

Abstract
Cancer immunotherapy has had a major impact on the established paradigms of drug development and clinical trial research. The innovative mechanism of action of these compounds has resulted in new patterns of response and safety profiles, which pose challenges for the classical trial methodology. In this review we report on the search for the maximum tolerated dose, the recommended phase II dose and the appropriate target population in phase I trials. We provide some statistical considerations on the choice of endpoints for phase II and III trials and the limitations of frequently used trial designs in the presence of a delayed treatment effect, which may be induced by the immune modulating effect of the checkpoint inhibitors. We summarize the currently available data on the safety profile of these new compounds, which can guide protocol safety recommendations. Finally, we report on the current evidence of biomarker development.
Keywords: Immunotherapy, checkpoint inhibitors, immune-related responses, immune-related adverse effects, biomarkers, delayed treatment effect
Introduction
Immunotherapy has been a major focus area for cancer treatment, prompting an exciting paradigm shift in oncology from targeting the cancer cell to targeting immune cells. By enhancing the innate powers of the immune system to fight cancer cells, it represents the most promising new cancer treatment approach since the development of the first chemotherapy drugs and, more recently, targeted treatments.
The new generation of immunotherapeutic agents corresponds to antibodies that block specific immune checkpoint compounds, such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1) and its ligand PD-L1. Several immunostimulatory monoclonal antibodies (imAbs) have now been approved based on their remarkable efficacy, after an expedited development process that has challenged key standards of drug development established in an era of conventional cytotoxic therapies and to some extent molecularly targeted agents (Table 1). In this article we review the current clinical methodology set up and the challenges that immunotherapy poses on the classical paradigms.
Table 1. Overview of currently approved compounds.
| Compound | Organization | Disease | Indication |
|---|---|---|---|
| Ipilimumab (1,2) | FDA, EMA | Cutaneous melanoma | Advanced unresectable melanoma |
| FDA | Adjuvant treatment of patients with melanoma with involvement of regional lymph nodes, after complete resection including total lymphadenectomy | ||
| Nivolumab (1,2) | EMA, FDA | Cutaneous, melanoma | Advanced disease; unresectable or metastatic |
| EMA | NSCLC | Advanced disease squamous histology | |
| FDA | Renal cell carcinoma, NSCLC | Advanced disease | |
| Advanced disease, non squamous and squamous histology | |||
| Pembrolizumab (1,2) | EMA, FDA | Melanoma | Advanced disease: unresectable or metastatic |
| FDA | NSCLC | Metastatic, PD-L1 expressing, with disease progression on or after platinum-containing chemotherapy | |
| Nivolumab + ipilimumab (1) | FDA | Melanoma | Advanced disease |
FDA, Food and Drug Administration; EMA, European Medicines Agency; NSCLC, non-small cell lung cancer.
Phase I trials
For more than a decade now, the development of molecularly targeted agents has had a major impact on the design and organization of phase I clinical trials. Whereas traditionally phase I trials were dedicated to any type of cancer, targeted agents have moved the focus to a more biomarker-driven approach. With the introduction of immune checkpoint inhibitors, phase I methodology might need to be revised once again, as the attention is now moving from the cancer cell to the immune system and the concept of immune tolerance.
Is maximum tolerated dose (MTD) still the mainstay?
The classical concept of identifying the MTD and establishing the recommended phase II dose (RP2D) of novel agents has been challenging, as in many phase I trials evaluating imAbs no MTD could be identified. In their comprehensive review on the early clinical development of anti-CTLA-4 and anti-PD-1/PD-L1 compounds, Postel-Vinay et al. assessed the outcome of 13 phase I trials (3). Among them, only one trial was able to identify per-protocol defined dose-limiting toxicities (DLTs) during the first cycles of treatment (4); in most other trials the maximum administered dose was used to guide the selection of the RP2D. This is not surprising given that the current classes of imAbs appear to be less prone to cause acute or cumulative toxicities, other than anaphylactic infusion reactions. In addition, immune-related adverse events (irAEs) are usually not observed during the first cycle of treatment, but are reported to occur at any time, as of approximately 8-10 weeks after starting treatment, regardless of grade (5). Consequently, late-onset DLTs may not be taken into account in the standard dose recommendation process. Furthermore, despite not being considered DLTs, irAEs can rapidly evolve into potentially life-threatening conditions (6,7), so that grade 2 events might often prompt temporary treatment interruption until properly managed.
Therefore, important considerations for the dose recommendation process of future phase I trials should be (1) to consider as DLTs also any immune-related toxicities, which would lead to a systematic decrease in drug exposure (thus impacting dose intensity) (7-9) and (2) to take into account toxicities scoring as DLTs, as they also occur beyond the first cycle of treatment (3,9).
An additional challenge in establishing the RP2D has also been the observation that, contrary to the classical model, an increase in the dose of the current classes of imAbs does not necessarily result in an increased rate of activity or toxicity (5,6,10-12). Therefore, rather than looking for the MTD, the objective of a phase I could shift to identify a minimal immunologically active dose, for example by looking for a dose that causes a saturation phenomenon in specific pharmacodynamic (PD) and/or pharmacokinetic (PK) parameters. This could be especially informative when the best administration schedule is unclear (e.g., single injection or continuous exposure, IV or IT administration, duration of the treatment). However PD evaluation still presents a lot of unknowns: examples are the minimal degree of target modulation that is pharmacologically meaningful (3) and whether there is a role for immunologic monitoring (e.g., CD4+ or CD8+ T-cells) as a potentially interesting biomarker of immune response (13).
The common IgG backbone of imAbs explains the relative similar PK profile, including a dose-dependent Cmax and area under the curve, and a median half-life of 16 days (9–21 days). Differences, however, exist according to their isotype: most imAbs are IgG1 (such as ipilimumab, atezolizumab or durvalumab), however some are IgG2 or IgG4 (such as tremelimumab and nivolumab, respectively). This can have a dramatic impact on bioactivity, as IgG1 and IgG3 are classically more prone to cause natural killer cell-mediated antibody-dependent cell-mediated cytotoxicity and IgG4 are more efficient in the activation of the alternative complement pathway (3).
Unfortunately, so far PK/PD data have only been reported in a limited number of studies.
The role of expansion cohorts
As imAbs seem to induce delayed immune responses, life expectancy of patients considered for the phase I study requires careful consideration. In addition, prior treatment with chemotherapy or radiotherapy may potentially have an impact on induced immune responses of cancer immunotherapy. This raises the question, whether selection criteria for immunotherapeutic phase I trials should not be revised to allow patients with a better outcome due to e.g., lower tumor burden, or with only minimal lesions in whom host immune responses have been maintained (3). On the other hand, activity and clinical benefit have been observed in patients with brain metastases (14), a population of patients who are often excluded from clinical trials. However, this would better take place in specific phase II trials or, as is more often the case nowadays, in dedicated expansion cohorts, once the toxicity profile of the imAb has been well characterized.
Note that safety expansion cohorts are increasingly important for the safety assessment of imAbs, with MTDs less likely to be identified in the dose-escalation phase and irAEs expected to occur beyond the first cycle (15). As the development of pembrolizumab has shown, expansion cohorts have also evolved into an opportunity to collect in parallel efficacy data on different dosages and histotypes, thus bypassing the cost and administrative burden of performing several separate trials. With efficacy data on hundreds of patients treated in its expansion cohorts, pembrolizumab received the Food and Drug Administration (FDA) breakthrough designation therapy, which allowed its commercialization less than 4 years after the beginning of the phase I trial (12), i.e., considerably faster than the historic average of 10 years. However, expansion cohorts are unfortunately not always accompanied by a clear justification for their sample size, as illustrated by a single center review of 522 phase I trials with expansion cohorts opened between 1988 and 2012 (16). Sixty percent of trials with three or more cohorts provided no statistical justification of the sample size, even though the majority stated efficacy as an objective. Such expansion cohorts should be held to the same standards as phase II studies, which involve considerations to control the possibility of false-positive or false-negative results, and stopping rules for lack of activity to avoid exposing a high number of patients to the risk of unethical treatments at inadequate doses (3,17).
Phase II and III: have the objectives changed?
Response assessment
Assessment of tumor burden is an integral part of clinical trial methodology in oncology. The Response Evaluation Criteria in Solid Tumors (RECIST), implemented in 2000 and updated in 2009 (18,19), provide a widely accepted tool for uniform assessment of tumor burden in multicenter clinical trials across multiple solid tumor types. Progression according to RECIST is defined as a significant increase in tumor burden (indicated by a 20% increase in the sum of the longest diameter of some selected measurable target lesions), unequivocal progression of non-targeted disease and/or the development of new lesions. While patients have responded to treatment with immunotherapy with immediate tumor shrinkage or stable disease, other patterns of response have also been documented (20). In some patients, a decrease in lesion size occurred after an initial increase, confirmed by biopsy as inflammatory cell infiltrates or necrosis (21). In others, a reduction in total tumor burden during or after the appearance of new lesions was observed, found to be associated with edema and infiltrates of immune cells and transient increases in baseline tumor lesions. Both cases would have led to a classification of progressive disease using RECIST and therefore the discontinuation of experimental treatment, from which a patient was still deriving clinically benefit. This has raised concerns about the use of classical response assessment tools to study the activity of imAbs and has resulted in the development of a set of alternative, immune-related response criteria (irRC) for the evaluation of immune therapy activity in solid tumors (20), with a proposal for simplification, to fit better with the RECIST framework added recently (22,23). The original irRC were evaluated in 227 advanced melanoma patients treated with ipilimumab (10 mg/kg), with so-called immune-related responses in 10% (22) of the patients. However, they remain to be validated beyond the setting of melanoma and ipilimumab. So far, only limited data is available on whether similar trends are seen for immunotherapies with other mechanisms of actions or for other solid tumor types, and the reported incidence seems to be very low (24). Therefore, response according to irRC remains an exploratory endpoint.
Progression-free survival
Approval of ipilimumab, nivolumab and pembrolizumab in advanced lung cancer and melanoma was based on overall survival (OS). However, the increasing availability of more effective immunotherapies and the subsequent increase in survival will likely refuel the debate on the most appropriate primary endpoint for future clinical trials. Although OS remains the gold standard, OS comparisons can be confounded by crossover within a trial, by subsequent treatments and by competing non-cancer related events. Therefore, progression-free survival (PFS) may be explored more in the development and registration of new imAbs.
Although regulators have accepted clinical trials where treatment is continued beyond RECIST progressive disease, immune-related PFS (irPFS), i.e., PFS by irRC, is not yet a commonly used endpoint as some challenges remain to be overcome. First, as already pointed out by Chiou and Burotto (24), so far there is only limited data available on irRC outcomes outside of the field of melanoma. Second, allowing treatment beyond (pseudo-) progression could be considered unethical for classical chemotherapy. An endpoint, which accommodates for different approaches in treatment arms according to their class of action can therefore be biased to favor, by definition, one treatment arm over the other. This should be avoided. Note that so far only few trials comparing imAbs to classical chemotherapy have considered immune-related progression-free survival (irPFS) as an endpoint. In addition, only two (similar) randomized phase II trials [SCLC and non-small cell lung cancer (NSCLC)] have looked at a combination of paclitaxel and carboplatin with or without ipilimumab, using irPFS as their primary endpoint (25,26). Of note, the currently ongoing follow-up phase III uses OS as primary endpoint and only standard PFS is considered as a secondary objective (NCT01285609).
Finally, in multicenter clinical trials it is important to use an endpoint, which is objectively and uniformly assessed across the participating sites. The call for a pseudo-progression, and therefore the decision to continue or not treatment, is often referred to clinical judgment, as there are currently no objective criteria to evaluate this. With clinical research starting to explore combination regimens of immune checkpoint inhibitors with chemotherapy or molecularly targeted agents, this will become even more difficult. The few preclinical data available hamper a solid choice of treatment schemes and schedules, and leave the treating physician with more uncertainty as to whether and how to treat beyond the first radiological signs of progressive disease. This may impact the outcome in terms of efficacy for the patient, but may also affect his/her safety and quality of life (QoL).
Even though not explored so far, the role of an endpoint combining efficacy and safety/QoL should be explored, as it would be clinically meaningful and relevant from a regulatory point of view. However such endpoints are particularly challenging to use; QoL in particular suffers from lack of compliance (27).
Delayed treatment effect
For now OS will remain the gold standard endpoint for showing a clinical benefit of the new immune modulating compounds currently being developed and tested. However, showing a survival benefit with immunotherapy is subject to its own statistical challenges. Several studies with treatments of melanoma patients have reported a delay in clinical effect of the compounds being investigated (28), visible by a temporary overlap of the Kaplan-Meier (KM) survival curves at the start of the study, as illustrated in Figure 1.
Figure 1.
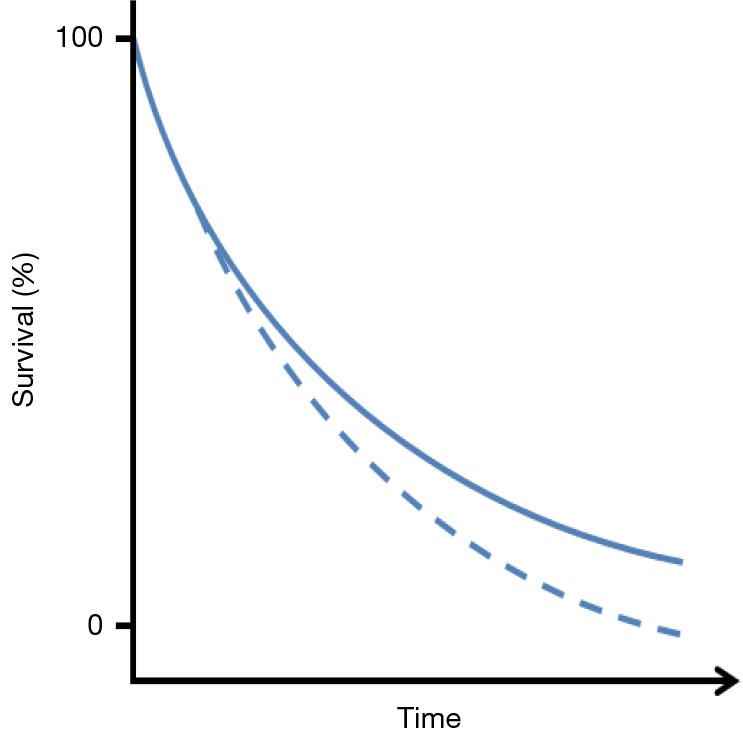
Graphical illustration of Kaplan-Meier survival curves with a delayed treatment effect.
Such a delay is in fact a violation of the assumption of proportional hazards that is often used for the sample size calculation of randomized clinical trials and will lead to a loss of differential power of the commonly used log-rank test during the final analysis (28-30). In addition, some of these agents have induced long-term survival in a small subset of patients. This adds an additional layer of complexity to the statistical analysis, as it can delay (or even prohibit) the occurrence of the required number of events and therefore considerably lengthen the study duration. Alternative methods should therefore be considered, to calculate the required size of a clinical trial when this is expected. Simulations can be used to study the impact of the delayed treatment effect on the power of the classical log-rank test and to adjust the sample size accordingly, or one could model and test the delayed effect assuming piecewise proportional hazard ratios and using a weighted log-rank test [e.g., as described by Fine (31) or Hasegawa (32)]. Note that these approaches may not necessarily result in smaller trials and that they require a reliable estimate of the timing of the separation of the two KM curves.
Alternatively, one could look at milestone survival, as a potential new efficacy endpoint for imAbs in late-stage drug development (33). Milestone survival is the survival probability at a prespecified time point, which could be chosen at a sufficient amount of time after the expected onset of the delayed treatment effect. As such it could potentially mitigate the challenge of accelerating the drug development process when the strength of this class of agents is derived from long-term follow-up (33).
Finally, it’s important to be aware that the impact of a delayed treatment effect extends beyond the choice of an appropriate endpoint and statistical test for the primary analysis of a protocol. It also affects interim evaluations for efficacy or futility. If the interim look is done too soon it will unlikely result in stopping earlier for a positive outcome, whereas a futility interim look, which is planned too soon, will likely increase the chance of (erroneous) early termination of the development of an active agent (33). The latter might have resulted in the premature closure of a phase III trial investigating the role of tremelimumab for the treatment of advanced melanoma (34). Therefore, careful planning of the timing of an interim look will need to take into account information on survival kinetics related to the compound being investigated. Exploratory randomized phase II trials will be needed, as the knowledge obtained from these studies will be essential to guide and improve the planning of subsequent confirmatory randomized phase III studies.
Immunotherapy, is it really so safe?
The unlocking of the mechanisms of immune control on normal tissue implies a number of irAEs, such as dermatologic, gastrointestinal, hepatic, endocrine and other less common inflammatory events, with notable differences between PD-1 and CTLA-4 blockade (35). IrAEs occur in up to 90% of patients treated with an anti-CTLA-4 antibody and in 70% of patients treated with a PD-1/PD-L1 antibody (36). The rates of grade 3/4 toxicity with immune checkpoint blockade (approximately 10% to 20%) are not higher than those seen with many standard chemotherapy or targeted therapy regimens (37). Unlike other oncology treatments, these toxicities do not appear to be cumulative over time (38). Moreover, it is unknown whether the development of irAEs is an inherent component of checkpoint blockade or if the rates of these events can be decreased by modifying the dosage, by using lower-than-approved doses in combination or in sequence, or by using immunologic outcomes in place of clinical outcomes to monitor response (39).
Recommendations for treating irAEs come from general clinical consensus, because no prospective trials have been conducted to specifically test whether one management strategy is superior. The management of irAEs includes early close monitoring, as the majority of immune-mediated reactions occur during the initial stages of treatment, temporary immunosuppression with corticosteroids or mycophenolate mofetil (35). Prompt recognition and initiation of appropriate management, usually in the form of immunosuppression, commonly results in complete reversibility, but failing to do so can lead to severe toxicity or even death.
Colitis is perhaps the most clinically relevant irAE, more commonly observed in patients treated with ipilimumab than in those treated with anti-PD-1 therapy. For example, in the phase III KEYNOTE-006 study, grade 3/4 colitis occurred in 7% of the patients in the ipilimumab group compared to only 1.4% to 2.5% in the pembrolizumab group (40). Moreover, it is typically dose-dependent and presents after approximately 6 weeks of treatment (35,41). Unfortunately, this adverse event has resulted in treatment-related deaths: diarrhea of any grade was reported in approximately 30% of the 511 patients treated with ipilimumab in a phase III melanoma trial, with less than 10% grade 3/4 events. Of the 511 patients, five (1%) developed intestinal perforation, four (0.8%) died as a result of complications and 26 (5%) were hospitalized for severe enterocolitis (42). The treatment of colitis depends on the severity of the reaction, the etiology and the speed of symptom resolution. For moderate reactions, temporary treatment interruption, antidiarrheal therapy (e.g., loperamide, diphenoxylate/atropine) and oral corticosteroids are the management standards. Patients with severe symptoms or those refractory to oral corticosteroids may need hospitalization for intravenous corticosteroids and hydration. Also, infliximab (5 mg/kg once every 2 weeks) can be helpful (43,44). Endoscopic evaluation with flexible sigmoidoscopy is also preferable to prove autoimmune colitis if symptoms persist for >1 week, prior to initiating steroids (41).
Immune checkpoint blockade may also cause endocrinopathies affecting the pituitary, adrenal and thyroid glands. Clinical symptoms may vary but often involve fatigue, headache and nausea. Diagnosis is usually made by laboratory findings and/or radiographic changes, such as enlargement of the pituitary gland (45,46). Because the pituitary gland also regulates the ovary/testes, pituitary dysfunction may contribute to gonadal dysfunction regardless of the age. The precise incidence of endocrinopathy is difficult to ascertain because endocrinopathies have been variably monitored and diagnosed in clinical trials. In the KEYNOTE-006 study, for example, hypo- and hyperthyroidism (of any grade) were the most frequent irAEs in the pembrolizumab group (10.1% and 6.5%, respectively) compared to colitis (8.2%) and hypophysitis (2.3%, any grade) in the ipilimumab group. Long-term data suggest that the rate of hypothyroidism is 7.5% (0.2%, grade 3/4) and the one of hyperthyroidism is 2.3% (0.3%, grade 3/4) (38,47). Endocrinopathies may require treatment with corticosteroids or longer-term hormone supplementation with levothyroxine or replacement hydrocortisone (37). Therefore, at baseline it is advisable to assess the thyroid but also the liver function (mainly AST, ALT and total bilirubin), with follow-up testing before each infusion or more frequently if needed. As a matter of fact, the development of hepatitis is mostly asymptomatic; with CTLA-4 blockade, grade 3/4 transaminase level increase is believed to occur less than 10% of the time (34,42), and grade 3/4 transaminase level increase with PD-1 blockade is rare (4,48). Treatment includes oral corticosteroids or mycophenolate mofetil (500 mg twice daily) in those who are refractory to steroids (35). Infliximab should not be used because it may contribute to hepatotoxicity.
The most common irAE for both CTLA-4 and PD-1 therapy is dermatologic toxicity (50% with ipilimumab), which is also typically the one with the earliest onset (3–17 weeks) (3,41). Physical examination findings can uncover a reticular, maculopapular, erythematous rash on the extremities or trunk (49). Perhaps more unique to the PD-1 experience, although in a small percentage of patients, is oral mucositis and/or dry mouth (38). For rashes topical corticosteroids or antipruritics, such as hydroxyzine and diphenhydramine can often be used successfully (35). More rarely (<1%), severe rashes, such as Stevens-Johnson syndrome and toxic epidermal necrolysis have been reported, and if suspected, hospitalization for intravenous corticosteroids, fluid and electrolyte monitoring are required (35). Vitiligo was reported to occur in both CTLA-4 and PD-1 inhibitor clinical trials (11%). This side-effect can be permanent, but does not require interruption of immune checkpoint inhibitor therapy or toxicity treatment (50).
Other rare irAEs related to checkpoint blockade include episcleritis, conjunctivitis, uveitis (51), neurologic adverse effects (52), increase of amylase and lipase, diabetes, pancreatitis (21,41), hematologic adverse effects (i.e., cytopenia, red cell aplasia, autoimmune neutropenia, acquired haemophilia A) (36,53,54), nephritis, glomerular lupus-like nephropathy and renal dysfunction (<1%, presenting after 10 months) (41,55) and pneumonitis. Pneumonitis is notable, because rare treatment-related deaths have been experienced in early clinical studies of PD-1 blockade (4). It presented after 5 months and can be deceptive and non-specific, so that new cough or dyspnea in patients treated with these agents warrants evaluation with pulmonary function tests, radiographic imaging and eventually even bronchoscopy. For grade 2 or higher pulmonary symptoms requiring medical intervention or limiting instrumental ADLs, admission to the hospital and pulmonary consultation is warranted together with intravenous corticosteroids and interruption of the treatment (41). Rare disorders reported in <1% of the patients include red cell aplasia, thrombocytopenia, hemophilia A, Guillain-Barre syndrome, myasthenia gravis, posterior reversible encephalopathy syndrome, aseptic meningitis and transverse myelitis (41).
Whether irAEs or other exacerbations of autoimmunity occur at a higher rate among patients with underlying autoimmune disorders is unknown, because patients with autoimmune disorders were not included in clinical trials of CTLA-4 and PD-1/PD-L1 agents. Nonetheless, several cases have recently been reported of patients successfully being treated with ipilimumab without exacerbation of their underlying autoimmune disorder (56). In this patient population, the potential benefits of CTLA-4 and PD-1/PD-L1 therapy in treating life-threatening malignancies should therefore be weighed against the theoretical risk of exacerbating an underlying autoimmune disorder (35).
In cases of prolonged immunosuppression, often required to treat irAEs, there is a risk of predisposing patients to opportunistic infections. In one case report, a patient treated with ipilimumab who required corticosteroids and infliximab for colitis ultimately developed Aspergillus pneumonia (57). Given this risk, prophylaxis against infectious organisms, such as Pneumocystis jirovecii should be considered (36,58).
Biomarkers
The design of biomarker-tailored or biomarker enriched studies has already been extensively discussed in the existing literature [see e.g., Freidlin and Korn (59) for a contemporary review]. Essential to this type of design is good knowledge on the performance of the biomarker, i.e., its ability to discriminate between patients and the operating characteristics of the associated assay. The complexity of immune-mediated responses has made biomarker evaluation difficult. The dynamic nature of the immune system constantly evolves during immune responses, making it difficult to identify a single biomarker to predict responses (60). Moreover, the efficacy of checkpoint inhibitors varies among tumor types, so that a better understanding of these differences is needed to enhance their efficacy and avoid safety being compromised.
PD-1, a CD28 receptor family member, is an inducible immune modulatory receptor. Upon interaction with its ligands B7 homolog 1 (PD-L1) and B7-DC (PD-L2), PD-1 plays important roles in negative regulation of T cell responses to antigen stimulation and maintaining peripheral tolerance. In addition to the inducible expression pattern on conventional T cells, PD-1 is also found on regulatory T cells, follicular T and B cells and antigen-presenting cells, including activated dendritic cells and monocytes. However, the underlying mechanisms of how PD-1 modulates B cell activation and affects antibody production remain largely uncertain (61).
Across all studies in multiple tumor types, it seems that patients whose tumors express PD-L1, as detected by immunohistochemical assays, have numerically higher response rates to PD-1/PD-L1 blockade than those who don’t (5,62). Although high expression of PD-L1 in tumor tissue prior to treatment is most likely indicative of a strong endogenous anti-tumor response (35), PD-L1 is inducible and can be upregulated in response to infiltrating immune cells and, possibly, genetic changes within the tumor. This might explain the varying response rates to PD-1 blockade among different tumor types (63-65). This might also explain why patients whose tumors do not express PD-L1 can have impressive responses to PD-1 blockade (66) and should therefore be considered eligible for PD-1-blocking approaches.
In 2014, both pembrolizumab and nivolumab (both targeting PD-1) obtained regulatory approval for the treatment of patients with advanced melanoma. No companion diagnostic was linked to their use, as clinical studies showed similar treatment outcomes, irrespectively of PD-L1 expression status. On the other hand, in NSCLC, two assays to detect PD-L1 expression in formalin-fixed, paraffin-embedded (FFPE) NSCLC tissue, PD-L1 IHC 22C3 pharmDx (Dako) and PD-L1 IHC 28-8 pharmDx (Dako), linked to the use of pembrolizumab (Keytruda, Merck Sharp & Dohme) (67) and nivolumab (Opdivo, Bristol-Myers Squibb) (68), respectively, were approved by FDA, although the latter only as a kind of ‘complementary test’ and not as a companion diagnostic (69).
Data on the not yet approved atezolizumab (Roche/Genentech PD-L1 checkpoint inhibitor) and its PD-L1 IHC expression test with the SP142 antibody assay (Ventana) showed superiority in the unselected intent to treat population independently of the PD-L1 expression, but a greater benefit in the highest biomarker expression group (70). In this study, also tumor-infiltrating immune cells (TILs) were analysed prospectively and expression of PD-L1 was identified, suggesting that positivity should be determined as a combination of expression of the receptor in tumor and also immune cells. This potential biomarker seems to have a positive prognostic influence, while high levels of tumor infiltrating lymphocytes determine an increase in PD-L1 expression (71), but their role still needs further investigation.
Other potentially relevant biomarkers currently being explored include mutational load, which may allow for an increased immune recognition of neo-epitopes and subsequent targeting with checkpoint blockade (72,73), and adequate assessment of the tumor inflammatory microenvironment to investigate the possible role of tumor heterogeneity (74).
In summary, PD-L1 expression is currently the most advanced biomarker in development, but its predictive role has yet to be established (35). One of the limiting factors may be the fact that, in the absence of a generally accepted standard assay, currently each company developing a PD-L1 inhibitor has done so with its own accompanying diagnostic assay, looking into different IHC antibody clones, staining protocols and platforms, clinical decision points and assessment and scoring methods. This is challenging for the setup and implementation of diagnostic PD-L1 testing in a pathology department, as this might potentially lead to heterogeneity staining results and lecture assessment and final reports in real clinical life. However, in oncology, we have met similar difficulty previously with the HER2 receptor for breast cancer in order to determine positivity for administering anti-HER2 treatment. Studies have proven the significant benefit of anti-HER2 therapy for the treatment of HER2-positive breast cancer (75,76). Thus, accurate HER2 testing was needed to ensure appropriate treatment of these patients. Finally on 2007, a joint expert panel convened by the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) developed guidelines for how to test for HER2 receptor overexpression following a globally accepted algorithm that includes IHC and in situ hybridization assays (ISH). IHC analysis is usually used as the primary assay and reflex ISH in specific subsets of IHC results (e.g., 2+), although many laboratories use as the primary test ISH technologies lately (77). This algorithm has been implemented in clinical practice for breast cancer and is regularly updated (78). One of the key challenges in the development of these assays was the lack of recognized absolute standards to define HER2 expression to benchmark the accuracy and validity of methods currently implemented. Over the last 15 years the accuracy and reproducibility of these assays has improved and the challenge on whether we should from now on further improve these assays or develop new better assays is the current focus of researchers in the field (79). Further optimization and interpretation is constantly required for more accurate assessment, revealing the intense research over the years, while it remains to be seen if such practice will eventually become established for standardization of PD-L1 expression testing, as well.
Conclusions
For over 50 years now, surgery, radiotherapy and chemotherapy have been the physician’s main weapons against cancer. The development and approval of targeted agents, directed against specific cancer cells, and now the immune checkpoint inhibitors, targeting the immune cells and promoting the immune response against cancer cells, has changed the landscape of cancer treatment, the prognosis of cancer patients and the mechanism and timings of regulatory bodies and reimbursement agencies. Checkpoint inhibitors, in particular, have revolutionized the cancer treatment strategy, thus leading to fast-track approval of two compounds already.
The focus of our review has been to highlight that the mechanism of action of these compounds, as well as the limited background data, still represent a challenge for their development from a methodological point of view. None of the phase I data helped identifying an MTD, thus raising questions on the optimal dose, schedule and treatment duration, both when administered alone and in combination. Immune-related responses, delayed treatment effects and the lack of validated biomarkers provide challenges for the ensuing phase II and III trials, when trying to identify the correct endpoint to measure and the population most likely to benefit. What seems to be reassuring so far is the safety profile of these compounds, provided attention is paid and a close follow-up is applied.
The next hurdles to overtake will be the cost assessment of imAbs and the economic challenge of reimbursement by health regulatory authorities and/or health insurances. There is now room for equivalence studies, to compare several doses, schedules and routes of administration, as well as identifying the population that might receive the greatest advantage from such treatments, in order to minimize the number of per-patient administrations required to obtain an optimal immunostimulatory effect.
Acknowledgements
This publication was supported by Fonds Cancer (FOCA) from Belgium.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Federal Drug Administration. [Accessed 24 February 2016]. Available online: www.fda.gov
- 2.European Medicines Agency. European public assessment reports. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124
- 3.Postel-Vinay S, Aspeslagh S, Lanoy E, et al. Challenges of phase 1 clinical trials evaluating immune checkpoint-targeted antibodies. Ann Oncol 2016;27:214-24. 10.1093/annonc/mdv550 [DOI] [PubMed] [Google Scholar]
- 4.Ribas A, Camacho LH, Lopez-Berestein G, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol 2005;23:8968-77. 10.1200/JCO.2005.01.109 [DOI] [PubMed] [Google Scholar]
- 5.Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012;30:2691-7. 10.1200/JCO.2012.41.6750 [DOI] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao J, He Q, Subudhi S, et al. Review of immune-related adverse events in prostate cancer patients treated with ipilimumab: MD Anderson experience. Oncogene 2015;34:5411-7. 10.1038/onc.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paoletti X, Le Tourneau C, Verweij J, et al. Defining dose-limiting toxicity for phase 1 trials of molecularly targeted agents: results of a DLT-TARGETT international survey. Eur J Cancer 2014;50:2050-6. 10.1016/j.ejca.2014.04.030 [DOI] [PubMed] [Google Scholar]
- 9.Postel-Vinay S, Collette L, Paoletti X, et al. Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents--dose-Limiting Toxicity and Toxicity Assessment Recommendation Group for Early Trials of Targeted therapies, an European Organisation for Research and Treatment of Cancer-led study. Eur J Cancer 2014;50:2040-9. 10.1016/j.ejca.2014.04.031 [DOI] [PubMed] [Google Scholar]
- 10.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65. 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res 2008;14:3044-51. 10.1158/1078-0432.CCR-07-4079 [DOI] [PubMed] [Google Scholar]
- 12.Patnaik A, Kang SP, Rasco D, et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin Cancer Res 2015;21:4286-93. 10.1158/1078-0432.CCR-14-2607 [DOI] [PubMed] [Google Scholar]
- 13.Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014;515:558-62. 10.1038/nature13904 [DOI] [PubMed] [Google Scholar]
- 14.Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 2012;13:459-65. 10.1016/S1470-2045(12)70090-6 [DOI] [PubMed] [Google Scholar]
- 15.Manji A, Brana I, Amir E, et al. Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single-agent phase I cancer trials. J Clin Oncol 2013;31:4260-7. 10.1200/JCO.2012.47.4957 [DOI] [PubMed] [Google Scholar]
- 16.Dahlberg SE, Shapiro GI, Clark JW, et al. Evaluation of statistical designs in phase I expansion cohorts: the Dana-Farber/Harvard Cancer Center experience. J Natl Cancer Inst 2014;106. pii: dju163. 10.1093/jnci/dju163 [DOI] [PubMed] [Google Scholar]
- 17.Iasonos A, O'Quigley J. Clinical trials: Early phase clinical trials-are dose expansion cohorts needed? Nat Rev Clin Oncol 2015;12:626-8. 10.1038/nrclinonc.2015.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205-16. 10.1093/jnci/92.3.205 [DOI] [PubMed] [Google Scholar]
- 19.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47. 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 20.Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009;15:7412-20. 10.1158/1078-0432.CCR-09-1624 [DOI] [PubMed] [Google Scholar]
- 21.Di Giacomo AM, Danielli R, Guidoboni M, et al. Therapeutic efficacy of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with metastatic melanoma unresponsive to prior systemic treatments: clinical and immunological evidence from three patient cases. Cancer Immunol Immunother 2009;58:1297-306. 10.1007/s00262-008-0642-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishino M, Giobbie-Hurder A, Gargano M, et al. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin Cancer Res 2013;19:3936-43. 10.1158/1078-0432.CCR-13-0895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishino M, Gargano M, Suda M, et al. Optimizing immune-related tumor response assessment: does reducing the number of lesions impact response assessment in melanoma patients treated with ipilimumab? J Immunother Cancer 2014;2:17. 10.1186/2051-1426-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiou VL, Burotto M. Pseudoprogression and Immune-Related Response in Solid Tumors. J Clin Oncol 2015;33:3541-3. 10.1200/JCO.2015.61.6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 2012;30:2046-54. 10.1200/JCO.2011.38.4032 [DOI] [PubMed] [Google Scholar]
- 26.Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol 2013;24:75-83. 10.1093/annonc/mds213 [DOI] [PubMed] [Google Scholar]
- 27.Long G, Atkinson V, Ascierto P, et al. Effect of nivolumab (NIVO) on quality of life (QoL) in patients (pts) with treatment-naïve advance melanoma (MEL): Results of a phase III study (CheckMate 066). J Clin Oncol 2015;33:abstr 9027.
- 28.Hoos A, Eggermont AM, Janetzki S, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst 2010;102:1388-97. 10.1093/jnci/djq310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen TT. Statistical issues and challenges in immuno-oncology. J Immunother Cancer 2013;1:18. 10.1186/2051-1426-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wason JM, Dentamaro A, Eisen TG. The power of phase II end-points for different possible mechanisms of action of an experimental treatment. Eur J Cancer 2015;51:984-92. 10.1016/j.ejca.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fine GD. Consequences of delayed treatment effects on analysis of time-to-event endpoints. Drug Information Journal 2007;41:535-9. [Google Scholar]
- 32.Hasegawa T. Sample size determination for the weighted log-rank test with the Fleming-Harrington class of weights in cancer vaccine studies. Pharm Stat 2014;13:128-35. 10.1002/pst.1609 [DOI] [PubMed] [Google Scholar]
- 33.Chen TT. Milestone Survival: A Potential Intermediate Endpoint for Immune Checkpoint Inhibitors. J Natl Cancer Inst 2015;107. pii: djv156. 10.1093/jnci/djv156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol 2013;31:616-22. 10.1200/JCO.2012.44.6112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 2015;33:1974-82. 10.1200/JCO.2014.59.4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54:139-48. 10.1016/j.ejca.2015.11.016 [DOI] [PubMed] [Google Scholar]
- 37.Agarwala SS. Practical Approaches to Immunotherapy in the Clinic. Semin Oncol 2015;42 Suppl 3:S20-7. 10.1053/j.seminoncol.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 38.Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020-30. 10.1200/JCO.2013.53.0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol 2006;90:297-339. 10.1016/S0065-2776(06)90008-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320-30. 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 41.Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res 2015;4:560-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23. 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pagès C, Gornet JM, Monsel G, et al. Ipilimumab-induced acute severe colitis treated by infliximab. Melanoma Res 2013;23:227-30. 10.1097/CMR.0b013e32835fb524 [DOI] [PubMed] [Google Scholar]
- 44.Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm 2009;24:321-5. 10.1089/cbr.2008.0607 [DOI] [PubMed] [Google Scholar]
- 45.Blansfield JA, Beck KE, Tran K, et al. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 2005;28:593-8. 10.1097/01.cji.0000178913.41256.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dillard T, Yedinak CG, Alumkal J, et al. Anti-CTLA-4 antibody therapy associated autoimmune hypophysitis: serious immune related adverse events across a spectrum of cancer subtypes. Pituitary 2010;13:29-38. 10.1007/s11102-009-0193-z [DOI] [PubMed] [Google Scholar]
- 47.Daud A, Ribas A, Robert C, et al. Long-term efficacy of pembrolizumab (pembro; MK-3475) in a pooled analysis of 655 patients with advanced melanoma enrolled in KEYNOTE-001. J Clin Oncol 2015;33:abstr 9005.
- 48.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134-44. 10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lacouture ME, Wolchok JD, Yosipovitch G, et al. Ipilimumab in patients with cancer and the management of dermatologic adverse events. J Am Acad Dermatol 2014;71:161-9. 10.1016/j.jaad.2014.02.035 [DOI] [PubMed] [Google Scholar]
- 50.Haanen JB, Thienen Hv, Blank CU. Toxicity patterns with immunomodulating antibodies and their combinations. Semin Oncol 2015;42:423-8. 10.1053/j.seminoncol.2015.02.011 [DOI] [PubMed] [Google Scholar]
- 51.Robinson MR, Chan CC, Yang JC, et al. Cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma: a new cause of uveitis. J Immunother 2004;27:478-9. 10.1097/00002371-200411000-00008 [DOI] [PubMed] [Google Scholar]
- 52.Wilgenhof S, Neyns B. Anti-CTLA-4 antibody-induced Guillain-Barré syndrome in a melanoma patient. Ann Oncol 2011;22:991-3. 10.1093/annonc/mdr028 [DOI] [PubMed] [Google Scholar]
- 53.Akhtari M, Waller EK, Jaye DL, et al. Neutropenia in a patient treated with ipilimumab (anti-CTLA-4 antibody). J Immunother 2009;32:322-4. 10.1097/CJI.0b013e31819aa40b [DOI] [PubMed] [Google Scholar]
- 54.Gordon IO, Wade T, Chin K, et al. Immune-mediated red cell aplasia after anti-CTLA-4 immunotherapy for metastatic melanoma. Cancer Immunol Immunother 2009;58:1351-3. 10.1007/s00262-008-0627-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Izzedine H, Gueutin V, Gharbi C, et al. Kidney injuries related to ipilimumab. Invest New Drugs 2014;32:769-73. 10.1007/s10637-014-0092-7 [DOI] [PubMed] [Google Scholar]
- 56.Pedersen M, Andersen R, Nørgaard P, et al. Successful treatment with Ipilimumab and Interleukin-2 in two patients with metastatic melanoma and systemic autoimmune disease. Cancer Immunol Immunother 2014;63:1341-6. 10.1007/s00262-014-1607-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kyi C, Carvajal RD, Wolchok JD, et al. Ipilimumab in patients with melanoma and autoimmune disease. J Immunother Cancer 2014;2:35. 10.1186/s40425-014-0035-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.National Comprehensive Cancer Network. Infections. Available online: http://www.nccn.org/professionals/physician_gls/pdf/infections.pdf
- 59.Freidlin B, Korn EL. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol 2014;11:81-90. 10.1038/nrclinonc.2013.218 [DOI] [PubMed] [Google Scholar]
- 60.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56-61. 10.1126/science.aaa8172 [DOI] [PubMed] [Google Scholar]
- 61.Yao S, Chen L. PD-1 as an immune modulatory receptor. Cancer J 2014;20:262-4. 10.1097/PPO.0000000000000060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grosso J, Horak CE, Inzunza D, et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO-4538). J Clin Oncol 2013;31:abstr 3016.
- 63.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064-74. 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 2014;20:3446-57. 10.1158/1078-0432.CCR-13-2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007;13:84-8. 10.1038/nm1517 [DOI] [PubMed] [Google Scholar]
- 66.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563-7. 10.1038/nature14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018-28. 10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 68.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 2015;373:1627-39. 10.1056/NEJMoa1507643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.US FDA. Premarket Approval (PMA) for PD-L1 IHC 28-8 pharmDx. [Accessed 24 February 2016]. Available online: http://www.accessdata.fda.gov/cdrh_docs/pdf15/P150025a.pdf
- 70.Vansteenkiste J, Fehrenbacher L, Spira IA, et al. Atezolizumab monotherapy vs docetaxel in 2L/3L non-small cell lung cancer: Primary analyses for efficacy, safety and predictive biomarkers from a randomized phase II study (POPLAR) 14LBA. Late breaking abstract presented at the European Cancer Congress. Vienna, Austria, 2015 Sep 25-29. [Google Scholar]
- 71.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-99. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214-8. 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J 2010;16:399-403. 10.1097/PPO.0b013e3181eacbd8 [DOI] [PubMed] [Google Scholar]
- 75.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 2005;353:1659-72. 10.1056/NEJMoa052306 [DOI] [PubMed] [Google Scholar]
- 76.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005;353:1673-84. 10.1056/NEJMoa052122 [DOI] [PubMed] [Google Scholar]
- 77.Pu X, Shi J, Li Z, et al. Comparison of the 2007 and 2013 ASCO/CAP evaluation systems for HER2 amplification in breast cancer. Pathol Res Pract 2015;211:421-5. 10.1016/j.prp.2014.09.010 [DOI] [PubMed] [Google Scholar]
- 78.Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997-4013. 10.1200/JCO.2013.50.9984 [DOI] [PubMed] [Google Scholar]
- 79.Perez EA, Cortés J, Gonzalez-Angulo AM, et al. HER2 testing: current status and future directions. Cancer Treat Rev 2014;40:276-84. 10.1016/j.ctrv.2013.09.001 [DOI] [PubMed] [Google Scholar]