

Abstract
Aims: Neuropeptide substance P (SP) is produced and released by a subset of peripheral sensory neurons that respond to tissue damage (nociceptors). SP exerts excitatory effects in the central nervous system, but peripheral SP actions are still poorly understood; therefore, here, we aimed at investigating these peripheral mechanisms. Results: SP acutely inhibited T-type voltage-gated Ca2+ channels in nociceptors. The effect was mediated by neurokinin 1 (NK1) receptor-induced stimulation of intracellular release of reactive oxygen species (ROS), as it can be prevented or reversed by the reducing agent dithiothreitol and mimicked by exogenous or endogenous ROS. This redox-mediated T-type Ca2+ channel inhibition operated through the modulation of CaV3.2 channel sensitivity to ambient zinc, as it can be prevented or reversed by zinc chelation and mimicked by exogenous zinc. Elimination of the zinc-binding site in CaV3.2 rendered the channel insensitive to SP-mediated inhibition. Importantly, peripherally applied SP significantly reduced bradykinin-induced nociception in rats in vivo; knock-down of CaV3.2 significantly reduced this anti-nociceptive effect. This atypical signaling cascade shared the initial steps with the SP-mediated augmentation of M-type K+ channels described earlier. Innovation: Our study established a mechanism underlying the peripheral anti-nociceptive effect of SP whereby this neuropeptide produces ROS-dependent inhibition of pro-algesic T-type Ca2+ current and concurrent enhancement of anti-algesic M-type K+ current. These findings will lead to a better understanding of mechanisms of endogenous analgesia. Conclusion: SP modulates T-type channel activity in nociceptors by a redox-dependent tuning of channel sensitivity to zinc; this novel modulatory pathway contributes to the peripheral anti-nociceptive effect of SP. Antioxid. Redox Signal. 25, 233–251.
Introduction
Substance P (SP) is an 11-amino-acid neuropeptide (RPKPQQFFGLM) of a tachykinin family that is produced in both peripheral and central nervous systems (CNS). SP is mainly an excitatory neuromodulator that is involved in pain transmission and smooth muscle contraction, and it modulates inflammatory and immune responses (69). Increased SP release or elevated expression of its receptors has been linked to many diseases such as acute and chronic inflammation and infections, asthma, affective and addictive disorders, and some forms of cancer [reviewed in Steinhoff et al. (62)]. In the peripheral somatosensory system, SP is abundantly produced by a subset of transient receptor potential cation channel subfamily V member 1 (TRPV1)-positive, pain-sensing (nociceptive) neurons and released in response to noxious stimulation both centrally and peripherally (40).
Peripheral SP released from nociceptive nerve endings causes so-called “neurogenic inflammation” (54); whereas in the spinal cord, SP functions as an excitatory co-transmitter alongside glutamate (12, 38). Intracellular effects of SP are mediated by the G-protein-coupled neurokinin (NK1 − 3) receptors. The strong excitatory role of SP in spinal nociceptive pathways has triggered a massive, industry-wide quest for new analgesics based on NK1 receptor antagonists. However, despite strong pre-clinical data, clinical trials of NK1 receptor antagonists failed (22, 59).
Innovation.
We discovered a novel endogenous pathway of “tuning” the activity of T-type Ca2+ channels via the modulation of its sensitivity to ambient zinc. We further suggest that simultaneous reciprocal modulation of two different ion channels by a single signaling cascade can sum up to produce a cumulative, endogenous anti-nociceptive effect. These findings describe and explain an unanticipated inhibitory effect of neuropeptide substance P (SP) in peripheral nociceptive pathways and shed new light on the mechanisms of endogenous analgesia.
The reasons for the lack of analgesic efficacy of the systemically applied NK antagonists are still unclear. However, one hypothesis is that, though excitatory in CNS, SP may exert paradoxical inhibitory/analgesic effects in the peripheral nociceptive pathways (32, 33). Thus, SP augments an inhibitory M-type K+ current in nociceptors via the atypical Gi/o-mediated signaling cascade that utilizes reactive oxygen species (ROS) as second-messenger molecules (32, 33).
Here, we discovered another component of the anti-excitatory action of SP in nociceptors: inhibition of T-type (CaV3) Ca2+ channels. CaV3 are encoded by CACNA1G, CACNA1H, and CACNA1I genes, which give rise to CaV3.1, CaV3.2, and CaV3.3 pore-forming α subunits, respectively (8). T-type channels are activated at voltages near or below −60 mV and may display a significant window current at the neuronal resting membrane potential (Erest) (51). CaV3s are abundantly expressed in cell bodies, axons, and peripheral terminals of nociceptive dorsal root ganglia (DRG) neurons, with CaV3.2 being the dominant isoform (9, 11, 18, 56, 63). Accordingly, peripherally applied T-type channel inhibition produces anti-nociceptive effects (2, 65).
Here, we show that (i) SP acutely inhibits T-type Ca2+ channels in DRG neurons via an ROS-dependent intracellular signaling cascade; (ii) this redox-mediated inhibition operates through the modulation of CaV3.2 channel sensitivity to ambient extracellular zinc; and (iii) a hind-paw injection of SP produces anti-nociceptive effects that can be reduced by the in vivo knock-down of CaV3.2 in DRG and partially mimicked by the peripheral injection of a specific T-type channel blocker. Thus, our study establishes a dual mechanism by which SP exerts its peripheral anti-nociceptive effect: a simultaneous inhibition of excitatory T-type Ca2+ current and an enhancement of inhibitory M-type K+ current.
Results
SP inhibits T-type Ca2+ current in sensory neurons via endogenous ROS generation
SP has been shown to trigger endogenous ROS release in nociceptive DRG neurons in an “atypical” Gi/o-mediated signaling cascade stimulating mitochondrial ROS generation (33). Since T-type Ca2+ channels are expressed in nociceptors and are redox sensitive (49, 65), we investigated whether SP affects the activity of low-voltage-activated (LVA, T-type) Ca2+ currents in DRG neurons.
Whole-cell recordings were made from cultured, small-diameter, and mostly TRPV1-positive neurons. In 301 of 710 (42%) such DRG neurons, depolarization to −40 mV induced LVA currents, with kinetic properties similar to those of recombinant CaV3.2 (Fig. 1A1 cf. Supplementary Fig. S1A1; Supplementary Data are available online at www.liebertpub.com/ars). These currents were sensitive to the selective T-type Ca2+-channel inhibitor, N-[[1-[2-(tert-butylamino)-2-oxoethyl]piperidin-4-yl]methyl]-3-chloro-5-fluorobenzamide (Z944; 1 μM; Fig. 1A1–A3), and the voltage-gated Ca2+ channel (VGCC) inhibitor mibefradil (MIB, 3 μM; Supplementary Fig. S1A1–D1). In our earlier experiments, we used MIB to confirm current identity but later switched to Z944, when it became available.
FIG. 1.

NK1 receptor activation inhibits T-type calcium channels in small-diameter DRG neurons. All recordings were performed from small (∼20 μm) DRG neurons with a mean whole-cell capacitance of 23.0 ± 0.9 pF (n = 100); these neurons are predominantly (∼70%) capsaicin sensitive (13, 35). (A) LVA Ca2+ current in DRG neurons is inhibited by an NK1-selective agonist S9SP. (A1) Example time course of the effects of 1 μM S9SP and 1 μM selective T-type channel inhibitor, Z944 on the LVA Ca2+ current recorded from small-diameter DRG neurons using whole-cell patch clamp. Plotted are peak LVA current amplitudes; periods of drug application are indicated by the vertical gray bars. Inset shows example current traces; current protocol is depicted above the traces. (A2) Similar to (A1) but 1 μM S9SP was co-applied with the 3 μM selective NK1 antagonist CP-99994 (after 30 min pre-incubation with the latter). (A3) Similar to (A1, A2) but another selective NK1 agonist, S9M11SP (1 μM) was applied instead of S9SP. (B) Scatter plot summarizing the experiments exemplified in (A1–A3). Each circle represents LVA current inhibition in a single neuron; the number of individual recordings is given above each group; horizontal gray bars are mean values for each group. ***Significantly different from the S9SP group, p < 0.001 (unpaired t-test). (C) Example experiment (C1) and summary (C2) of the experiments showing concentration dependency of the LVA current inhibition by S9SP in DRG neurons. Increasing concentrations of S9SP were applied during the periods indicated by horizontal black bars. (C2) The data from eight to nine experiments as in (C1) were fit to the one-component logistic function (see Materials and Methods section). Data are given as mean ± SEM. CP-99994, (2S,3S)-3-(2-Methoxybenzylamino)-2-phenylpiperidine dihydrochloride; DRG, dorsal root ganglion; LVA, low-voltage activated; NK1, neurokinin 1; S9M11SP, [Sar9, Met(O2)11]-Substance P; S9SP, [Sar9]-Substance P; SEM, standard error of the mean; Z944, N-[[1-[2-(tert-butylamino)-2-oxoethyl]piperidin-4-yl]methyl]-3-chloro-5-fluorobenzamide.
The mean LVA current amplitude was −161.7 ± 11.3 pA. The NK1-specific agonist [Sar9]-Substance P (S9SP; 1 μM) acutely inhibited LVA current in the majority of the LVA-expressing DRG neurons (Fig. 1A1, B); the mean inhibition was 22.5% ± 2.3% (n = 84; p < 0.001), although there was a degree of scattering in the magnitude of a response and approximately a third of neurons tested displayed minimal response (e.g., 28/84 or 33% neurons had <10% inhibition), which was suggestive of a low level of NK1 receptor expression in these neurons.
Analysis of the concentration dependency of S9SP effects (Fig. 1C) revealed that SP inhibited LVA current in DRG neurons, with IC50 of 6.2 ± 3.0 nM and maximal inhibition of 59.2% ± 4.2% (n = 8–9). NK1-selective antagonist (2S,3S)-3-(2-Methoxybenzylamino)-2-phenylpiperidine dihydrochloride (CP-99994) abolished the effect of S9SP (Fig. 1A2, B). Another NK1-selective agonist, [Sar9, Met(O2)11]-Substance P (S9M11SP; 1 μM) produced LVA current inhibition quantitatively similar to that of S9SP (Fig. 1A3, B).
Although NK1 receptor signaling is generally associated with the Gq/11 type of signaling cascade (37, 43), recent data suggest that at least some effects of SP in DRG neurons, such as ROS release and redox-dependent M channel augmentation, occur via the Gi/o-associated pathway (33). To test which signaling cascade is used in the S9SP-mediated inhibition of LVA current in DRG neurons, we used small interfering RNA (siRNA) knock-down of either Gi/o or Gq/11 Gα subunits in DRG cultures (Fig. 2A). Simultaneous knock-down of Gi1–3 and Go subunits abolished the S9SP-mediated LVA current inhibition, whereas simultaneous knock-down of Gq and G11 subunits was without an effect (as compared with non-targeting “scrambled” siRNA control; Fig. 2A, B). All siRNA oligos reduced the abundance of their corresponding target messenger RNA (mRNA) by 60%–80% (Fig. 2C).
FIG. 2.

NK1 receptors inhibit LVA currents in DRG neurons using a Gi/o-mediated signaling cascade. (A) Example time courses of the effects of S9SP in DRG neurons where either Gi and Go (A2) or Gq and G11 (A3) Gα subunits were knocked down using siRNA cocktails (see Materials and Methods section). Non-targeting “scrambled” siRNA oligonucleotides were used as negative control (A1). (B) Scatter plot summarizing the experiments exemplified in (A1–A3). Each circle represents LVA current inhibition in a single neuron; the number of individual recordings is given above each group; horizontal gray bars are mean values for each group. (C) Quantification of knock-down efficiency 48 h post-transfection from experiments as these shown in (A, B). Data are given as mean ± SEM; ***Significantly different from the scrambled oligo control, p < 0.001 (unpaired t-test). (D). The Gi/o inhibitor PTX but not the protein kinase C inhibitor BIM I inhibited NK1-induced LVA Ca2+ current inhibition. Example time courses of the effects of 1 μM S9SP and 1 μM Z944 on the LVA currents recorded from small-diameter DRG neurons after the PTX (400 ng/ml; overnight; D1) or BIM I (200 nM; 20 min; D2) pretreatment. Plotted are peak LVA current amplitudes; periods of drug application are indicated by the vertical gray bars; VGCC inhibitor MIB (3 μM) was used instead of Z944 in (D1). (E) Scatter plot summarizing the experiments exemplified in (D1, D2), similar to that shown in (B). ***Significantly different from the control, p < 0.001 (unpaired t-test). The data for the S9SP inhibition in control conditions (black circles) are the same as in Figure 1B and are included for comparison. BIM I, bisindolylmaleimide I; MIB, mibefradil; PTX, pertussis toxin; siRNA, small interfering RNA; VGCC, voltage-gated Ca2+ channel.
In accord with these findings, overnight pre-treatment of DRG cultures with the Gi/o inhibitor, pertussis toxin (PTX; 400 ng/ml) completely abolished the S9SP-mediated T-type current inhibition (Fig. 2D1, E). A protein kinase C inhibitor, bisindolylmaleimide I (BIM I, 200 nM) was without a significant effect (Fig. 2D2, E). These data indicate that, similar to their effect on the M channels, NK1 receptors use the Gi/o-mediated pathway to inhibit T-type channels in DRG neurons. Immunohistochemical localization of CaV3.2, NK1, and a major M channel subunit, KCNQ2 revealed that, indeed, CaV3.2 co-localizes with NK1 and KCNQ2 in many DRG neurons, particularly in those of a small diameter (Supplementary Fig. S2).
Washout of S9SP (up to 5 min) did not restore LVA current amplitude (not shown) but the inhibition was completely reversed by the reducing agent dithiothreitol (DTT; 1 mM), even in the continued presence of S9SP (Fig. 3A); DTT on its own did not significantly affect LVA current amplitude, but it rendered S9SP ineffective when both compounds were applied together (Fig. 3B).
FIG. 3.

SP inhibits T-type calcium channels using a redox-dependent mechanism. (A) S9SP-induced inhibition of LVA current in DRG neurons is reversed by the reducing agent DTT. (A1) Example time course of the effects of 1 μM S9SP, 1 mM DTT, and 3 μM MIB on the LVA Ca2+ current recorded from small-diameter DRG neurons. Inset shows example current traces; the current protocol was similar to that shown in Figure 1A1. (A2) Summarizes the effects. (B, B1, B2) DTT prevents the SP-induced LVA current inhibition. Experiments are similar to those shown in (A), but DTT is applied both before and during the S9SP application; layout and labeling are similar to (A). (C) Example time course of the effect of H2O2 (300 μM) on LVA Ca2+ currents in small-diameter DRG neurons. Note that DTT (1 mM) could almost completely reverse the inhibition. (C2) Summarizes the data for experiments shown in (C1). (D1, D2) Pre-application of H2O2 precludes the effect of S9SP on LVA Ca2+ currents in small-diameter DRG neurons. Layout and labeling are similar to (A–C). (E) Summarized data of the effect of the mitochondrial electron transport chain inhibitor, Ant.A (1 μM) on the LVA currents in DRG neurons. (E1) Ant.A inhibited LVA current, an effect that was completely reversed by DTT (1 mM). (E2) S9SP applied after the Ant.A produced only marginal further inhibition of LVA current. The number of experiments is shown within the bars. Asterisks indicate significant difference from the group indicated by the line connector with *p < 0.05, **p < 0.01, or ***p < 0.001 (paired t-test). In all bar charts, data are shown as mean ± SEM. Ant.A, antimycin A; DTT, dithiothreitol; SP, substance P.
S9SP had a similar effect on recombinant CaV3.2 (main T-type channel subunit in DRG) that was transiently overexpressed in human embryonic kidney (HEK293) cells together with NK1 receptors (Supplementary Fig. S1A). The inhibition of T-type channels by S9SP was accompanied by a modest leftward shift of the CaV3.2 voltage dependence (Supplementary Fig. S3A1) and a small but significant slowing of the inactivation kinetics (Supplementary Fig. S3A2). In accord with previous findings (58), in DRG neurons, S9SP had no significant effect on the high-voltage-activated (HVA) Ca2+ currents that are mainly conducted by N-, P/Q-, and L-type VGCCs (55) (Supplementary Fig. S3B).
Reversibility of the S9SP-induced T-type current inhibition by DTT suggested a redox-related mechanism. Thus, we investigated whether exogenous or endogenous ROS can produce action similar to S9SP on the channels. Bath application of H2O2 (300 μM) inhibited LVA current in cultured DRG neurons by ∼20% (Fig. 3C, D; scatter plots for inhibition in each individual neuron in this and other similar experiment series are shown in Supplementary Fig. S4). The effect of H2O2 was also reversed by DTT. Application of S9SP after (and in the presence of) H2O2 did not produce a significant further inhibition of the LVA current (Fig. 3D). H2O2 also inhibited recombinant CaV3.2, an effect that was reversed by DTT (Supplementary Fig. S1B).
The mitochondrial electron transport chain (ETC) is one of the main sources of intracellular ROS (70), and SP was shown to induce ROS release from mitochondria in DRG neurons (33). Inhibition of the ETC complex III with antimycin A has been shown to cause a burst release of mitochondrial ROS (76), and antimycin A-induced ROS release augmented the activity of redox-sensitive M-type K+ channels in DRG neurons (33). Consistently, application of antimycin A (1 μM) inhibited LVA current in DRG by ∼25% (Fig. 3E), and this effect was completely reversed by DTT (Fig. 3E1). S9SP applied after the pre-application and in the continuous presence of antimycin A only had a marginal further effect (Fig. 3E2).
These experiments suggest that a major component of S9SP-mediated T-type channel inhibition is mediated by endogenous ROS (although a contribution from additional mechanisms cannot be excluded).
SP acts by increasing CaV3.2 sensitivity to ambient zinc
Recently, we showed that hydrogen sulfide (H2S) inhibits CaV3.2 by increasing channel sensitivity to zinc (15), a trace metal that inhibits CaV3.2 at sub-micromolar concentrations and is present in sufficient amounts in biological fluids to affect CaV3.2 activity (27, 66). Therefore, we tested whether a similar mechanism might account for the SP-mediated T-type channel inhibition.
Application of 750 nM ZnCl2 caused inhibition of LVA current in DRG neurons by ∼25% (p < 0.001; n = 9; Fig. 3A), but when S9SP was applied in the presence of zinc, it had no further inhibitory effect (Fig. 4A). Furthermore, S9SP did not produce any effect in the presence of the zinc chelator N,N,N′,N′-Tetrakis(2-pyridylmethyl)ethylenediamine (TPEN; 10 μM; Fig. 4B and Supplementary Fig. S4). S9SP-induced inhibition was completely reversed by bath application of TPEN (still in the presence of S9SP, Fig. 4C and Supplementary Fig. S4). Similar to DRG neurons, SP- and H2O2-induced inhibitions of recombinant CaV3.2 were prevented by TPEN (Supplementary Fig. S1C, D).
FIG. 4.

Extracellular zinc mimics SP action on T-type Ca2+ channels. (A) Extracellular ZnCl2 inhibited LVA Ca2+ currents in DRG neurons. (A1) Example time course of the effect of ZnCl2 (750 nM), S9SP (1 μM), and MIB (3 μM) on the LVA Ca2+ current recorded form small-diameter DRG neurons using whole-cell patch clamp. Periods of drug application are indicated by the vertical gray bars; (A2) summarizes the effects. (B) Zinc chelator TPEN (10 μM) prevents SP-mediated LVA current inhibition in DRG neurons. (B1) Example time course of the effect of TPEN (10 μM), S9SP (1 μM), and MIB (3 μM) on the LVA current; (B2) summarizes the effects; layout and labeling are similar to (A). (C) TPEN reverses the SP-induced LVA current inhibition in DRG neurons. (C1) Example time course of the effect of S9SP (1 μM), TPEN (10 μM), and MIB (3 μM) on the LVA current; (C2) summarizes the effects; layout and labeling are similar to (A, B). Asterisks indicate significant difference from the group indicated by the line connector with **p < 0.01 or ***p < 0.001 (paired t-test). In all bar charts, data are shown as mean ± SEM. TPEN, N,N,N′,N′-Tetrakis(2-pyridylmethyl)ethylenediamine.
To accept a hypothesis that SP promotes T-type channel inhibition by ambient zinc and that this effect is mediated by a redox-dependent mechanism, two questions need to be addressed. (i) Is there enough ambient zinc to mediate the observed effects (as all our solutions are nominally zinc free, unless stated otherwise)? (ii) Does SP induce any changes to extracellular or intracellular zinc levels in DRG neurons?
First, we measured the zinc content of extracellular solutions and media using the atomic absorption spectroscopy (AAS; Fig. 5A). Both the DRG culture medium and the extracellular bath solution contained micromolar concentrations of zinc (∼10 μM in Dulbecco's modified Eagle medium [DMEM] and ∼5 μM in extracellular bath solution), which were similar to the reported plasma zinc levels in humans (44). There were no detectable changes in zinc content in the extracellular media after up to 10 min incubation of DRG cultures with S9SP (Fig. 5A). Notably, AAS detects total zinc content and the free zinc is likely to be significantly lower than the 5–10 μM; this can explain the significant inhibition of LVA current observed in the extracellular solution supplemented with 750 nM ZnCl2 (Fig. 4A).
FIG. 5.

SP does not change extracellular or intracellular zinc levels in DRG cultures. (A) Nominally zinc-free media contains micromolar levels of zinc. Atomic absorption spectroscopy measurements of total zinc in experimental media (as indicated); left eight bars correspond to the standard solutions of ZnCl2 in de-ionized water. Bars in the middle correspond to the levels of zinc in experimental media: DMEM is a DRG culture medium tested either on its own or collected from a DRG culture plate (“DMEM” and “DMEM+DRG”); EC is an extracellular solution used in patch-clamp and imaging experiments tested either on its own or collected from a DRG culture plate (“EC” and “EC+DRG”). Two bars on the right correspond to the experiments where zinc content has been analyzed in the EC of DRG cultures treated with 1 μM S9SP for 2 or 10 min (as indicated). (B) SP does not produce detectable rises in intracellular zinc levels in DRG neurons. Fluorescence imaging of cultured small-diameter DRG neurons loaded with zinc fluorophore, FluoZin™-3AM. Example time course of the effect of S9SP (1 μM), ZnCl2 (1.5 mM) and zinc ionophore, ZnPy (10 μM) on FluoZin-3 fluorescence. Corresponding images of the culture under basal conditions and after the application of ZnPy (as indicated) are shown on the right. (C) Increase in FluoZin-3 fluorescence induced by 10 μM ZnPy is completely reversed by 20 μM zinc chelator, TPEN but is unaffected by 1 mM DTT. (D, E) Summarize the experiments presented in (B, C), respectively. (D) Asterisks indicate significant difference from the basal fluorescence with ***p < 0.001 (paired t-test); (E) Asterisks indicate significant difference from the fluorescence in the presence of ZnPy with ***p < 0.001 (paired t-test). In all bar charts, data are shown as mean ± SEM. DMEM, Dulbecco's modified Eagle medium; ZnPy, zinc pyrithione. To see this illustration in color, the reader is referred to the web version of this article at www.liebertonline.com/ars
Next, we measured intracellular zinc levels using confocal fluorescence microscopy of DRG cultures loaded with the zinc indicator FluoZin™-3AM. Application of S9SP did not produce detectable changes in intracellular zinc levels; however, large responses were recorded in response to 1.5 mM ZnCl2 or zinc ionophore zinc pyrithione (ZnPy; 10 μM; Fig. 5B, D). In a separate experiment, we detected significant intracellular zinc transients in response to acidification (pH 5.5; Supplementary Fig. S5). Acidification has been previously shown to release intracellular zinc from cytosolic zinc-cysteine complexes (28) and, thus, acidification-induced zinc transients are likely to represent intracellular zinc release.
We then tested whether DTT can chelate zinc, as has been suggested (30, 48). We first loaded DRG cultures with zinc by the application of ZnPy and then applied, in sequence, 1 mM DTT and 20 μM TPEN (still in the presence of ZnPy; Fig. 5C, E). Cell-permeable (19) DTT did not alter the elevated zinc levels, whereas TPEN completely reversed the increase in fluorescence produced by ZnPy (Fig. 5C, E). This suggests that TPEN effectively competed for zinc binding with the zinc-sensitive dye at a molar ratio of 2:1, whereas DTT was unable to produce such an effect even at a molar ratio of 200:1. The experiments presented in Figure 5 and Supplementary Figure S5 established that (i) ambient zinc is present in nominally zinc-free solutions and media at levels that are sufficient to affect CaV3.2 activity (27, 66); (ii) we could not detect significant changes in intracellular or extracellular zinc levels in DRG neurons in response to SP; and (iii) we could not detect zinc chelator activity of DTT. Although this last observation cannot rule out zinc binding by DTT, in principle, it argues against zinc chelation by DTT as the reason underlying the reversibility of the SP effect on T-type channels by DTT in our experiments. Instead, we hypothesize that oxidation increases and reduction decreases channel sensitivity to zinc. We further suggest that SP induces T-type channel inhibition in DRG neurons by producing ROS-mediated modification of T-type channel protein, which, in turn, alters the channel sensitivity to zinc.
To directly test whether SP treatment changes zinc sensitivity of CaV3.2, we investigated concentration dependency of zinc effect on the recombinant CaV3.2 overexpressed in HEK293 cells together with NK1 receptors with and without S9SP treatment. We applied 10 μM TPEN in the nominally zinc-free bath solution at the beginning of each of such experiments to achieve near-zero extracellular free zinc levels and then applied increasing concentrations of ZnCl2 (now without TPEN; Fig. 6A, B).
FIG. 6.

SP increases zinc-mediated inhibition of CaV3.2 that is mediated by a high-affinity binding site. (A) Exemplary patch-clamp recording from HEK293 cell co-transfected with CaV3.2 and NK1 showing concentration dependency of the effect of extracellular ZnCl2 on CaV3.2 currents. TPEN (10 μM) was applied at the beginning of the experiment to record CaV3.2 current amplitude at a near-zero extracellular zinc concentration. Increasing concentrations of ZnCl2 were applied during the periods indicated by vertical gray bars. (B) Experiment similar to (A) but increasing ZnCl2 concentrations were applied after application of 1 μM S9SP; 1 μM S9SP was present in all ZnCl2 solutions. (C). The data from 6–12 experiments as in (A, B) were fit to two-component logistic function (see Materials and Methods section). The solution containing 10 μM TPEN and no added ZnCl2 was assigned a free zinc concentration of 0.05 nM. In control conditions, the IC50(1) was 0.25 ± 1.4 nM and the IC50(2) was 495 ± 326 nM. After the S9SP treatment, the IC50(1) was 0.35 ± 0.25 nM and the IC50(2) was 923 ± 582 nM. (C) Data are shown as mean ± SEM. HEK, human embryonic kidney.
The concentration dependency was best fit with the two-component logistic function (Fig. 6C; see Materials and Methods section). The data obtained in control conditions could be fit well with the one-component function as well [in agreement with previous reports (27, 66); not shown], as the inhibition at low nanomolar zinc concentrations was small. However, a far better fit was achieved with the two-component logistic function, which clearly separated two zinc effects (Fig. 6C). Such bi-phasic concentration dependence is likely to indicate that CaV3.2 uniquely possesses high-affinity (sub-micromolar) and low-affinity (micromolar) zinc binding sites. The latter is likely to be shared with other CaV3 subunits and other types of VGCC channels, as these are also inhibited by micromolar (but not nanomolar) zinc levels (6, 27, 66).
S9SP treatment strongly increased the efficacy of zinc-mediated inhibition via the high-affinity mechanism while having no effect on the inhibition at higher concentrations (Fig. 6C). The maximal inhibition at the high-affinity site in the control and S9SP-treated cells was 9.9% ± 11.3% and 44.1% ± 5.1%, respectively; the IC50 values for high- and low-affinity sites did not change significantly. We estimated that free zinc concentrations in our solutions are in the low nanomolar range, since S9SP inhibited CaV3.2 in nominally zinc-free solutions by ∼40% (Supplementary Fig. S1A and Fig. 6); however, TPEN produced only marginal augmentation of CaV3.2 currents in control conditions (Supplementary Fig. S1C, D and Fig. 6A, B). These experiments also explain why application of 750 nM extracellular zinc abolished the S9SP effect (Fig. 4A), as at concentrations higher than 100 nM the difference in zinc-induced inhibition between control and S9SP-treated cells started to disappear (Fig. 6C). The presence of low-nanomolar free zinc in nominally zinc-free solutions introduces a slight systematic error to the concentration-dependency measurements; however, we believe that the overall conclusion that SP treatment enhances inhibition of CaV3.2 by zinc acting at its high-affinity site is valid.
The unique high-affinity metal-binding site of CaV3.2 has been elucidated (27): It is composed of an Asp-Gly-His motif in the extracellular S3–S4 linker of the domain I and a separate Asp in the S1–S2 linker of the same domain (Fig. 7A1, A2); this site is also critical for channel modulation by redox agents (26, 27, 47, 48). His191 plays a central role in the zinc-mediated channel inhibition. This residue is absent in CaV3.1 and CaV3.3; accordingly, these channels are far less sensitive to zinc.
FIG. 7.

High-affinity zinc binding site is necessary for CaV3 channel sensitivity to SP. (A1) Depicts the amino-acid sequence alignment of the transmembrane segments S3 and S4 of domain I (with extracellular linker) of CaV3.1 and CaV3.2; non-conserved H191 of CaV3.2 is highlighted in red. (A2) Simplified schematic of domain I of CaV3 channels. (B) H191Q substitution in CaV3.2 renders the channel insensitive to SP inhibition. (B1) Example time course of the effect of S9SP (1 μM), DTT (1 mM), and MIB (3 μM) on the CaV3.2(H191Q) mutant recorded in HEK293 cell co-transfected with NK1 receptor; inset shows example current traces. (B2) Shows summary of the effects. (C) Q172H substitution in CaV3.1 renders the channel sensitive to SP inhibition. (C1, C2) Example time courses of the effects of S9SP (1 μM) and MIB (3 μM) on the wtCaV3.1 (C1) and CaV3.1(Q172H) mutant (C2) recorded in HEK293 cell co-transfected with NK1 receptor. Insets show example current traces; (C3) shows summary of the effects. The number of experiments is shown within the bars in (B2, C3). Asterisks indicate significant difference from the group indicated by the line connector with ***p < 0.001 (paired t-test). In all bar charts, data are shown as mean ± SEM. To see this illustration in color, the reader is referred to the web version of this article at www.liebertonline.com/ars
To test whether SP-mediated CaV3.2 channel inhibition requires this high-affinity zinc-binding site, we tested the SP sensitivity of two reciprocal mutants: CaV3.2 with H191 substituted by glutamine (CaV3.2H191Q) and CaV3.1 with H-to-Q substitution at the homologous position 172 (CaV3.1Q172H; Fig. 7A1, A2). Notably, CaV3.2H191Q displayed no sensitivity to SP (Fig. 7B); current amplitude in the presence of S9SP was 103.1% ± 9.8% of basal value (n = 8) versus 69.8% ± 6.8% for the wtCaV3.2 (p < 0.001; n = 13; Supplementary Fig. S1A). Similarly insensitive to S9SP was the wild-type CaV3.1 (Fig. 7C1); whereas CaV3.1Q172H mutant, in contrast, displayed small but significant inhibition by S9SP (10.4% ± 1.6%; p < 0.05; n = 8; Fig. 7C2, C3). In accord with previous reports, Q172H substitution in CaV3.1 only partially restored channel sensitivity to sub-millimolar zinc, as the entire binding site is required for the full effect (27).
Effects of novel T-type Ca2+ channel modulators on sensory neuron excitability
The T-type Ca2+ current is an important regulator of sensory neuron excitability (10, 17, 18, 20, 23, 24, 51, 71, 75). To evaluate the contribution of T-type Ca2+ channel modulation to the effects of SP on sensory neuron excitability both in vitro and in vivo, we used recently developed specific and selective T-type Ca2+ channel modulators: spiro[imidazo[1,2-a]pyridine-3,2-indan]-2(3H)-one (ST101), a highly active CaV3 potentiator (13, 45) with an EC50 in sub-nanomolar range, and Z944, a selective CaV3 inhibitor with IC50 ∼50 nM (67). Z944 (1 μM) inhibited LVA current in DRG neurons by 71.4% ± 2.7% (p < 0.001; n = 7), without a noticeable effect on HVA current (Fig. 8A); at this concentration, Z944 produced no significant effect on L- and N-type VGCCs, Nav1.5 and hERG channels (67).
FIG. 8.

Modulation of T-type channel activity in DRG neurons reveals the channel's role in control over the excitability. (A) Z944 potently inhibits LVA but not HVA Ca2+ currents in DRG neurons. (A1) Shows example whole-cell patch clamp recordings, and (A2) shows summarized data for the effects of 1 μM Z944 on LVA (left) and HVA (right) Ca2+ currents; voltage protocols are depicted above the traces. (B) ST101 potently augments LVA but not HVA Ca2+ currents in DRG neurons; layout and labeling are similar to (A1, A2); to inhibit HVA current, MIB was used at 10 μM. (A, B) HVA currents were recorded from neurons lacking the LVA current. *p < 0.05, ***p < 0.001; paired t-test. (C1, C2) Effect of Z944 (C1) and a T-type channel activator, ST101 (C2) on small DRG neuron excitability recorded in perforated patch-clamp current mode. Current injection protocol is depicted beneath the traces in (C). Extended analysis of the ST101 and Z944 effects on excitability is given in Supplementary Figure S7. (A, B) Data are shown as mean ± SEM. HVA, high-voltage activated; ST101, spiro[imidazo[1,2-a]pyridine-3,2-indan]-2(3H)-one. To see this illustration in color, the reader is referred to the web version of this article at www.liebertonline.com/ars
ST101 (1 nM) enhanced peak LVA current in DRG neurons by 50.6% ± 14.1% (p < 0.01; n = 7), also without an effect on HVA current (Fig. 8B); at 1 nM, ST101 had no effect on delayed-rectifier K+ current and voltage-gated Na+ current in DRG neurons (Supplementary Fig. S6). Although several selective blockers of T-type Ca2+ channels were reported (10, 17), selective T-type channel potentiators were not available until recently.
We next tested the effect of Z944 and ST101 on DRG neuron excitability using the perforated current clamp technique. Z944 decreased (Fig. 8C1) and ST101 increased (Fig. 8C2) small DRG neuron excitability; these effects are further analyzed in Supplementary Figure S7: Z944 significantly increased the rheobase, decreased action potential (AP) firing, and hyperpolarized small-diameter DRG neurons without significantly affecting AP amplitude or duration. In contrast, ST101 significantly increased AP firing and depolarized Erest; AP amplitude or duration was not affected (Supplementary Fig. S7A–C).
S9SP produced a pattern of effects similar to that of Z944 (Supplementary Fig. S7A–C), although only the effect on rheobase (Supplementary Fig. S7A) reached significance. Of note, mean values and the statistical significance of the effects shown in Supplementary Figure S7 are underestimated, since not all DRG neurons express all the necessary components of the signaling cascade. Thus, ∼40% of small DRG neurons in our cultures express LVA currents; ∼40% to 50% of small neurons express functional NK1 receptors [(33), present study]; and ∼70% of LVA-expressing neurons respond to S9SP. Therefore, in each series of experiments, there was a subpopulation of “unresponsive” neurons.
We further tested whether SP or a T-type Ca2+ channel blocker would attenuate the excitatory effect of the pro-algesic peptide bradykinin (BK) (36, 52). BK (250 nM) significantly reduced the rheobase, increased the AP number, and depolarized the Erest (Supplementary Fig. S7A–C). When co-applied with BK, both S9SP and Z944 completely reversed the effects of BK on the rheobase and the AP number (Supplementary Fig. S7A, B). The BK-induced depolarization in the presence of either S9SP or Z944 was more variable (Supplementary Fig. S7C), perhaps reflecting the complexity of the depolarizing effects of BK (25, 36, 52), but in both cases the effect of BK was no longer statistically significant.
To confirm that the effects of Z944 and ST101 on sensory neuron excitability are, indeed, mediated by T-type channels, we repeated some of the current clamp experiments in DRG neurons in which CaV3.2 expression has been downregulated with siRNA against Cacna1h. The procedure (see Materials and Methods section) reduced the Cacna1h transcript levels by ∼50% while having no effect on the housekeeping transcript (Gapdh; Supplementary Fig. S8A, B). In the Cacna1h knocked-down neurons, neither Z994 nor ST101 produced any significant effect on Erest, AP number, or rheobase (Supplementary Fig. S8C–E); whereas in scrambled oligonucleotide-transfected neurons, both drugs were still effective (cf. Supplementary Figs. S7 and S8C–E).
Taken together, the experiments reported in Figure 8 and Supplementary Figures S7 and S8 suggest that (i) T-type channels modulate excitability of many small-diameter DRG neurons; (ii) acute inhibition of T-type channels produces an anti-excitatory effect that can partially offset the excitatory action of BK; (iii) SP produces an anti-excitatory effect that is qualitatively similar to that produced by a T-type channel blocker; and (iv) effects of Z944 and ST101 on DRG neuron excitability are, indeed, mediated by T-type Ca2+ channels.
T-type Ca2+ channel inhibition contributes to the peripheral anti-nociceptive effect of SP in vivo
We next investigated the influence of T-type channels on peripheral sensory afferents in vivo. First, we tested whether T-type channel augmentation is, indeed, pro-nociceptive. An injection of ST101 (50 μl; 0.001–10 nmol/site) into the rat hind paw induced a moderate but significant protective effect (“nocifensive” behavior, see Materials and Methods section) that was indicative of moderate pain. The pro-algesic effect of ST101 was smaller than that induced by BK (50 μl; 10 nmol/site), but it was significantly stronger than that of saline (Fig. 9A).
FIG. 9.

T-type Ca2+ channel contributes to the peripheral analgesic effect of SP. (A) Plantar hind-paw injection of ST101 (50 μl; 0.001–10 nmol/site) and BK (50 μl; 10 nmol/site) induced nocifensive behavior in rats. Nocifensive behavior (time spent licking, flinching, and biting the paw) was quantified over the period of 30 min. Extended analysis of the data is given in Supplementary Figure S9. (B) Effects of plantar hind-paw injections of ST101 (0.001 nmol/site), M channel opener, RTG (50 μl; 0.1–10 nmol/site), Z944 (50 μl; 0.005–0.5 nmol/site), or saline on the paw withdrawal latency on exposure to noxious heat (Hargreaves test). (C) Effects of plantar hind-paw injections of ST101 (50 μl; 0.001 nmol/site), Z944 (50 μl; 0.5 nmol/site), S9SP (50 μl; 0.5 nmol/site), or saline on the response rate on application of von Frey filaments of varying bending force to the plantar surface of the hind paw. Vertical dotted line indicates 60% withdrawal threshold of saline-injected rats. (D) Effects of plantar hind-paw injections of ST101 (50 μl; 0.001 nmol/site), Z944 (50 μl; 0.5 nmol/site), S9SP (50 μl; 0.5 nmol/site), or saline on the sensitivity to gentle touch in cotton-swab test. (E) Plantar hind-paw injection of S9SP (50 μl; 0.5–5 nmol/site) failed to induce nocifensive behavior in rats (in contrast to BK). #, *, **, ***Significantly different from the control group (C) or from the group indicated by the line connector (all other panels) with p < 0.05, p < 0.01, or p < 0.001; Kruskal–Wallis ANOVA and Mann–Whitney test. All data are shown as mean ± SEM. ANOVA, analysis of variance; BK, bradykinin; RTG, retigabine. To see this illustration in color, the reader is referred to the web version of this article at www.liebertonline.com/ars
Time courses of the nociceptive effects of BK and ST101 are shown in Supplementary Figure S9. The nocifensive behavior induced by BK usually tailed out after 15 min post-injection (Supplementary Fig. S9A); whereas the effects of ST101 showed a distinct pattern, with lower concentrations causing a delayed response and higher doses producing more immediate and shorter effects (Supplementary Fig. S9B). Such different kinetic patterns may indicate that excitations produced by BK and ST101 have distinct mechanisms.
We then evaluated the effects of Z944, ST101, and an M-type K+ channel activator, retigabine (RTG), on animal sensitivity to noxious heat using the Hargreaves’ method. RTG has well-known peripheral analgesic activity (13, 21, 36, 50), and M-channel potentiation also contributes to the endogenous anti-nociceptive effect of SP (33). A peripheral hind-paw injection of Z944 (50 μl; 0.005–0.5 nmol/site) significantly increased paw withdrawal latency on radiant heat exposure, suggesting decreased heat sensitivity (Fig. 9B). RTG (50 μl; 0.1–10 nmol/site) produced a similar effect. In contrast, ST101 significantly reduced withdrawal latency, indicating increased heat sensitivity (Fig. 9B); time courses of these effects of Z944 and RTG are given in Supplementary Figure S9C.
CaV3.2 is expressed in subpopulations of Aδ and C low-threshold mechanoreceptors (LTMRs) and plays a role in tuning sensitivity to light touch (18). Thus, we tested the effects of local application of Z944, ST101, and S9SP on a range of mechanical stimulations.
In von Frey test (see Materials and Methods section), saline-injected rats had a 60% withdrawal threshold at 9.2 ± 1.0 g (n = 25). Injections of Z944 (50 μl; 0.5 nmol/site) and S9SP (50 μl; 0.5 nmol/site) significantly impaired their ability to respond to stimulation, ranging from innocuous to noxious; Z944 was more efficacious in the noxious range (Fig. 9C). In contrast, an injection of ST101 (50 μl; 0.001 nmol/site) greatly increased response rates (Fig. 9C). The cotton swab assay (see Materials and Methods section) also showed significantly reduced sensitivity to light touch after the S9SP injection (50 μl; 0.5 nmol/site); Z944 (50 μl; 0.5 nmol/site) produced a similar effect (Fig. 9D). An injection of ST101, however, was ineffective in this type of test.
We then tested the contribution of T-type channel modulation to the peripheral effect of S9SP. Consistent with previous reports (33), a local hind-paw injection of S9SP (0.5–5 nmol/site) did not produce measurable nocifensive behavior; whereas BK induced a strong nocifensive response (Fig. 9E).
We then pre-injected RTG, Z944, or S9SP into the rat hind paw 5 min before the BK injection (together with the same pre-injection drug; Fig. 10A, D). RTG served as a positive control, as it was shown to offset excitatory effects of BK and to reduce the BK-induced nociception (33, 34, 36). Pre-injections of both RTG (50 μl; 1–10 nmol/site) and Z944 (at 0.5 nmol/site but not at 0.05 nmol/site) significantly attenuated the BK-induced nociception. A pre-injection of S9SP (0.5 nmol/site) also strongly suppressed the BK-induced nocifensive behavior (Fig. 10A). Importantly, a mixture of RTG (1 nmol/site) and Z944 (0.5 nmol/site) was as efficacious as S9SP in reducing BK-induced nocifensive behavior and showed a tendency to be more efficacious than RTG or Z944 injected alone (p = 0.059 vs. RTG alone; Fig. 10A); this finding is consistent with a hypothesis that M-channel augmentation and T-type channel inhibition can be cumulative components of peripheral anti-nociceptive effects of SP.
FIG. 10.
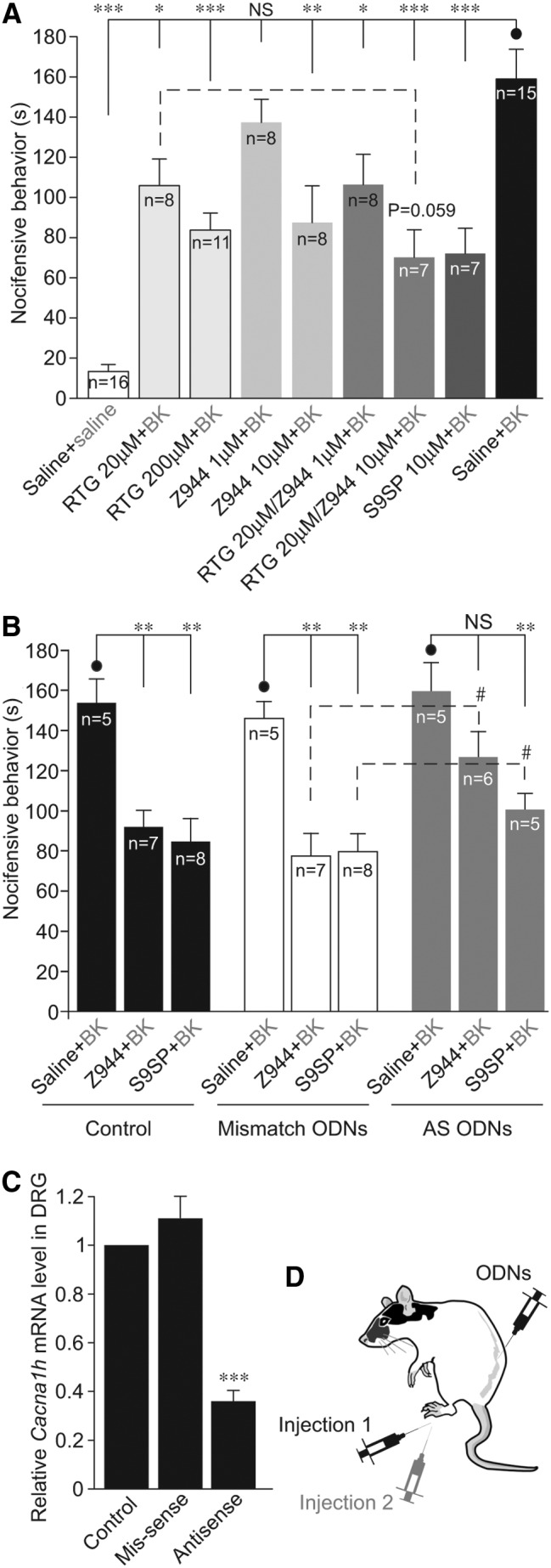
NK1 receptor-mediated T-type Ca2+ channel inhibition is a likely contributor to the anti-nociceptive effect of SP. (A) Plantar hind-paw injections of S9SP (50 μl; 0.5 nmol/site), RTG (50 μl; 1–10 nmol/site), Z944 (50 μl; 0.05–0.5 nmol/site) or a combination of RTG+Z944 solutions (at concentrations indicated) were administered 5 min before the injection of BK (10 nmol/site) and mixed together with the pre-injection drug or drug mixture in 50 μl saline [schematic of the double-injection protocol is shown in (D)]. The number of animals in each group is shown within the bars. (B) Intrathecal knock-down of CaV3.2 in lumbar DRG reduces anti-nociceptive effect of Z994 and S9SP. Animals with intrathecal injection of saline, CaV3.2 AS ODNs, or non-targeting “mismatch” ODNs were investigated. Hind-paw injections of S9SP (50 μl; 0.5 nmol/site) or Z944 (50 μl; 0.5 nmol/site) were administered 5 min before the injection of BK (10 nmol/site), similar to experiments shown in (A). (C) Quantification of knock-down efficiency; the analysis was performed on the second day after the last intrathecal injection. The CaV3.2 transcript levels in lumbar DRGs of AS ODN- and mismatch ODN-injected animals were normalized to those of saline-injected animals in the same experimental run; one to three animals per run, three independent runs. *, **, ***Significantly different from the group indicated by the line connector with p < 0.05, p < 0.01, or p < 0.001; Kruskal–Wallis ANOVA and Mann–Whitney test. All data are shown as mean ± SEM. AS, antisense; ODNs, oligodeoxynucleotides.
Finally, we performed in vivo knock-down of CaV3.2 in DRG neurons by an intrathecal injection of antisense (AS) phosphorodiester oligodeoxynucleotides (ODNs) using the protocol that had been successfully used earlier for the same purpose [(3, 41); Materials and Methods section]. In saline- and mismatch ODN-injected animals, both Z944 (0.5 nmol/site) and S9SP (0.5 nmol/site) still significantly attenuated nocifensive behavior produced by the hind-paw BK injection. However, in CaV3.2 AS ODN-injected animals, the nocifensive behavior produced by both Z944 and S9SP was significantly reduced (Fig. 10B). The levels of CaV3.2 transcript in lumbar DRGs of CaV3.2 AS ODN-injected animals were reduced by ∼70% as compared with the saline-injected controls and mismatch ODN-injected animals (Fig. 10C).
Discussion
Here, we described a hitherto unknown endogenous signaling pathway linking NK1 receptors with CaV3.2 channel activity and demonstrated that this pathway is involved in the paradoxical peripheral anti-nociceptive effect of SP. T-type Ca2+ channels are sensitive to redox modulation; oxidizing agents or ROS inhibit channel activity (47, 65), whereas reducing agents such as L-cysteine or DTT reverse such inhibition and can augment channel activity above the basal level in some cases [(46, 65); Fig. 3 and Supplementary Fig. S1].
SP induces ROS release in immune (64) and epithelial (60) cells and in DRG neurons (33). Accordingly, SP-induced inhibition of T-type Ca2+ channels observed in this study displayed the following features: (i) It was fully reversed or prevented by DTT (Fig. 3); (ii) mimicked by exogenously applied H2O2 (Fig. 3 and Supplementary Fig. S1); and (iii) stimulated by endogenous ROS release (Fig. 3E).
The mechanism of the redox sensitivity of T-type Ca2+ channels is not entirely understood, but it requires the extracellular His191 in CaV3.2; the other CaV3 isoforms do not have a corresponding histidine and are largely redox insensitive (47). The same H191 mediates CaV3.2 inhibition by sub-micromolar concentrations of zinc (26, 27). H191 is a general metal-binding site, and it was hypothesized that it could participate in the metal-catalyzed oxidation (MCO) reaction accelerated by transition metals (47, 61).
It is also possible that an oxidative modification (possibly but not necessarily at H191) can promote zinc binding or potentiate its action on the channel, thus sensitizing it to ambient concentrations of this trace metal. The following observations are in favor of this latter mechanism. (i) SP induced a large increase in zinc-mediated inhibition at the high-affinity site in CaV3.2 (Fig. 6). (ii) A mutation of H191 in CaV3.2 rendered the channels insensitive to SP (Fig. 7). (iii) CaV3.1 is insensitive to SP but insertion of His into the position equivalent to H191 in CaV3.2 renders the CaV3.1 channel sensitive to SP (Fig. 7). (iv) The zinc chelator TPEN prevents or reverses SP-induced T-type Ca2+ current inhibition (Fig. 4 and Supplementary Fig. S1). (v) Extracellular zinc inhibits LVA currents in DRG neurons, an effect that is similar in amplitude to that produced by SP (Fig. 4). (vi) Extracellular zinc occludes any further inhibitory actions of SP (Fig. 4). (vii) We could not detect any changes in intracellular or extracellular zinc levels induced by SP (Fig. 5 and Supplementary Fig. S5). (viii) Nominally zinc-free media contain low-nanomolar concentrations of free zinc, which are sufficient to affect channel activity when the channel is sensitized by SP (Fig. 6); however, these levels are too low to significantly inhibit T-type channels in their tonic state. The last observation would explain why TPEN produced only a marginal effect on T-type currents under basal conditions (Figs. 4B and 6B and Supplementary Fig. S1C, D).
Despite previous suggestions (30, 48), we could not detect zinc chelation by DTT under our experimental conditions (Fig. 5). Therefore, we suggest that the reversal of the SP-mediated T-type channel inhibition by DTT is due to the reducing activity of the latter.
Interestingly, there are three extracellular cysteines (residues that can be easily modified by redox mechanisms) in the IS1-IS2 linker of CaV3.2, a domain that contributes to the high-affinity zinc-binding site (27). We hypothesize that cumulative oxidation of such extracellular cysteines may result in a graded increase in channel sensitivity to zinc. In such a scenario, CaV3.2 tonically exists in a partially “oxidized” state with intermediate zinc sensitivity, which is, however, too low for inhibition by ambient zinc. This could be why in our hands TPEN or DTT had only marginal effects on channel activity under control conditions.
The partially oxidized state can then be converted to a fully oxidized state, with the highest zinc sensitivity by SP or ROS, or to a fully reduced state, with the lowest zinc sensitivity by DTT. This could account for the augmentation of tonic CaV3.2 currents by DTT (65) and TPEN (15) that are observed in some labs, as different experimental conditions may result in different “tonic” redox states of the channel (e.g., DTT or TPEN would potentiate fully oxidized channels). This hypothesis requires further verification.
The levels of NK receptor expression in peripheral sensory neurons are reportedly low. Thus, one immunohistochemical study found little evidence for NK receptor immunoreactivity in rat sensory neurons (5); whereas recent RNAseq experiments found very low levels of NK receptor transcripts in mouse DRG (39, 68), although the levels were higher in human DRG (16). However, in both mouse (39) and human (16) DRG, the NK receptor transcript levels were comparable (or even higher) to those of the brain or lung, tissues where prominent SP signaling is well characterized. Moreover, the DRG NK receptor levels are comparable to those of BK B2 receptors (16, 39), which exert robust effects in DRG neurons [reviewed in Petho and Reeh (52)].
Perhaps low transcript levels and expression in only specific cell populations are sufficient for functional activity of such G-protein-coupled receptors. In support of this notion, in vitro and, particularly, in vivo evidence presented here and elsewhere (32, 33, 73) clearly demonstrate the presence of functional SP signaling in DRG. In addition, NK1 receptor immunoreactivity was found throughout DRG neuron somata and often co-localized with that of CaV3.2, especially in small-diameter neurons (Supplementary Fig. S2).
There is also some discrepancy regarding cellular effects of SP in cultured DRG neurons. Thus, some potentially excitatory effects, including depolarization (1, 74), potentiation of Nav1.8 Na+ channels (7), L- and N-type Ca2+ channels (58), and sensitization of TRPV1 (33, 57, 73), were reported. However, there is no acute pain produced by local SP injections [(33); present study]. Application of SP to human skin by dermal microdialysis produces no painful sensation or primary afferent excitation (72). Thus, at present, the excitatory effects of SP in cultured neurons are difficult to reconcile with the lack of behavioral responses to local applications of SP. However, excitatory actions of SP may underlie responses of neurons innervating some internal organs, for example, bladder (74).
Opposing modulation of T-type channel activity by the latest generation of selective T-type channel modulators, ST101 (potentiator) and Z944 (inhibitor) confirmed the role of this channel in the control of sensory neuron excitability (Fig. 8 and Supplementary Figs. S7 and S8). Accordingly, a peripheral injection of ST101 induced moderate “spontaneous” nociception and increased sensitivity to both light and noxious touch, whereas Z944 reduced heat and touch sensitivity (Fig. 9); these effects are consistent with the analgesic efficacy of Z944 (31).
Importantly, we now show that both S9SP and Z944 significantly attenuate nociception induced by a peripheral injection of BK and in vivo knock-down of CaV3.2 in DRG reduces such attenuation (Fig. 10). Co-application of Z944 with M-channel opener RTG produced a similar anti-nociceptive effect as that of S9SP (Fig. 10).
Since both M-type (33) and T-type (present study) channels are modulated by SP (although in opposite directions) via a signaling cascade that shares common initial steps, we suggest that both M-channel augmentation and T-type channel inhibition contribute to the redox-mediated anti-nociceptive action of SP in peripheral nerves (Fig. 11). We hypothesize that since peptidergic nociceptive fibers release SP locally in response to noxious stimulation, this neuropeptide may exert local endogenous analgesia in an autocrine/paracrine fashion by simultaneous inhibition of T-type Ca2+ channels and augmentation of M channels in nociceptors.
FIG. 11.

Schematic of the proposed peripheral anti-nociceptive signaling via NK1 receptors. Local tissue damage excites peptidergic nociceptors that release SP into the tissue damage area. Acting at the NK1 receptors expressed in the neighboring fiber endings (potentially including the SP-releasing fibers as well), SP triggers mitochondrial ROS release and redox-dependent inhibition of T-type Ca2+ channels and potentiation of M-type K+ channels, resulting in hyperpolarization and reduced excitability of the fibers. ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertonline.com/ars
Although we have previously shown that SP does induce mitochondrial ROS release in DRG neurons (33), the release of ROS from other, non-neuronal NK1-expressing cells surrounding the site of tissue damage cannot be excluded (particularly in the in vivo experiments), especially given the fact that the redox-sensitive site in T-type channels (but not M-type channels) is located extracellularly. Additionally, activation of NK receptors in DRG may result in the release of other substances that can modulate T-type channel activity, such as thioredoxin (4) or H2S (14, 15). Interestingly, both factors inhibit CaV3.2 channels by interfering with their extracellular zinc-binding site (4, 14, 15).
Finally, although we could not detect any changes in intracellular or extracellular zinc levels in response to SP in vitro, one cannot exclude that some degree of redox-mediated zinc liberation from intracellular ligands, such as metallothioneins (28), or even extracellular zinc-bound proteins could occur (especially in the in vivo situation). Such zinc release could potentially contribute to the SP-mediated T-type channel inhibition, although in the case of intracellular zinc, it would need to be transported to the extracellular space to be able to affect T-type channel activity.
Clearly, further research is needed to address these intriguing questions; nevertheless, the present study firmly establishes redox-dependent modulation of T-type Ca2+ channels in nociceptive neurons as a mechanism contributing to the acute anti-nociceptive action of SP in the peripheral nociceptive system.
Materials and Methods
Cell culture, transfections, complementary DNA constructs, and chemicals
DRG neurons from adult male Sprague–Dawley rats (170–180 g) were dissociated as previously described (13, 29) and cultured in DMEM supplemented with GlutaMax I, 10% fetal calf serum, penicillin (50 U/ml), and streptomycin (50 μg/ml) on glass coverslips that were coated with poly-d-lysine for 2–5 days in a humidified incubator (37°C, 5% CO2). No growth factors were added to the culture media. HEK 293 cells were cultured in the same DMEM media; cells were passaged every 2 days. Human NK1 receptor (GenBank accession No. AY462098) complementary DNA (cDNA) was from the Missouri Science and Technology cDNA Resource Center. WtCaV3.1 (GenBank accession No. AF027984), wtCaV3.2 (GenBank accession No. AF051946), CaV3.1Q172H, and CaV3.2H191Q were kindly provided by Dr E. Perez-Reyes (University of Virginia). HEK293 cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. ST101 was synthesized in house; Z944 was from Toronto Research Chemicals; PTX was from Enzo Life Science; and FluoZin-3AM was from Life Technologies. All other chemicals were from Sigma.
siRNA gene silencing
To knock down CaV3.2, DRGs were dissociated as described (13); immediately after dissociation, DRG pellets were resuspended in 900 μl DMEM (without GlutaMax I, serum or antibiotics), to which a mixture of 9 μl Lipofectamine RNAiMAX (Invitrogen) and either a mix of three anti-Cacna1h Stealth siRNAs (RSS350285, RSS350286, RSS350287; Life Technologies; 200 nM each) or a scrambled control oligo (Stealth RNAi™ siRNA Negative Control; Life Technologies; 200 nM) were added immediately before resuspension. Cultures were than plated in 50 μl droplets onto glass coverslips that were coated with poly-d-lysine and incubated for 5–6 h; the transfection media were then replaced with full DRG culture medium. Patch-clamp recordings were conducted 24–48 h after the transfection. The following primers were used for reverse transcription polymerase chain reaction (RT-PCR) analysis:
Cacna1h sense: 5′-TGCCCACGGAGTCTATGAGT-3′
Cacna1h AS: 5′-GTTGTAGGGGTTCCGGATGT-3′
Gapdh sense: 5′-GACATGCCGCCTGGAGAAAC-3′
Gapdh AS: 5′-AGCCCAGGATGCCCTTTAGT-3′
To knock down Gi/o or Gq/11 Gα subunits, Silencer Select RNAi siRNA (Invitrogen) oligonucleotides were used; the DRG dissociation and transfection were performed as described earlier. A mixture of the siRNA against Gq and G11 or against Gi1-3 and Go was co-transfected. The following siRNA oligos were used:
Anti-Gnaq sense: 5′-UGACUCUACCAAAUACUAUtt-3′
Anti-Gnaq AS: 5′-AUAGUAUUUGGUAGAGUCAga-3′
Anti-Gna11 sense: 5′-GCAUCGCUACAGUAGGCUAtt-3′
Anti-Gna11 AS: 5′-UAGCCUACUGUAGCGAUGCgg-3′
Anti-Gnai1 sense: 5′-GAAGCUGUUCGAUAGCAUAtt-3′
Anti-Gnai1 AS: 5′-UAUGCUAUCGAACAGCUUCat-3′
Anti-Gnai2 sense: 5′-CAGAGUGACUAUAUCCCUAtt-3′
Anti-Gnai2 AS: 5′-UAGGGAUAUAGUCACUCUGtg-3′
Anti-Gnai3 sense: 5′-CAAUGAUUCUGCUUCAUAUtt-3′
Anti-Gnai3 AS: 5′-AUAUGAAGCAGAAUCAUUGag-3′
Anti-Gnao sense1: 5′-GCAGAUGAAGAUCAUCCAUtt-3′
Anti-Gnao AS1: 5′-AUGGAUGAUCUUCAUCUGCtt-3′
Anti-Gnao sense2: 5′-CCUCCACUUCAGGCUGUUUtt-3′
Anti-Gnao AS2: 5′-AAACAGCCUGAAGUGGAGGtt-3′
The following primers were used for RT-PCR analysis:
Gnaq sense: 5′-TCCCAGAATATGATGGACCCC-3′;
Gnaq AS: 5′-CGGATGTTCTCCGTGTCTGT-3′.
Gna11 sense: 5′-ACAAGGCCAATGCACTCCTG-3′;
Gna11 AS: 5′-GGTCCACGTCCGTCAAGTAG-3′.
Gnai1 sense: 5′-GGATGATGCTCGCCAACTCT-3′;
Gnai1 AS: 5′-CATTCAGGTAGTACGCCGCC-3′.
Gnai2 sense: 5′-TACACAGGGGCCAACAAGTAT-3′;
Gnai2 AS: 5′-CTCACAGGTAGTGGGGAGCA-3′.
Gnai3 sense: 5′-TGTAGGTGGCCAAAGATCCG-3′;
Gnai3 AS: 5′-TGCATTCGGTTCATTTCCTCG-3′.
Gnao1 sense: 5′-AAAGCAAAAACCGCTCACCC-3′;
Gnao1 AS: 5′-CATGAAGCAGTCAAATAGGTTGC-3′.
Electrophysiology
All recordings were made using Multiclamp 700B amplifier in combination with pCLAMP 10.4 software (Molecular Devices). Voltage-clamp recordings from DRG neurons were made using whole-cell patch clamp. The standard bath solution contained the following (in mM): 150 TEA-Cl; 2.5 CsCl; 2.5 CaCl2; 10 HEPES; 0.5 MgCl2; and 10 glucose (pH 7.4 adjusted with CsOH). The standard pipette solution contained the following (in mM): 110 CsCl; 3 MgCl2; 10 EGTA; 10 HEPES; 3 Mg-ATP; and 0.6 GTP (pH 7.4 adjusted with CsOH). The access resistance was typically within 4–8 MΩ. T-type calcium currents (LVA) were measured by 50 ms square voltage pulses to −40 mV from a holding potential of −90 mV. HVA currents were measured by 200 ms square voltage pulses to 0 mV from a holding potential of −90 mV.
In recordings from HEK 293 cells, amphotericin B-perforated patch-clamp technique was used (13). The pipette solution contained the following (in mM): 155 CsCl; 10 HEPES; 1 EGTA; and 4 MgCl2 supplemented with amphotericin B (250 μg/ml), pH 7.4 adjusted with CsOH. The bath solution was the same as for DRG neuron recordings. The access resistance was typically within 8–12 MΩ.
Voltage-clamp recordings were sampled at 4 kHz. Series resistance was compensated online by 50%; the voltage error due to series resistance in our recordings did not exceed 6 mV and typically was within ∼2 mV.
In experiments with H2O2, 3% agar-salt (3 M KCl) bridges were used. Concentration dependence of CaV3.2 channel inhibition by zinc was fit to the two-component logistic function:
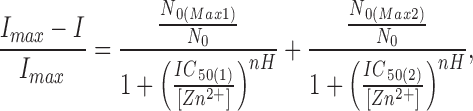 |
where N0 is the total amount of zinc-binding sites, N0(max1) is the amount of high-affinity binding sites, N0(max2) is the amount of low-affinity binding sites, Imax is the maximal CaV3.2 peak current amplitude, IC50 is the [Zn2+] necessary for half-maximal inhibition, and nH is the Hill coefficient. The concentration dependency of the S9SP effect was fit to the one-component logistic function:
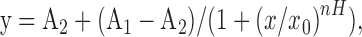 |
where y is the current inhibition; A1 and A2 are the maximum and minimum effects, respectively; x is the S9SP concentration; and nH is the Hill coefficient.
Neuronal excitability was studied using the amphotericin B-perforated patch-clamp current technique (13). The extracellular solution contained the following (in mM): 160 NaCl, 2.5 KCl, 5 CaCl2, 1 MgCl2, 10 HEPES, and 8 Glucose. The intracellular solution contained the following (in mM): 145 KCl, 15 NaCl, 5 MgCl2, 10 HEPES, and 250 μg/ml Amphotericin B. The access resistance was typically within 8–12 mΩ. APs were generated by an injection of 200 pA current for 1 s from a holding current of 0 pA. Rheobase was determined from a train of current steps from 0 to 400 pA delivered in 20-pA increments. Current clamp recordings were sampled at 12.5 kHz. Bridge balance adjustments were not applied, since only relative changes in Erest, AP number, and rheobase were investigated.
Measurement of zinc concentrations
The concentrations of zinc in experimental solutions were determined using an automated Ion Channel Reader 8000 flame atomic absorption spectrometer (Aurora Biomed) according to the manufacturer's instructions. To test whether SP induces zinc release from the cultured DRG cells, DRG cultures were grown on 10 mm round coverglass in 24-well plates; cultures were washed with extracellular solution (EC) and incubated in 0.5 ml EC that was supplemented with 1 μM S9SP for 2–10 min; and supernatants were collected and analyzed for zinc content.
Zinc imaging
Changes in the intracellular zinc levels were evaluated using fluorescence imaging. DRG cultures were loaded with FluoZin-3AM (5 μM for 30 min at 37°C in the presence of 0.02% pluronic F-127). Cells were washed with extracellular bath solution composed of the following (in mM): 160 NaCl; 2.5 KCl; 2 CaCl2; 1 MgCl2; and 10 HEPES (pH 7.4 adjusted with NaOH); they were imaged using a Leica SP5 fluorescence imaging system assembled on a Leica DMI6000 microscope and illuminated with 488 nm light for 1200 ms (400 Hz) with a 2 s interval. Solutions were applied using a gravity perfusion system.
Behavioral assays
Sprague–Dawley rats (body weight, 170–180 g) were grouped randomly and allowed to acclimate for at least 30 min in a transparent observation chamber before the experiment. Animals received a 50-μl intraplantar injection of ST101 (0.001–10 nmol/site), BK (10 nmol/site), RTG (1–10 nmol/site), Z944 (0.05–5 nmol/site), SP (0.5–5 nmol/site), or saline, respectively, into the right hind paw. For evaluation of the acute “spontaneous” pain, animals were video-recorded for 30 min after the injection and protective (“nocifensive”) behavior was analyzed as time spent licking, biting, lifting, and flinching the injected paw. The analyses were performed by an observer who was unaware of treatment allocations. In the pre-injection experiments, the right hind paw of the animal received a pre-injection of 50 μl Z944 (0.05–0.5 nmol/site), RTG (1–10 nmol/site), SP (0.5 nmol/site), Z944 (0.05–0.5 nmol/site)+RTG (1 nmol/site), or saline; 5 min later, on the same paw, the animal received a further 50-μl injection of BK (10 nmol/site) together with the pre-injection drug mix. Nocifensive behavior was analyzed as described earlier.
For measurement of thermal hyperalgesia, the change in the latency of hind-paw withdrawal in response to noxious heat was recorded using the Hargreaves’ plantar method (Ugo Basile) at 8 min after the hind-paw injection of drugs; the heat source was set to 25% of the maximal intensity.
For measurement of sensitivity to gentle touch and mechanical hyperalgesia, two methods were used. (i) Cotton swab test was performed as previously described (53). Briefly, rats were allowed to acclimate in a cage with a wire mesh bottom for at least 30 min and then were manually probed with a “cotton swab” pulled from a Q-tip (to approximately three times the original size). A swab was applied in a sweeping motion underneath the rats’ paws, and the withdrawal responses were recorded. Five sweeps were applied in each test, with at least 10 s between each sweep; withdrawal score was calculated as a percentage of positive responses. (ii) The von Frey test was performed using a calibrated range of filaments with different bending forces (2, 4, 6, 8, 10, and 15 g; Stoelting Co.). A single filament was applied perpendicularly to the plantar surface of the hind paw for five times, with an interval of 5 s, and it was scored for withdrawal responses. Each force was probed five times, the number of withdrawals out of five trials was counted, and the percentage of withdrawal responses was calculated. Both types of assays were performed 8 min after the hind-paw drug injection.
Intrathecal administration of ODNs targeting CaV3.2 subunit
The in vivo CaV3.2 knock-down was performed as described earlier (3) by using the same ODN sequences that were synthesized by SBS Genetech. AS ODNs had the following sequence: CCACCTTCTTACGCCAGCGG. The mismatch ODNs of the following sequence were used as a negative control: TACTGTACTTGCGAGGCCAC. The mismatch ODN was not complementary to any known nucleotide sequence. ODNs were reconstituted in saline before injection. ODNs (12.5 μg/rat in 10 μl of saline) or saline were injected intrathecally (i.t.) between the L5 and L6 dorsal spinous processes as described earlier (3, 42) under isoflurane anesthesia. The treatment was repeated twice daily for 4 days. The pain tests were performed on the next day after the last injection. After behavioral testing, animals were humanly sacrificed; lumbar DRGs were extracted and analyzed for the knock-down efficiency by comparing the Cacna1h mRNA levels in lumbar (L4 to L6) DRGs of the saline-injected, mismatch ODN-injected, and AS ODN-injected animals using RT-PCR. The same Cacna1h and Gapdh primers as in siRNA experiments were used for RT-PCR.
Reagents
Signaling peptide bradykinin (Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg; BK; Sigma); NK1-selective tachykinin receptor agonists [Sar9]-Substance P (Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Sar-Leu-Met-NH2; S9SP; Sigma) and [Sar9, Met(O2)11]-Substance P (Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Sar-Leu-Met[O2]-NH2; S9M11SP; synthesized by SBS Genetech); NK1-selective antagonist CP-99994 (Sigma); reducing agent DTT (Sigma); Gi/o pathway inhibitor PTX (Sigma); M-type channel opener RTG (synthesized in house); VGCC blocker MIB (Sigma); T-type specific Ca2+ channel blocker Z944 (synthesized in house); cognitive enhancer and T-type Ca2+ channel activator ST101 (synthesized in house); zinc chelator TPEN (Sigma); and protein kinase C inhibitor, BIM I were used as reagents.
Statistics
All data are given as mean ± standard error of the mean. Differences between groups were assessed by Student's t test (paired or unpaired, as appropriate) or one-way analysis of variance (ANOVA) with Bonferroni correction. Behavioral experiments were analyzed using Kruskal–Wallis ANOVA; difference between groups was analyzed with Mann–Whitney test. The effects of the drugs on neuronal excitability were analyzed using either paired t test or paired Wilcoxon signed ranks test (for data sets that failed normality test). The differences were considered significant at p ≤ 0.05. Statistical analyses were performed using Origin 8.6 (OriginLab Corporation).
Supplementary Material
Abbreviations Used
- ANOVA
analysis of variance
- AP
action potential
- AS
antisense
- BIM I
bisindolylmaleimide I
- BK
bradykinin
- CNS
central nervous system
- CP-99994
(2S,3S)-3-(2-Methoxybenzylamino)-2-phenylpiperidine dihydrochloride
- DMEM
Dulbecco's modified Eagle medium
- DRG
dorsal root ganglion
- DTT
dithiothreitol
- Erest
resting membrane potential
- ETC
electron transport chain
- H2S
hydrogen sulfide
- HEK
human embryonic kidney
- HVA
high-voltage activated
- LVA
low-voltage activated
- MIB
mibefradil
- mRNA
messenger RNA
- NK
neurokinin
- ODN
oligodeoxynucleotide
- PTX
pertussis toxin
- ROS
reactive oxygen species
- RTG
retigabine
- RT-PCR
reverse transcription polymerase chain reaction
- S9M11SP
[Sar9, Met(O2)11]-Substance P
- S9SP
[Sar9]-Substance P
- SP
substance P
- ST101
spiro[imidazo[1,2-a]pyridine-3,2-indan]-2(3H)-one
- TPEN
N,N,N′,N′-Tetrakis(2-pyridylmethyl)ethylenediamine
- TRPV1
transient receptor potential cation channel subfamily V member 1
- VGCC
voltage-gated Ca2+ channel
- Z944
N-[[1-[2-(tert-butylamino)-2-oxoethyl]piperidin-4-yl]methyl]-3-chloro-5-fluorobenzamide
- ZnPy
zinc pyrithione
Acknowledgments
The authors thank Han Hao and Yuan Wang for expert technical assistance, Hongchao Men for help with immunohistochemistry, and Bingcai Guan for advice on electrophysiology. This work was supported by the National Natural Science Foundation of China (Grant Nos. 31270882 to H.Z., 31571088 to X.D., and 81201642 to H.G.), the National Basic Research Program (Grant No. 2013CB531302 to H.Z.), and MRC (Grant Nos. G1002183 to N.G. and MR/K021303/1 to C.P. and N.G.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abdulla FA, Stebbing MJ, and Smith PA. Effects of substance P on excitability and ionic currents of normal and axotomized rat dorsal root ganglion neurons. Eur J Neurosci 13: 545–552, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Ayoola C, Hwang SM, Hong SJ, Rose KE, Boyd C, Bozic N, Park JY, Osuru HP, DiGruccio MR, Covey DF, Jevtovic-Todorovic V, and Todorovic SM. Inhibition of CaV3.2 T-type calcium channels in peripheral sensory neurons contributes to analgesic properties of epipregnanolone. Psychopharmacology (Berl) 231: 3503–3515, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, and Nargeot J. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 24: 315–324, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boycott HE, Dallas ML, Elies J, Pettinger L, Boyle JP, Scragg JL, Gamper N, and Peers C. Carbon monoxide inhibition of Cav3.2 T-type Ca2+ channels reveals tonic modulation by thioredoxin. FASEB J 27: 3395–3407, 2013 [DOI] [PubMed] [Google Scholar]
- 5.Brown JL, Liu H, Maggio JE, Vigna SR, Mantyh PW, and Basbaum AI. Morphological characterization of substance P receptor-immunoreactive neurons in the rat spinal cord and trigeminal nucleus caudalis. J Comp Neurol 356: 327–344, 1995 [DOI] [PubMed] [Google Scholar]
- 6.Busselberg D, Michael D, Evans ML, Carpenter DO, and Haas HL. Zinc (Zn2+) blocks voltage gated calcium channels in cultured rat dorsal root ganglion cells. Brain Res 593: 77–81, 1992 [DOI] [PubMed] [Google Scholar]
- 7.Cang CL, Zhang H, Zhang YQ, and Zhao ZQ. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol Pain 5: 33, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catterall WA, Perez-Reyes E, Snutch TP, and Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57: 411–425, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA, Hill JA, and Campbell KP. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science 302: 1416–1418, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, and Todorovic SM. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol 80: 900–910, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coste B, Crest M, and Delmas P. Pharmacological dissection and distribution of NaN/Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol 129: 57–77, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Biasi S. and Rustioni A. Glutamate and substance P coexist in primary afferent terminals in the superficial laminae of spinal cord. Proc Natl Acad Sci U S A 85: 7820–7824, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du X, Hao H, Gigout S, Huang D, Yang Y, Li L, Wang C, Sundt D, Jaffe DB, Zhang H, and Gamper N. Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain 155: 2306–2322, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elies J, Scragg JL, Boyle JP, Gamper N, and Peers C. Regulation of the T-type Ca channel Cav3.2 by hydrogen sulfide: Emerging controversies concerning the role of H S in nociception. J Physiol 2016. [Epub ahead of print]; DOI: 10.1113/JP270963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elies J, Scragg JL, Huang S, Dallas ML, Huang D, MacDougall D, Boyle JP, Gamper N, and Peers C. Hydrogen sulfide inhibits Cav3.2 T-type Ca2+ channels. FASEB J 28: 5376–5387, 2014 [DOI] [PubMed] [Google Scholar]
- 16.Flegel C, Schobel N, Altmuller J, Becker C, Tannapfel A, Hatt H, and Gisselmann G. RNA-Seq analysis of human trigeminal and dorsal root ganglia with a focus on chemoreceptors. PLoS One 10: e0128951, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, Uebele VN, Renger JJ, Eschalier A, Ardid D, and Bourinet E. State-dependent properties of a new T-type calcium channel blocker enhance CaV3.2 selectivity and support analgesic effects. Pain 154: 283–293, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noel J, Wood JN, Moqrich A, Pongs O, and Bourinet E. The low-threshold calcium channel Cav3.2 determines low-threshold mechanoreceptor function. Cell Rep pii: , 2015 [DOI] [PubMed] [Google Scholar]
- 19.Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, and Shapiro MS. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J 25: 4996–5004, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, Francois A, Bourinet E, and Zamponi GW. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83: 1144–1158, 2014 [DOI] [PubMed] [Google Scholar]
- 21.Hayashi H, Iwata M, Tsuchimori N, and Matsumoto T. Activation of peripheral KCNQ channels attenuates inflammatory pain. Mol Pain 10: 15, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill R. NK1 (substance P) receptor antagonists – Why are they not analgesic in humans? Trends Pharmacol Sci 21: 244–246, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Iftinca MC. and Zamponi GW. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci 30: 32–40, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Jevtovic-Todorovic V. and Todorovic SM. The role of peripheral T-type calcium channels in pain transmission. Cell Calcium 40: 197–203, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA, Zhang H, and Gamper N. Activation of the Cl− channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci Signal 6: ra73, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang HW, Park JY, Jeong SW, Kim JA, Moon HJ, Perez-Reyes E, and Lee JH. A molecular determinant of nickel inhibition in Cav3.2 T-type calcium channels. J Biol Chem 281: 4823–4830, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Kang HW, Vitko I, Lee SS, Perez-Reyes E, and Lee JH. Structural determinants of the high affinity extracellular zinc binding site on Cav3.2 T-type calcium channels. J Biol Chem 285: 3271–3281, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiedrowski L. Proton-dependent zinc release from intracellular ligands. J Neurochem 130: 87–96, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirton HM, Pettinger L, and Gamper N. Transient overexpression of genes in neurons using nucleofection. Methods Mol Biol 998: 55–64, 2013 [DOI] [PubMed] [Google Scholar]
- 30.Krezel A, Lesniak W, Jezowska-Bojczuk M, Mlynarz P, Brasun J, Kozlowski H, and Bal W. Coordination of heavy metals by dithiothreitol, a commonly used thiol group protectant. J Inorg Biochem 84: 77–88, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Lee M. Z944: A first in class T-type calcium channel modulator for the treatment of pain. J Peripher Nerv Syst 19Suppl 2: S11–S12, 2014 [DOI] [PubMed] [Google Scholar]
- 32.Lin CC, Chen WN, Chen CJ, Lin YW, Zimmer A, and Chen CC. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc Natl Acad Sci U S A 109: E76–E83, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linley JE, Ooi L, Pettinger L, Kirton H, Boyle JP, Peers C, and Gamper N. Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc Natl Acad Sci U S A 109: E1578–E1586, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linley JE, Pettinger L, Huang D, and Gamper N. M channel enhancers and physiological M channel block. J Physiol 590: 793–807, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linley JE, Rose K, Patil M, Robertson B, Akopian AN, and Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: A signaling pathway mediating inflammatory nociception. J Neurosci 28: 11240–11249, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, and Gamper N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J Clin Invest 120: 1240–1252, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macdonald SG, Dumas JJ, and Boyd ND. Chemical cross-linking of the substance P (NK-1) receptor to the alpha subunits of the G proteins Gq and G11. Biochemistry 35: 2909–2916, 1996 [DOI] [PubMed] [Google Scholar]
- 38.Malcangio M. and Bowery NG. Peptide autoreceptors: Does an autoreceptor for substance P exist? Trends Pharmacol Sci 20: 405–407, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Manteniotis S, Lehmann R, Flegel C, Vogel F, Hofreuter A, Schreiner BS, Altmuller J, Becker C, Schobel N, Hatt H, and Gisselmann G. Comprehensive RNA-Seq expression analysis of sensory ganglia with a focus on ion channels and GPCRs in Trigeminal ganglia. PLoS One 8: e79523, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMahon SB, Bennet DLH, and Bevan S. Inflammatory mediators and modulators. In: Wall and Melzack's Textbook of Pain, edited by McMahon SB. and Koltzenburg M. Edinburgh: Elsevier Churchill Livingstone, 2006, pp. 49–72 [Google Scholar]
- 41.Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM, and Jevtovic-Todorovic V. In vivo silencing of the CaV3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 145: 184–195, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mestre C, Pelissier T, Fialip J, Wilcox G, and Eschalier A. A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods 32: 197–200, 1994 [DOI] [PubMed] [Google Scholar]
- 43.Meza U, Thapliyal A, Bannister RA, and Adams BA. Neurokinin 1 receptors trigger overlapping stimulation and inhibition of CaV2.3 (R-type) calcium channels. Mol Pharmacol 71: 284–293, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Moran VH, Stammers AL, Medina MW, Patel S, Dykes F, Souverein OW, Dullemeijer C, Perez-Rodrigo C, Serra-Majem L, Nissensohn M, and Lowe NM. The relationship between zinc intake and serum/plasma zinc concentration in children: A systematic review and dose-response meta-analysis. Nutrients 4: 841–858, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moriguchi S, Shioda N, Yamamoto Y, Tagashira H, and Fukunaga K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J Neurochem 121: 44–53, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Nelson MT, Joksovic PM, Perez-Reyes E, and Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci 25: 8766–8775, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson MT, Joksovic PM, Su P, Kang HW, Van Deusen A, Baumgart JP, David LS, Snutch TP, Barrett PQ, Lee JH, Zorumski CF, Perez-Reyes E, and Todorovic SM. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J Neurosci 27: 12577–12583, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez-Reyes E, Lee JH, Shin HS, and Todorovic SM. Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J Neurosci 27: 8250–8260, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orestes P, Bojadzic D, Lee J, Leach E, Salajegheh R, Digruccio MR, Nelson MT, and Todorovic SM. Free radical signalling underlies inhibition of CaV3.2 T-type calcium channels by nitrous oxide in the pain pathway. J Physiol 589: 135–148, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Passmore GM, Reilly JM, Thakur M, Keasberry VN, Marsh SJ, Dickenson AH, and Brown DA. Functional significance of M-type potassium channels in nociceptive cutaneous sensory endings. Front Mol Neurosci 5: 63, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117–161, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Petho G. and Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev 92: 1699–1775, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, Mathur J, Begay V, Coste B, Mainquist J, Wilson AJ, Francisco AG, Reddy K, Qiu Z, Wood JN, Lewin GR, and Patapoutian A. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516: 121–125, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richardson JD. and Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther 302: 839–845, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Rola R, Szulczyk PJ, and Witkowski G. Voltage-dependent Ca2+ currents in rat cardiac dorsal root ganglion neurons. Brain Res 961: 171–178, 2003 [DOI] [PubMed] [Google Scholar]
- 56.Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic-Todorovic V, and Todorovic SM. Immunohistological demonstration of CaV3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250: 263–274, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sculptoreanu A, Aura Kullmann F, and de Groat WC. Neurokinin 2 receptor-mediated activation of protein kinase C modulates capsaicin responses in DRG neurons from adult rats. Eur J Neurosci 27: 3171–3181, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sculptoreanu A, and de Groat WC. Protein kinase C is involved in neurokinin receptor modulation of N- and L-type Ca2+ channels in DRG neurons of the adult rat. J Neurophysiol 90: 21–31, 2003 [DOI] [PubMed] [Google Scholar]
- 59.Sindrup SH, Graf A, and Sfikas N. The NK1-receptor antagonist TKA731 in painful diabetic neuropathy: A randomised, controlled trial. Eur J Pain 10: 567–571, 2006 [DOI] [PubMed] [Google Scholar]
- 60.Springer J, Groneberg DA, Dinh QT, Quarcoo D, Hamelmann E, Braun-Dullaeus RC, Geppetti P, Anker SD, and Fischer A. Neurokinin-1 receptor activation induces reactive oxygen species and epithelial damage in allergic airway inflammation. Clin Exp Allergy 37: 1788–1797, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci 928: 22–38, 2001 [DOI] [PubMed] [Google Scholar]
- 62.Steinhoff MS, von Mentzer B, Geppetti P, Pothoulakis C, and Bunnett NW. Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiol Rev 94: 265–301, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, and Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19: 1895–1911, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanabe T, Otani H, Bao L, Mikami Y, Yasukura T, Ninomiya T, Ogawa R, and Inagaki C. Intracellular signaling pathway of substance P-induced superoxide production in human neutrophils. Eur J Pharmacol 299: 187–195, 1996 [DOI] [PubMed] [Google Scholar]
- 65.Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney JW, and Zorumski CF. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron 31: 75–85, 2001 [DOI] [PubMed] [Google Scholar]
- 66.Traboulsie A, Chemin J, Chevalier M, Quignard JF, Nargeot J, and Lory P. Subunit-specific modulation of T-type calcium channels by zinc. J Physiol 578: 159–171, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, Eduljee C, Jiang X, Smith P, Morrison JL, Jones NC, Braine E, Rind G, Fee-Maki M, Parker D, Pajouhesh H, Parmar M, O'Brien TJ, and Snutch TP. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med 4: 121ra19, 2012 [DOI] [PubMed] [Google Scholar]
- 68.Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, Linnarsson S, and Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18: 145–153, 2015 [DOI] [PubMed] [Google Scholar]
- 69.von Bohlen und Halbach O. and Dermietzel R. Neurotransmitters and Neuromediators. Handbook of Receptors and Biological Effects. Weinheim: Wiley-Vch Verlag Gmbh & Co., 2006, p. 386 [Google Scholar]
- 70.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, and Cheng H. Superoxide flashes in single mitochondria. Cell 134: 279–290, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Waxman SG. and Zamponi GW. Regulating excitability of peripheral afferents: Emerging ion channel targets. Nat Neurosci 17: 153–163, 2014 [DOI] [PubMed] [Google Scholar]
- 72.Weidner C, Klede M, Rukwied R, Lischetzki G, Neisius U, Skov PS, Petersen LJ, and Schmelz M. Acute effects of substance P and calcitonin gene-related peptide in human skin – A microdialysis study. J Invest Dermatol 115: 1015–1020, 2000 [DOI] [PubMed] [Google Scholar]
- 73.Zhang H, Cang CL, Kawasaki Y, Liang LL, Zhang YQ, Ji RR, and Zhao ZQ. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: A novel pathway for heat hyperalgesia. J Neurosci 27: 12067–12077, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Pietra C, Lovati E, and de Groat WC. Activation of neurokinin-1 receptors increases the excitability of guinea pig dorsal root ganglion cells. J Pharmacol Exp Ther 343: 44–52, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Qin W, Qian Z, Liu X, Wang H, Gong S, Sun YG, Snutch TP, Jiang X, and Tao J. Peripheral pain is enhanced by insulin-like growth factor 1 through a G protein-mediated stimulation of T-type calcium channels. Sci Signal 7: ra94, 2014 [DOI] [PubMed] [Google Scholar]
- 76.Zorov DB, Filburn CR, Klotz LO, Zweier JL, and Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192: 1001–1014, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.