, session effect ShA (one-way ANOVA for repeated measures): p = n.s.]. In contrast to ShA cocaine self-administration, LgA cocaine self-administration resulted in an escalation of the drug intake during the first hour [Fig. 2B; access × session interaction (two-way ANOVA): F(1,40) = 10.5, p = 0.002; session effect LgA (one-way ANOVA): F(1,20) = 19.91, p < 0.001; session effect ShA (one-way ANOVA): p = n.s.]. In addition to the differences in cocaine intake under an FR1 schedule of reinforcement (Fig. 2A,B), cocaine intake under a PR schedule of reinforcement was also found to be larger in LgA than ShA rats [Fig. 2C; access effect (one-way ANOVA): F(1,20) = 4.75, p = 0.042].
Figure 2.
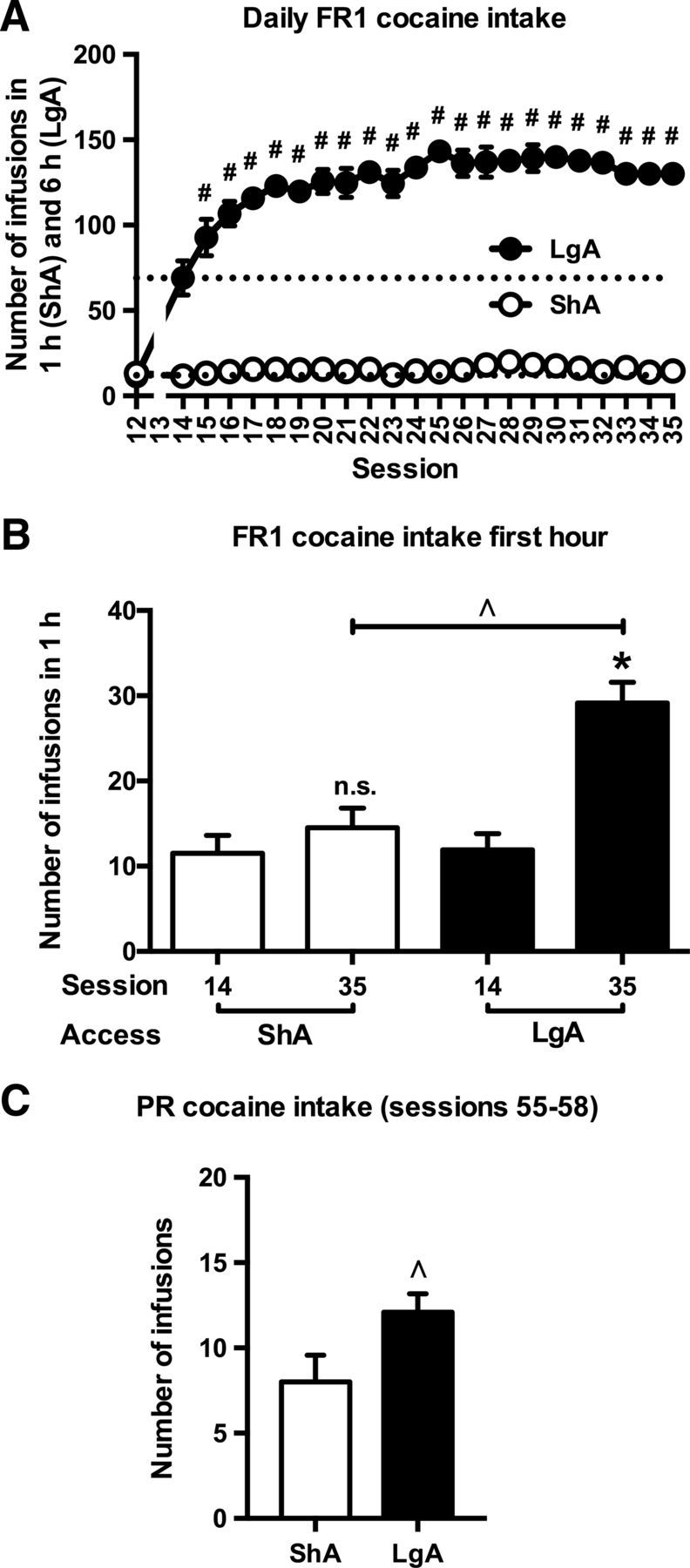
A, Cocaine self-administration in LgA rats, but not ShA rats increased over time. #Significant increase vs session 14 (t test, p < 0.05). B, C, The initial drug loading (B) and the motivation to work for cocaine (C) were increased in LgA rats compared with ShA rats. ∧Significant difference (one-way ANOVA, p < 0.05) compared with ShA (B, C). *Significant difference (one-way ANOVA, p < 0.05) compared with session 14 (B); n.s., No change compared with session 14 (B). ShA, n = 11; LgA, n = 11.
The control peptide of TC, labeled TE, did not alter cocaine self-administration under both ShA [Fig. 3A; treatment effect and treatment × session interaction (two-way ANOVA for repeated measures): p = n.s.] and LgA [Fig. 3B; treatment effect and treatment × session interaction (two-way ANOVA for repeated measures): p = n.s.] conditions. The TrkB inhibitor TC dose-dependently reduced cocaine intake under both ShA [Fig. 3C; dose effect (one-way ANOVA for repeated measures): F(4,40) = 23.5, p < 0.001] and LgA [Fig. 3D; dose effect: F(4,40) = 7.29, p < 0.001, dose × time interaction (two-way ANOVA for repeated measures): F(20,200) = 2.88, p < 0.001] conditions. The intermediate dose of 7.5 mg/kg TC decreased PR responding in both ShA [Fig. 3E; treatment effect (one-way ANOVA): F(1,18) = 17.60, p < 0.001] and LgA [Fig. 3F; treatment effect (one-way ANOVA): F(1,20) = 7.33, p = 0.014] animals. By setting the maximum duration to reach the next PR criterion to 30 min (see Materials and Methods), TC-treated rats reached their breakpoints within the 90 min interval during which the TrkB antagonist was effective (ShA, 57 ± 8 min; LgA, 71 ± 8 min).
Figure 3.
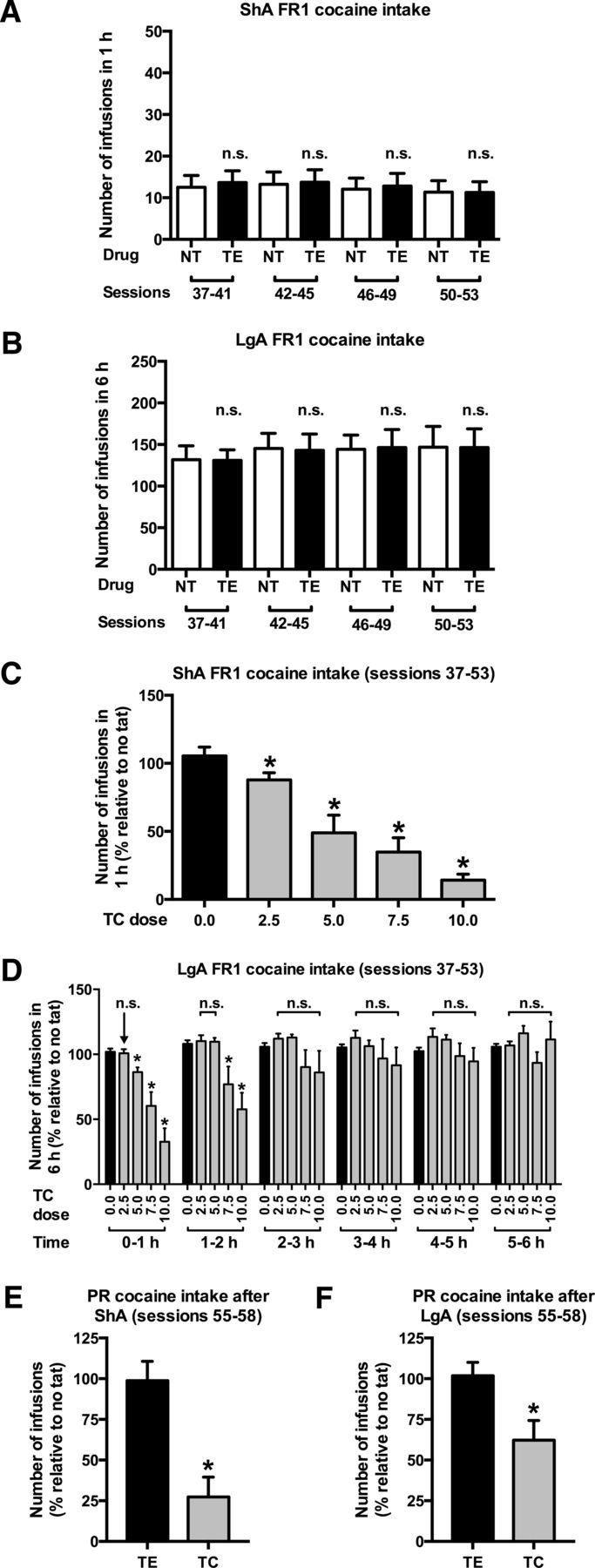
A, B, Intravenous administration of the control peptide TE did not alter cocaine self-administration under an FR1 schedule of reinforcement in both ShA (A) and LgA (B) rats. C, D, Systemic administration of the TrkB antagonist TC dose-dependently reduced ShA (C) and LgA (D) cocaine intake. E, F, The dose of 7.5 mg/kg TC also reduced the motivation to work for cocaine under a PR schedule of reinforcement in both ShA (E) and LgA (F) rats. *Significant decrease (LSD, p < 0.05) vs TC 0.0 mg/kg (C, D), or significant decrease (one-way ANOVA, p < 0.05) vs the control peptide TE (E, F); n.s., No change compared with NT days (A, B) or TC 0.0 mg/kg (D). C–F, Values are normalized to preceding NT sessions. ShA: n = 11 (A–C) or n = 10 (E); LgA: n = 11 (B, D, F).
Systemic injection of a brain-penetrant TrkB antagonist does not impact responding for a natural reinforcer
After the cocaine self-administration experiments, rats were briefly trained to press for a glucose/saccharin solution [Fig. 4A; session effect (two-way ANOVA for repeated measures): F(3,57) = 26.1, p < 0.001]. The oral consumption of this highly palatable reinforcer was not different between rats with an ShA and LgA history of cocaine [Fig. 4A; access effect and access × session interaction (two-way ANOVA for repeated measures): p = n.s.]. TC did not change the responding for glucose/saccharin [Fig. 4B; treatment effect and treatment × access interaction (two-way ANOVA): p = n.s.].
Figure 4.

A, Similar self-administration of a sweet solution in rats with a history of ShA and LgA to cocaine. #Significant increase vs session 60 (t test, p < 0.05). B, Intravenous TC did not alter glucose/saccharin intake in these animals. n.s., No change vs control peptide (TE). B, Values are normalized to preceding NT sessions. ShA, n = 10; LgA, n = 11.
Systemic injection of a brain-penetrant TrkB antagonist reduces cocaine-induced reinstatement of drug-taking behavior
To analyze the effects of TC on drug-induced reinstatement of cocaine intake, rats were first re-escalated to previous cocaine intake levels [Fig. 5A; session effect: F(7,119) = 5.56, p < 0.001; access × session interaction (two-way ANOVA for repeated measures): F(7,119) = 6.50, p < 0.001], followed by an extinction period. LgA rats returned to less than 3 infusions per h later than ShA rats [Fig. 5B; session effect: F(12,204) = 12.2, p < 0.001; access × session interaction (two-way ANOVA for repeated measures): F(12,204) = 4.19, p < 0.001].
Figure 5.

A, Re-escalation of cocaine intake in rats with a history of LgA, but not in rats with a history of ShA, to cocaine. #Significant increase vs session 70 (t test, p < 0.05). B, Slower extinction of responding in rats with a history of LgA cocaine self-administration. #Significant increase (t test, p < 0.05) vs average of sessions 89–91 (= 10 infusions in 4 h = less than 3 infusions per h). ShA, n = 9; LgA, n = 10.
As expected, cocaine-induced reinstatement of drug taking was larger in LgA than in ShA rats [Fig. 6A; reinstatement × access interaction (two-way ANOVA): F(1,34) = 4.31, p = 0.046; reinstatement effect of ShA (one-way ANOVA): F(1,16) = 6.71, p = 0.020; reinstatement effect of LgA (one-way ANOVA): F(1,18) = 17.82, p = 0.002]. The intermediate dose of 7.5 mg/kg TC reduced the reinstatement of cocaine intake in both ShA rats [Fig. 6B; treatment effect (one-way ANOVA): F(1,16) = 7.01, p = 0.018] and LgA rats [Fig. 6C; treatment effect (one-way ANOVA): F(1,18) = 5.39, p = 0.032]. The latency to the first cocaine self-infusion was shorter in rats primed with cocaine compared with rats primed with saline [Fig. 6D; reinstatement effect (two-way ANOVA): F(1,34) = 58.60, p < 0.001; reinstatement × access interaction (two-way ANOVA): p = n.s.] and the latency of the first self-infusion postcocaine prime was shorter in LgA than in ShA rats [Fig. 6D; access effect (one-way ANOVA): F(1,17) = 6.29, p = 0.023]. Interestingly, TC increased this latency [Fig. 6E (ShA); treatment effect (one-way ANOVA): F(1,16) = 47.86, p < 0.001; Fig. 6F (LgA); treatment effect (one-way ANOVA): F(1,18) = 5.20, p = 0.035], and did so to a greater extent in LgA rats than in ShA rats [Fig. 6, compare E, F; treatment × access interaction (two-way ANOVA): F(1,34) = 3.90, p = 0.054].
Figure 6.
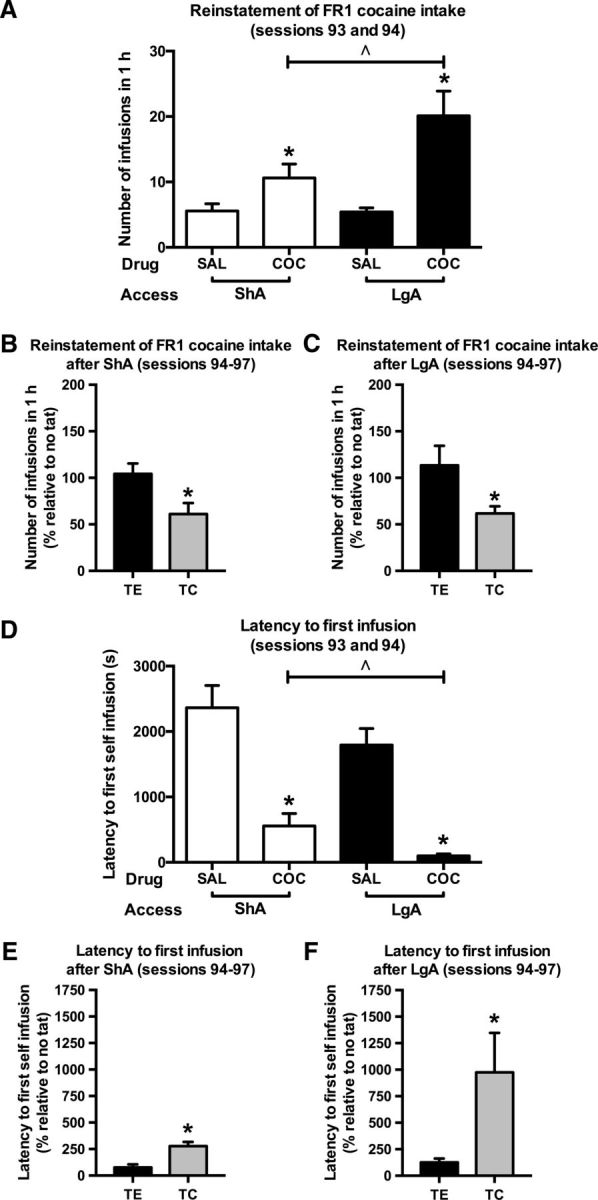
A, Greater drug-induced reinstatement of cocaine (COC) intake in LgA compared with ShA rats. B, C, Intravenous TC reduced drug-induced reinstatement of cocaine intake in both ShA (B) and LgA (C) rats. D–F, The latency to the first self-infusion was shorter in rats primed with a cocaine infusion than in rats primed with a saline infusion (D), and TC increased this latency more strongly in LgA than ShA rats (E vs F). *Significant difference (one-way ANOVA, p < 0.05) compared with saline (SAL; A, D) or control peptide TE (B, C, E, F). ∧Significant difference (one-way ANOVA, p < 0.05) compared with ShA (A, D). In B, C, E, and F, values are normalized to preceding NT sessions. ShA, n = 9; LgA, n = 10. Note: cocaine intake remained stable throughout the four reinstatement sessions (B, C, TE bar is not different from 100%; one-sample t test, p = n.s.), which suggest that the motivational drive for responding was not substantially affected by session repetition.
Effects of a brain-penetrant TrkB antagonist on active and inactive lever presses and timeout responses
The above-mentioned effect of TC on cocaine self-administration under FR1 and PR schedules in both ShA and LgA rats was driven by a selective decrease in active lever presses (Table 1, group A, left column), which was not accompanied by changes in timeout responding (Table 1, group A, middle column) or inactive lever presses (Table 1, group A, right column). TC did not change active (Table 1, group B, left column), timeout (Table 1, group B, middle column), or inactive (Table 1, group B, right column) responses for glucose/saccharin in either group of animals. Finally, TC reduced the number of active presses (Table 1, group C, left column) and timeout presses (Table 1, group C, middle column) during reinstatement, which was not accompanied by a change in inactive lever presses (Table 1, group C, right column).
Systemic injection of a brain-penetrant TrkB antagonist reverses cocaine self-administration-induced changes in TrkB signaling in the nucleus accumbens and prefrontal cortex
A separate cohort of LgA rats pretreated with TC was used to analyze TrkB signaling in the nucleus accumbens, medial prefrontal cortex, and ventral tegmental area. Cocaine intake in this second cohort was comparable to the intake of the first cohort used for behavioral characterization [first hour intake: 28 ± 5.5 infusions (second cohort) vs 29 ± 1.5 infusions (first cohort); cohort effect (one-way ANOVA): p = n.s.]. In addition, systemic administration of 7.5 mg/kg TC reduced cocaine intake in this second cohort [treatment effect (one-way ANOVA): F(1,23) = 23.44, p < 0.001] to the same extent as in the first cohort [reduction during first hour of the session: 60 ± 7.0% (second cohort) vs 40 ± 10.6% (first cohort); cohort effect (one-way ANOVA): p = n.s.]. This TC-induced reduction in cocaine self-administration was again driven by a selective decrease in active lever presses [treatment effect (one-way ANOVA): F(1,23) = 9.63, p = 0.005], whereas TC did not affect timeout responding or inactive lever pressing [treatment effect (one-way ANOVA): p = n.s.].
In the nucleus accumbens, cocaine self-administration increased the protein levels of TrkB [Fig. 7A; treatment effect (one-way ANOVA): F(2,32) = 6.99, p = 0.003; cocaine effect (t test): p = 0.006] and pTrkB [Fig. 7B; treatment effect (one-way ANOVA): F(2,32) = 3.93, p = 0.030; cocaine effect (t test): p = 0.030]. It also increased the total and phosphorylated levels of Akt [Fig. 7C; treatment effect (one-way ANOVA): F(2,32) = 11.08, p < 0.001; cocaine effect (t test): p < 0.001; and Fig. 7D; treatment effect (one-way ANOVA): F(2,32) = 4.38, p = 0.021; cocaine effect (t test): p = 0.014], but did not alter ERK (Fig. 7E) and phosphorylated (p) ERK (Fig. 7F) levels [treatment effect (one-way ANOVA): p = n.s.]. Conversely, in the prefrontal cortex, cocaine self-administration reduced pTrkB levels [Fig. 8B; treatment effect (one-way ANOVA): F(2,32) = 3.64, p = 0.038; cocaine effect (t test): p = 0.009] and pAkt levels [Fig. 8D; treatment effect (one-way ANOVA): F(2,32) = 8.49, p = 0.001; cocaine effect (t test): p = 0.003], without affecting TrkB total protein levels [Fig. 8A: treatment effect (one-way ANOVA): F(2,32) = 3.30, p = 0.050; cocaine effect (t test): p = n.s.] and Akt total protein levels [Fig. 8C; treatment effect (one-way ANOVA): p = n.s.]. pERK and ERK were also unaltered in the prefrontal cortex [Fig. 8E,F; treatment effect (one-way ANOVA): p = n.s.].
Figure 7.
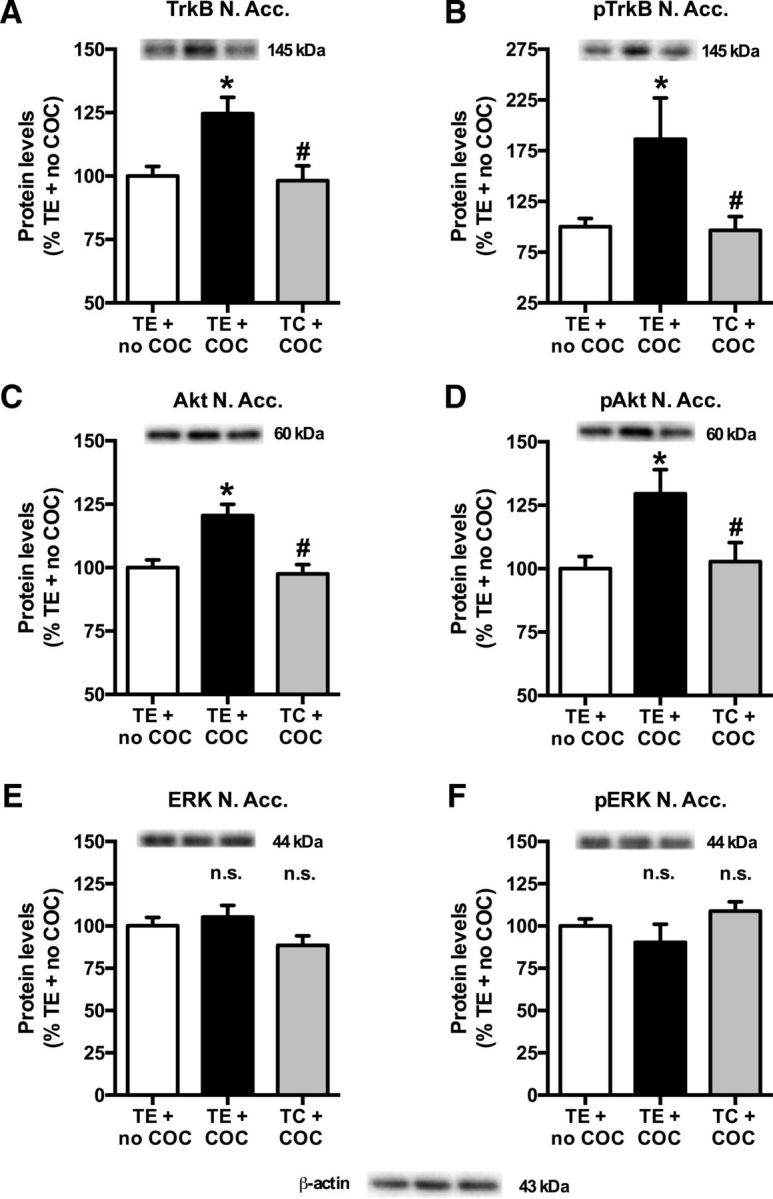
A–F, Cocaine (COC)-induced increase in TrkB (A), pTrkB Y706 (B), Akt (C), and pAkt S473 (D), but not in ERK1 (E) and pERK1 T202/Y204 (F) protein levels in the nucleus accumbens (N. Acc.). *Significant increase (t test, p < 0.05) vs TE + no COC (A–D). TC prevented the cocaine-induced increase in protein levels. #Significant reduction (t test, p < 0.05) vs TE + COC (A–D). n.s., No difference between TE + COC and TE + no COC or no difference between TC + COC and TE + COC (E, F). In each panel, values are normalized to TE + no COC and pictures show representative Western blot bands of the corresponding proteins (left lane, TE + no COC; middle lane, TE + COC; right lane, TC + COC). TE + no COC, n = 10; TE + COC, n = 12; TC + COC, n = 13.
Figure 8.
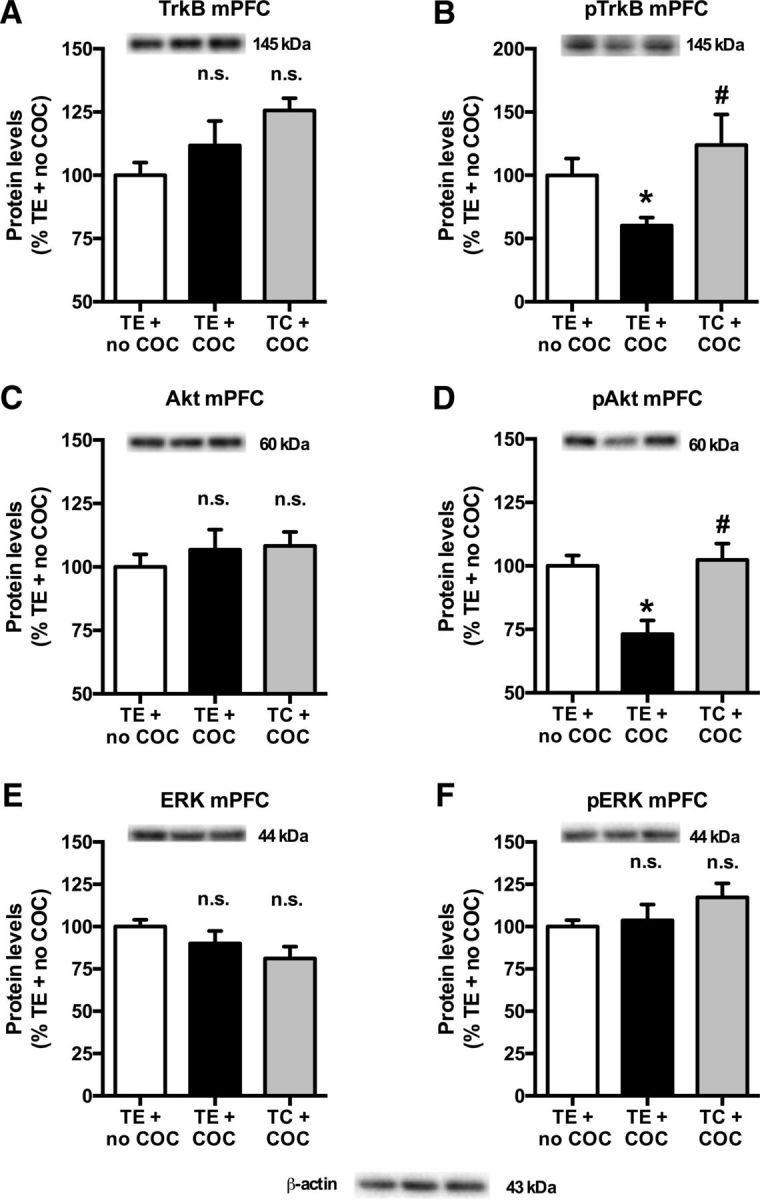
A–F, Cocaine (COC)-induced reduction in pTrkB Y706 (B) and pAkt S473 (D), but not in TrkB (A), Akt (C), ERK1 (E), and pERK1 T202/Y204 (F) protein levels in the medial prefrontal cortex (mPFC). *Significant decrease (t test, p < 0.05) vs TE + no COC (B, D). TC prevented the cocaine-induced decrease in protein levels. #Significant increase (t test, p < 0.05) vs TE + COC (B, D). n.s., No difference between TE + COC and TE + no COC or no difference between TC + COC and TE + COC (A, C, E, F). In each panel, values are normalized to TE + no COC, and pictures show representative Western blot bands of the corresponding proteins (left lane, TE + no COC; middle lane, TE + COC; right lane, TC + COC). TE + no COC, n = 10; TE + COC, n = 12; TC + COC, n = 13.
TC reversed the effects of cocaine self-administration on TrkB, pTrkB, Akt, and pAkt in the nucleus accumbens [TC effect (t test): TrkB (Fig. 7A), p = 0.002; pTrkB (Fig. 7B), p = 0.016; Akt (Fig. 7C), p < 0.001; pAkt (Fig. 7D), p = 0.017], as well as on pTrkB and pAkt in the prefrontal cortex [TC effect (t test): pTrkB (Fig. 8B), p = 0.012; pAkt (Fig. 8D), p < 0.001].
Systemic injection of a brain-penetrant TrkB antagonist does not affect cocaine self-administration-induced TrkB activation in the ventral tegmental area
In the ventral tegmental area, cocaine self-administration increased the protein levels of TrkB [Fig. 9A; treatment effect (one-way ANOVA): F(2,32) = 4.23, p = 0.025; cocaine effect (t test): p = 0.016] and pTrkB [Fig. 9B; treatment effect (one-way ANOVA): F(2,32) = 7.59, p = 0.003; cocaine effect (t test): p = 0.005], and these effects were not altered by pretreatment with TC [TC effect (t test): TrkB (Fig. 9A) and pTrkB (Fig. 9B): p = n.s.].
Figure 9.

A, B, No effect of TC on the cocaine (COC)-induced increase in TrkB (A) and pTrkB Y706 (B) protein levels in the ventral tegmental area (VTA). *Significant increase (t test, p < 0.05) vs TE + no COC (A, B). n.s., No difference between TC + COC and TE + COC (A, B). In each panel, values are normalized to TE + no COC, and pictures show representative Western blot bands of the corresponding proteins (left lane, TE + no COC; middle lane, TE + COC; right lane, TC + COC). TE + no COC, n = 10; TE + COC, n = 12; TC + COC, n = 13.
Discussion
Cocaine self-administration
Consistent with previous studies, cocaine self-administration under LgA, but not ShA, conditions resulted in an escalation of drug intake over time (Ahmed and Koob, 1998; Orio et al., 2009; Wee et al., 2012). Compared with ShA animals, LgA animals showed increased motivation for cocaine intake, as indicated by increased breakpoints during a PR schedule of reinforcement. After 10 d of abstinence, the intake of cocaine re-escalated only in animals that had LgA to cocaine. During extinction, responding diminished more slowly in LgA than in ShA rats. During reinstatement, the latency to first self-infusion was shorter and subsequent cocaine intake was larger in rats that had LgA to the drug. The finding that ShA and LgA rats did not differ in their glucose/saccharin intake corroborates the notion that the mechanisms mediating self-administration of palatable solutions and cocaine are not identical (Vendruscolo et al., 2010).
Tat-cyclotraxin-B reduces cocaine self-administration
Systemic administration of the brain-penetrant TrkB antagonist TC dose-dependently decreased cocaine self-administration in both ShA and LgA rats. TC also effectively reduced the motivation to work for cocaine as well as the drug-induced reinstatement of cocaine taking in both groups of animals. In contrast to repeated postsession intra-accumbal infusions of BDNF antiserum that produced a long-lasting reduction in various measures of cocaine dependence (Graham et al., 2007), the acute presession systemic injections of TC decreased cocaine self-administration for <2 h. Differential half-lives of systemically administered TC and locally infused BDNF antiserum may explain this discrepancy. In addition, an action of TC outside of the nucleus accumbens may also shorten the duration of its inhibitory effect on cocaine self-administration. Long-term treatment with TC, before or after the cocaine self-administration sessions, may lead to longer-lasting behavioral effects. The finding that rats repeatedly treated with the control peptide TE showed cocaine self-administration levels similar to those in NT-treated rats suggests that the fusion peptide does not produce major side effects on operant behavior. In addition, TC did not alter the intake of a highly rewarding sweet solution, demonstrating that the effect of TC on self-administration is specific for cocaine reward. These results support the hypothesis that BDNF signaling in the mesolimbic dopamine pathway is not recruited by sweet reinforcers (Grimm et al., 2003; Graham et al., 2009), and further indicate that TC-induced inhibition of cocaine self-administration cannot be attributed to a reduction of motor performance. Altogether, the data suggest that BDNF–TrkB signaling is not engaged upon moderate activation of the brain reward system by a palatable sweet solution, but is recruited upon excessive activity of this system by cocaine. In addition, our data suggest that BDNF–TrkB signaling is activated by cocaine before the transition into dependence because we observed similar effects of TC in both ShA and LgA rats.
Brain region-specific changes in TrkB signaling after cocaine self-administration
At the molecular level, the increases in TrkB signaling that we observed in the nucleus accumbens and ventral tegmental area of cocaine self-administering rats are consistent with the increases in BDNF protein levels previously reported in these brain regions following cocaine self-administration (Grimm et al., 2003; Pu et al., 2006; Graham et al., 2007, 2009; Schmidt et al., 2012; Fumagalli et al., 2013; Li et al., 2013). The decrease in TrkB signaling that we observed in the prefrontal cortex is more surprising given that the BDNF protein level in this brain region was previously reported to be increased by cocaine (Fumagalli et al., 2007, 2013; Lu et al., 2010; McGinty et al., 2010; Sadri-Vakili et al., 2010). However, in these previous studies, BDNF upregulation in the prefrontal cortex was detected several hours to days after the last cocaine exposure, while we collected brain samples immediately after the last cocaine self-administration session. In addition, the decrease in TrkB signaling we observed could represent a postsynaptic adaptation independent of BDNF synthesis and release.
Tat-cyclotraxin-B normalizes cocaine self-administration-induced changes in TrkB signaling
We found that pretreatment with TC reverses the effects of cocaine self-administration on TrkB signaling in the nucleus accumbens and prefrontal cortex. Cyclotraxin-B is a potent and specific inhibitor of TrkB, and the TC administration procedure used in the present study was previously shown to reduce the phosphorylation of central TrkB receptors by ∼50% (Cazorla et al., 2010). Combining our results with the existing literature on the effects of BDNF system modulation in the mesocorticolimbic dopaminergic pathway (Horger et al., 1999; Lu et al., 2004, 2009; Graham et al., 2007, 2009; Bahi et al., 2008; Li et al., 2013), we propose that the reduction in TrkB signaling induced by TC in the nucleus accumbens drives the reduction in cocaine self-administration. We further hypothesize that the reversal of the cocaine-induced decrease in cortical TrkB signaling by TC indirectly results from the blunting of the cocaine-induced TrkB activation in the nucleus accumbens. According to this framework, the inhibitory effect of LgA cocaine self-administration on TrkB signaling in the prefrontal cortex would occur secondary to the increased signaling observed in the nucleus accumbens. Such antagonistic feedback from the nucleus accumbens to the prefrontal cortex could be relayed via the ventral tegmental area, which receives inhibitory GABAergic inputs from the nucleus accumbens and sends dopaminergic projections to the prefrontal cortex (Russo and Nestler, 2013), or via cortico–striatal–pallidal–thalamic loops that provide a basal ganglia–cortical feedback (Haber et al., 2000). Reduction of cocaine self-administration by TC may ultimately result from the combined reversal of cocaine effects on TrkB signaling in the nucleus accumbens and prefrontal cortex, as both reduced TrkB signaling in the nucleus accumbens and increased TrkB signaling in the prefrontal cortex have previously been found to reduce cocaine seeking (Horger et al., 1999; Lu et al., 2004, 2009; Berglind et al., 2007, 2009; Graham et al., 2007, 2009; Bahi et al., 2008; Sadri-Vakili et al., 2010; Whitfield et al., 2011; Li et al., 2013). In both brain regions, changes in TrkB signaling were accompanied by similar changes in Akt, but not in ERK, indicating that the behavioral effects of cocaine and TC are intracellular pathway specific.
An alternative interpretation of our molecular data would be that TC-induced changes in TrkB/Akt signaling result from, rather than drive, the reduced cocaine intake. However, the fact that TC did not affect TrkB and pTrkB levels in the ventral tegmental area of cocaine self-administering rats does not support this interpretation. The latter finding suggests that the effects of TC on TrkB signaling result from a brain region-specific action of TC, rather than being an indirect consequence of lower levels of cocaine exposure throughout the brain. Although the mechanism underlying this regional specificity of TC activity remains to be determined, it is worth noting that ANA-12, another brain-penetrant TrkB antagonist that, similar to TC, displays anxiolytic-like properties, exerts more potent TrkB inhibition in striatal areas than in cortical areas (Cazorla et al., 2011).
Perspectives
Given that dopaminergic signaling in the nucleus accumbens and prefrontal cortex plays a key role in the positive reinforcing properties of cocaine (Everitt and Robbins, 2005; Koob and Volkow, 2010; George et al., 2012), the inhibitory effect of TC on the cocaine intake of ShA rats probably results from a disruption of the complex interplay between BDNF and dopamine signaling in these dopamine-innervated regions. In addition, an action of TC on the glutamatergic neurons regulating accumbal dopamine release may also contribute (Berglind et al., 2009). The inhibitory effect of TC on the cocaine intake of LgA rats raises the possibility that incentive salience driven by negative reinforcement also involves BDNF signaling. Accordingly, genetic deletion of BDNF in ventral tegmental area dopaminergic neurons reduces social aversion elicited by repeated aggression (Berton et al., 2006) and systemic administration of the brain-penetrant TrkB inhibitors TC and ANA-12 exerts anxiolytic-like and antidepressant-like properties in naive mice (Cazorla et al., 2010, 2011). Therefore, these findings suggest that the effects of TC on relieving the negative emotional state associated with withdrawal may also contribute to the reduction in cocaine intake in LgA rats. Altogether, our study validates systemic TrkB antagonism as a potential new strategy to curb cocaine use disorders.
Footnotes
This research was supported by the Zardi-Gori Foundation (to F.F.) and National Institutes of Health Grants AA020913 and AA024198 (to C.C.). M.M.M.V., J.R.H., G.F.K., and C.C. were supported by a joint program of the Netherlands Organization for Scientific Research and U.S. National Institute on Drug Abuse (NIDA) Project 31180005. M.M.M.V. was supported by a European College of Neuropsychopharmacology Research Grant for Young Scientists and a NIDA INVEST Drug Abuse Research Fellowship. L.F.V. and G.F.K. were supported by the NIDA Intramural Research Program. We thank Kiki Rink and Famke Ouwerkerk (Department of Cognitive Neuroscience, Radboud University Nijmegen, Nijmegen, The Netherlands) for technical assistance.
The authors declare no competing financial interests.
References
- Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- Altar CA, Boylan CB, Jackson C, Hershenson S, Miller J, Wiegand SJ, Lindsay RM, Hyman C. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci U S A. 1992;89:11347–11351. doi: 10.1073/pnas.89.23.11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi A, Boyer F, Chandrasekar V, Dreyer JL. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology (Berl) 2008;199:169–182. doi: 10.1007/s00213-008-1164-1. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Jouvenceau A, Rose C, Guilloux JP, Pilon C, Dranovsky A, Prémont J. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corominas M, Roncero C, Ribases M, Castells X, Casas M. Brain-derived neurotrophic factor and its intracellular signaling pathways in cocaine addiction. Neuropsychobiology. 2007;55:2–13. doi: 10.1159/000103570. [DOI] [PubMed] [Google Scholar]
- Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35:47–56. doi: 10.1016/j.tins.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Filip M, Faron-Górecka A, Kuśmider M, Golda A, Frankowska M, Dziedzicka-Wasylewska M. Alterations in BDNF and trkB mRNAs following acute or sensitizing cocaine treatments and withdrawal. Brain Res. 2006;1071:218–225. doi: 10.1016/j.brainres.2005.11.099. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Repeated exposure to cocaine differently modulates BDNF mRNA and protein levels in rat striatum and prefrontal cortex. Eur J Neurosci. 2007;26:2756–2763. doi: 10.1111/j.1460-9568.2007.05918.x. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Caffino L, Racagni G, Riva MA. Repeated stress prevents cocaine-induced activation of BDNF signaling in rat prefrontal cortex. Eur Neuropsychopharmacol. 2009;19:402–408. doi: 10.1016/j.euroneuro.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Moro F, Caffino L, Orrù A, Cassina C, Giannotti G, Di Clemente A, Racagni G, Riva MA, Cervo L. Region-specific effects on BDNF expression after contingent or non-contingent cocaine i.v. self-administration in rats. Int J Neuropsychopharmacol. 2013;16:913–918. doi: 10.1017/S146114571200096X. [DOI] [PubMed] [Google Scholar]
- George O, Le Moal M, Koob GF. Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol Behav. 2012;106:58–64. doi: 10.1016/j.physbeh.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotti G, Caffino L, Calabrese F, Racagni G, Riva MA, Fumagalli F. Prolonged abstinence from developmental cocaine exposure dysregulates BDNF and its signaling network in the medial prefrontal cortex of adult rats. Int J Neuropsychopharmacol. 2014;17:625–634. doi: 10.1017/S1461145713001454. [DOI] [PubMed] [Google Scholar]
- Goggi J, Pullar IA, Carney SL, Bradford HF. Signalling pathways involved in the short-term potentiation of dopamine release by BDNF. Brain Res. 2003;968:156–161. doi: 10.1016/S0006-8993(03)02234-0. [DOI] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Graham DL, Krishnan V, Larson EB, Graham A, Edwards S, Bachtell RK, Simmons D, Gent LM, Berton O, Bolanos CA, DiLeone RJ, Parada LF, Nestler EJ, Self DW. Tropomyosin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biol Psychiatry. 2009;65:696–701. doi: 10.1016/j.biopsych.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. 2001;411:86–89. doi: 10.1038/35075076. [DOI] [PubMed] [Google Scholar]
- Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med. 2007;13:443–448. doi: 10.1016/j.molmed.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–2382. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodos W. Progressive ratio as a measure of reward strength. Science. 1961;134:943–944. doi: 10.1126/science.134.3483.943. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yeh CM, Wu MY, Chang AY, Chan JY, Chan SH, Hsu KS. Cocaine withdrawal impairs metabotropic glutamate receptor-dependent long-term depression in the nucleus accumbens. J Neurosci. 2011;31:4194–4203. doi: 10.1523/JNEUROSCI.5239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Diaz J, Sokoloff P. A single cocaine exposure increases BDNF and D3 receptor expression: implications for drug-conditioning. Neuroreport. 2005;16:175–178. doi: 10.1097/00001756-200502080-00022. [DOI] [PubMed] [Google Scholar]
- Li X, DeJoseph MR, Urban JH, Bahi A, Dreyer JL, Meredith GE, Ford KA, Ferrario CR, Loweth JA, Wolf ME. Different roles of BDNF in nucleus accumbens core versus shell during the incubation of cue-induced cocaine craving and its long-term maintenance. J Neurosci. 2013;33:1130–1142. doi: 10.1523/JNEUROSCI.3082-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Wang X, Wu P, Xu C, Zhao M, Morales M, Harvey BK, Hoffer BJ, Shaham Y. Role of ventral tegmental area glial cell line-derived neurotrophic factor in incubation of cocaine craving. Biol Psychiatry. 2009;66:137–145. doi: 10.1016/j.biopsych.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fardon R, Weiss F. N-(2-methyl-6-benzoxazolyl)-N′-1,5-naphthyridin-4-yl urea (SB334867), a hypocretin receptor-1 antagonist, preferentially prevents ethanol seeking: comparison with natural reward seeking. Addict Biol. 2014;19:233–236. doi: 10.1111/j.1369-1600.2012.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Iverson MT, Todd KG, Altar CA. Brain-derived neurotrophic factor and neurotrophin-3 activate striatal dopamine and serotonin metabolism and related behaviors: interactions with amphetamine. J Neurosci. 1994;14:1262–1270. doi: 10.1523/JNEUROSCI.14-03-01262.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DM, Brown AN, Bhide PG. Regulation of BDNF expression by cocaine. Yale J Biol Med. 2012;85:437–446. [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio L, Edwards S, George O, Parsons LH, Koob GF. A role for the endocannabinoid system in the increased motivation for cocaine in extended-access conditions. J Neurosci. 2009;29:4846–4857. doi: 10.1523/JNEUROSCI.0563-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Richardson NR, Roberts DC. Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods. 1996;66:1–11. doi: 10.1016/0165-0270(95)00153-0. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Kumaresan V, Schmidt HD, Famous KR, Chawla P, Vassoler FM, Overland RP, Xia E, Bass CE, Terwilliger EF, Pierce RC, Cha JH. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Sangrey GR, Darnell SB, Schassburger RL, Cha JH, Pierce RC, Sadri-Vakili G. Increased brain-derived neurotrophic factor (BDNF) expression in the ventral tegmental area during cocaine abstinence is associated with increased histone acetylation at BDNF exon I-containing promoters. J Neurochem. 2012;120:202–209. doi: 10.1111/j.1471-4159.2011.07571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo LF, Gueye AB, Darnaudéry M, Ahmed SH, Cador M. Sugar overconsumption during adolescence selectively alters motivation and reward function in adult rats. PLoS One. 2010;5:e9296. doi: 10.1371/journal.pone.0009296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij MM, de Mulder EL, De Leonibus E, van Loo KM, Cools AR. Rats that differentially respond to cocaine differ in their dopaminergic storage capacity of the nucleus accumbens. J Neurochem. 2008;105:2122–2133. doi: 10.1111/j.1471-4159.2008.05323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Specio SE, Koob GF. Effects of dose and session duration on cocaine self-administration in rats. J Pharmacol Exp Ther. 2007;320:1134–1143. doi: 10.1124/jpet.106.113340. [DOI] [PubMed] [Google Scholar]
- Wee S, Vendruscolo LF, Misra KK, Schlosburg JE, Koob GF. A combination of buprenorphine and naltrexone blocks compulsive cocaine intake in rodents without producing dependence. Sci Transl Med. 2012;4:146ra110. doi: 10.1126/scitranslmed.3003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield TW, Jr, Shi X, Sun WL, McGinty JF. The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci. 2011;31:834–842. doi: 10.1523/JNEUROSCI.4986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]