

Abstract
Background
Neuronal loss and alpha‐synuclein (α‐syn) pathology are diagnostic of PD in the appropriate clinical context. However, some Parkinson's disease (PD) patients have comorbid Alzheimer's disease (AD) pathology on autopsy, including amyloid‐beta (Aβ) plaques and neurofibrillary tangles. Florbetapir (18F) is a PET ligand that detects Aβ pathology. We hypothesized that florbetapir (18F) imaging could detect Aβ pathology in PD dementia (PDD) patients before death. The aim of this study was to determine the utility of florbetapir (18F) PET imaging in detecting Aβ pathology in patients with autopsy‐confirmed PDD.
Methods
Five participants with PDD had florbetapir (18F) PET imaging before death as a part of a longitudinal research study of cognitive decline in PD. PET scans were evaluated by expert raters blinded to clinical and neuropathological information. At autopsy, all 5 participants underwent semiquantitative assessments of regional Aβ and tau immunohistochemistry.
Results
All participants met neuropathological criteria for PD. Two had both positive florbetapir (18F) scans and Aβ‐positive plaques in multiple brain regions. Regional florbetapir (18F) binding correlated with regional semiquantitative Aβ pathology in these cases. Three cases had negative florbetapir (18F) scans. Two of these had significant tau pathology without Aβ pathology, consistent with PSP in 1 case and argyrophilic grain disease in the other. The last case had a low level of AD neuropathological change.
Conclusions
Florbetapir (18F) Aβ imaging can detect the presence of Aβ neuropathology in patients with PDD. This imaging technique may aid the clinical evaluation of PDD patients to determine whether cognitive decline is occurring in the setting of Aβ accumulation.
Keywords: neuroimaging, autopsy, parkinsonism, neurodegeneration, tauopathy
Approximately 80% of Parkinson's disease (PD) patients develop functionally significant cognitive decline during their disease course,1 leading to significant morbidity.2 The neuropathological underpinning of cognitive decline in PD is heterogeneous, and a combination of alpha‐synuclein (α‐syn) aggregates, amyloid‐β (Aβ) plaque deposits, tau‐positive neurofibrillary tangles, and cerebrovascular changes appear to contribute to varying degrees in individual cases.3, 4 Several lines of evidence point to accumulation of Aβ pathology in addition to α‐syn as one contributor to dementia in PD.3, 4, 5, 6, 7
Florbetapir (18F) (herein, florbetapir) is a PET imaging radioligand that can detect fibrillar Aβ pathology in vivo in patients with cognitive decline,8 as well as in some cognitively healthy older individuals.9 In Alzheimer's disease (AD), florbetapir imaging accurately reflects Aβ pathology observed at autopsy.10, 11, 12 Aβ amyloid PET imaging with Pittsburgh compound B (PiB) during life was concordant with Aβ pathology on autopsy in a small sample of PD patients13 and dementia with Lewy body (DLB) patients.14 However, analogous studies using florbetapir PET imaging in PD have not been performed.
We hypothesized that Aβ amyloid imaging with florbetapir PET would correlate with Aβ amyloid neuropathology in PD patients with dementia. In this study, we present the neuro‐ and clinicopathological evaluations for 5 patients with clinical diagnoses of PDD who had florbetapir imaging before death.
Patients and Methods
Patients
We recruited patients from the Parkinson Disease and Movement Disorders Center of the University of Pennsylvania as a cohort within our Morris K. Udall Center for Parkinson Disease Research (John Q. Trojanowski, principal investigator). Five patients with clinical PD (according to the UK Brain Bank Criteria15) and had annual motor and cognitive assessments as part of our ongoing longitudinal study of cognitive decline in PD. All 5 participants were male and had gradually progressive parkinsonism accompanied subsequently by cognitive impairment. At death, each had clinically significant dementia as determined by an internal, blinded consensus conference, composed of three to five pairs of trained physician raters (movement disorders specialists or psychiatrists with expertise in PD cognition). Demographic, neuropsychological, and clinical data were reviewed for this determination, in keeping with the recommendations of the International Parekinson and Movement Disorder Society Task Forces for mild cognitive impairment (MCI)16 and dementia.17 Additional details of each participant's clinical course were retrospectively obtained from the medical record. All study protocols, including florbetapir PET imaging and neuropathological examination, were approved by the University of Pennsylvania Institutional Review Board. Informed consent was obtained before enrolling each participant in the study.
Clinical Assessments
Motor function was rated with the UPDRS and assessment of modified H & Y score. Motor phenotype (tremor‐dominant [TD] vs. postural instability gait dysfunction [PIGD]) was determined using a previously published formula.18 Briefly, item 10 from UPDRS part 2 and items 15, 16, 17, and 18 from UPDRS part 3 were averaged for the numerator. Items 12 and 13 from UPDRS part 2 and items 10, 11, and 12 from UPDRS part 3 were averaged for the denominator. A ratio of ≥1.15 was consistent with TD whereas a ratio of ≤0.90 was consistent with PIGD. Cognitive performance was assessed by the Mattis Dementia Rating Scale‐2 (DRS‐2) as a global cognitive measure and a comprehensive neuropsychological test battery covering attention, executive functions, memory, visuospatial functions, and language. Total DRS‐2 scores range from 0 to 144. Test performance at 1.5 standard deviations (SDs) or below the normative mean was considered abnormal.17 All testing was performed in the patient's on state. The presence of falls, depression, and hallucinations at scan time were determined by responses on the UPDRS, the 15‐item Geriatric Depression Scale, and the neuropsychiatric inventory questionnaire and then confirmed by chart review.
Aβ Amyloid Imaging
Brain neuritic Aβ plaque burden was assessed by PET neuroimaging with florbetapir. Briefly, participants underwent a 10‐minute PET scan 50 to 60 minutes after an intravenous bolus of 370 MBq (10 mCi) of florbetapir. Images were acquired with a 128 × 128 matrix (zoom 2) and reconstructed using iterative or row action maximization likelihood algorithms. Readers experienced in the evaluation of florbetapir PET images visually assessed the images according to current guidelines19 and published methods.11 Briefly, scans that evidenced increased retention of tracer in cortical gray matter, as shown by the apparent loss of contrast in gray or white matter in any two cortical regions, or intense uptake in at least one cortical region, were classified as Aβ amyloid positive. The majority rating of the three experts was used to classify the scans as either Aβ− or Aβ+. Expert readers were blinded to demographic, clinical, and neuropathological data, including postmortem diagnoses. Aβ amyloid burden was also expressed quantitatively as a global cortical/cerebellar standardized uptake value (SUV) ratio (SUVr) as previously described.11 The whole cerebellum, including gray and white matter, was used for reference. To determine the correlation between florbetapir PET imaging and Aβ‐positive deposits at autopsy, we analyzed regional SUVr values in the context of regional Aβ immunoreactivity using a semiquantitative analysis, as described below.
Neuropathological Assessments
All autopsies were performed at the hospital of the University of Pennsylvania within 15 hours of death. Gross neuropathological evaluation of fresh tissue included brain weight, depigmentation of SN and locus ceruleus, and patterns of cortical atrophy, as previously described.20 Brains were sectioned and fixed in neutral‐buffered formalin. Neuronal loss and gliosis were assessed by hematoxylin stain. As previously described,20 senile Aβ plaques and neurofibrillary tau tangles were assessed by thioflavin S staining and immunohistochemistry (see a previous work19 and references therein for monoclonal antibodies used for Aβ, phosphorylated tau, α‐syn, and transactive response DNA‐binding protein 43 kDa [TDP‐43]). Semiquantitative assessment of microscopic sections was performed as previously described.3 Immunostained slide sections were imaged using a Lamina Multilabel Scanner (PerkinElmer, Waltham, MA) at 20× magnification. All neuropathological diagnoses were finalized by a clinical neuropathologist at the University of Pennsylvania Center for Neurodegenerative Disease Research for the medical record. No radiological or other biomarker data were used in establishing postmortem diagnoses.
Statistical Analyses
The effect of expert scan rating on cognitive performance was determined by t test, for both total DRS‐2 score and for five DRS‐2 subscores (attention, initation/perseveration, construction, conceptualization, and memory). A linear mixed‐effects model21 was used to determine the contribution of multiple factors to raw SUV. This procedure accounts for correlations among raw SUV measures across different regions within a subject. A random intercept was included in the mixed‐effects model. Expert scan rating and individual cortical region were included as independent variables and treated as fixed effects. All coefficients (β) are expressed as the value (standard error) along with Z score and P value. Statistical tests were two sided and statistical significance was set at <0.05 level. All statistical analyses were performed using Stata/IC software (13.1; StataCorp LP, College Station, TX).
Results
Participant Characteristics
Demographics, clinical data, and neuropathological data are summarized in Table 1. Average disease duration was 10.8 years (SD, 3.1), and average age of onset and death were 62.6 (SD, 4.1) and 73.4 years (SD, 4.7), respectively. Scans were performed between 3.5 and 41 months before death (Table 1). Except for case 2, all 5 had DRS‐2 scores typical of PDD at time of scan and were felt to have PDD by consensus conference. Case 2 met criteria for PD‐MCI at scan time, but converted to PDD before death. At scan time, the 2 participants with the lowest total DRS‐2 (cases 1 and 4) also had the most severe motor phenotype as measured by modified H & Y score (4) and had the shortest scan–death intervals (3.5 and 5.5 months, respectively). Two participants with intermediate total DRS‐2 (cases 3 and 5) also had intermediate modified H & Y score (3) and longer scan–death intervals (41 and 32 months, respectively). TD versus PIGD motor phenotypes were determined using a previously published calculator (see Patients and Methods).18 Cases 1 to 4 were of the PIGD motor phenotype at time of scan, and case 5 was TD.
Table 1.
Demographic, clinical, imaging, and neuropathological data for study participants
| Case | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Demographic and clinical data | |||||
| Sex | M | M | M | M | M |
| Age of onset, years | 58 | 59 | 65 | 68 | 63 |
| Age at death, years | 73 | 67 | 78 | 78 | 71 |
| Scan–death interval, m | −3.5 | −36 | −41 | −5.5 | −32 |
| DRS‐2 scores at scan time | |||||
| Total | 92 | 134 | 114 | 106 | 117 |
| Attention | 25 | 36 | 31 | 31 | 30 |
| Initiation | 18 | 37 | 30 | 24 | 29 |
| Construction | 5 | 6 | 6 | 6 | 4 |
| Conceptualization | 36 | 36 | 32 | 30 | 35 |
| Memory | 8 | 19 | 15 | 15 | 19 |
| Diagnosis at scan time | PDD | PD‐MCI | PDD | PDD | PDD |
| Modified H & Y at scan time | 4 | 2 | 3 | 4 | 3 |
| Motor subtype at scan time | PIGD | PIGD | PIGD | PIGD | TD |
| Falls at scan time | + | − | − | + | − |
| Depression at scan time | + | − | − | + | − |
| Hallucinations at scan time | + | − | + | + | + |
| Florbetapir PET data | |||||
| Expert visual rating | Aβ− | Aβ− | Aβ− | Aβ+ | Aβ+ |
| Cortical average SUV | 472 ± 17 | 523 ± 26 | 526 ± 27 | 626 ± 23 | 842 ± 23 |
| Cortical:cerebellar SUVr | 0.94 | 0.99 | 1.46a | 1.19 | 1.55 |
| Cortical/centrum semiovale SUVr | 0.58 | 0.63 | 0.59 | 0.74 | 0.82 |
| Neuropathology data | |||||
| Brain weight, g | 1459 | 1367 | 1390 | 1474 | 1439 |
| Ventricular enlargement | None | Severe | Severe | None | Mild |
| SN gross depigmentation | Severe | Normal | Mild | Severe | Severe |
| Locus ceruleus gross depigmentation | Severe | Normal | Normal | Normal | Severe |
| Neuropathological diagnosis no. 1 | LBD, diffuse neocortical | LBD, limbic (transitional) | LBD, brainstem predominant | LBD, diffuse neocortical | LBD, diffuse neocortical |
| Neuropathological diagnosis no. 2 | PSP | Low likelihood of AD | AGD | Intermediate likelihood of AD | Intermediate likelihood of AD |
| Thal phase | 0 (A0) | 1 (A1) | 0 (A0) | 3 (A2) | 4 (A3) |
| Braak tau stage | V–V1 (B3) | II (B1) | 0 (B0) | III–IV (B2) | IV (B2) |
| CERAD stage | C0 | C0 | C0 | C2 | C3 |
All demographic and clinical data were obtained at or nearest to scan time. + = present; − = absent.
SUVr discordant with both pathologist's diagnosis and majority reading.
Case 1 presented with right‐hand tremor that spread to the left hand and chin after around 1 year. Dopamine replacement with levodopa therapy and a dopamine agonist were very helpful for motor symptoms. Seven years after motor onset, he had noticeable short‐term memory loss to family members. His course was complicated by visual hallucinations, significant sleep reversal, vivid dreaming, and dream re‐enactment behaviors, although a polysomnogram was not consistent with rapid eye movement sleep behavior disorder (RBD). His gait remained largely intact in the first 10 years without frequent falling. He complained of diplopia 13 years after the tremor began, although no objective oculomotor abnormalities were observed. Cognitive impairment was transiently responsive to acetylcholinesterase inhibitors. Case 2 presented with a 3‐year history of asymmetric bradykinesia that was preceded by anxiety and insomnia. He had no symptoms of RBD. Symptoms were dopamine responsive, followed several years later by motor fluctuations, including both dyskinesia and wearing off. He developed dementia 8 years after motor onset, with intermittent confusion, fluctuating short‐term memory, and agitation. He did not have frequent falls, hallucinations, or depression, but functional impairment resulting from cognitive deficits necessitated admission to an assisted living facility. Case 3 presented with asymmetric leg tremor, which was mildly dopaminergic responsive. Over time, the motor disease slowly progressed whereas the cognitive impairment contributed more to overall morbidity. He developed visual hallucinations and memory loss approximately 3 years after the motor symptoms and frank dementia approximately 10 years after motor onset. He did not have symptoms of RBD or depression. Case 4 presented with asymmetric cogwheel rigidity and relatively mild tremor, both of which were dopamine responsive. Cognitive impairment also emerged 3 years after motor onset. Similar to the previous case, formal dementia was not reached until 10 years after motor symptom onset. At time of scan, he had frequent falls and hallucinations, as well as vivid dreams without frank dream enactment. Case 5 presented with asymmetric arm tremor and difficulty with grasping in the right hand. He remained tremor dominant throughout his disease course. His family noted a pervasive sadness and stress in the year preceding diagnosis, but he was not diagnosed with depression. He was not dopamine responsive. Postural righting reflexes were relatively spared. Cognitive impairment began 2 years after motor onset and progressed to involve incoherent speech, poor attention, and forgetfulness. There were visual hallucinations, but no symptoms of RBD. An initial response to acetylcholinesterase inhibitors waned closer to death.
Florbetapir Aβ Imaging
Representative views from the florbetapir PET scans are depicted in Figure 1. Three of the five cases (cases 1, 2, and 3) were unanimously negative for Aβ amyloid, as determined by three expert raters. The remaining cases (cases 4 and 5) demonstrated delayed retention of florbetapir binding diffusely in cortical regions consistent with a positive Aβ amyloid scan designation. Cases with positive florbetapir scans (cases 4 and 5) did not have higher total DRS‐2 scores as compared to cases with negative scans (cases 1, 2, and 3) by t test (t(3) = 0.1133; P = 0.917). Individual DRS‐2 subscores (Table 1) were also not significantly different in positive cases versus negative cases (data not shown).
Figure 1.
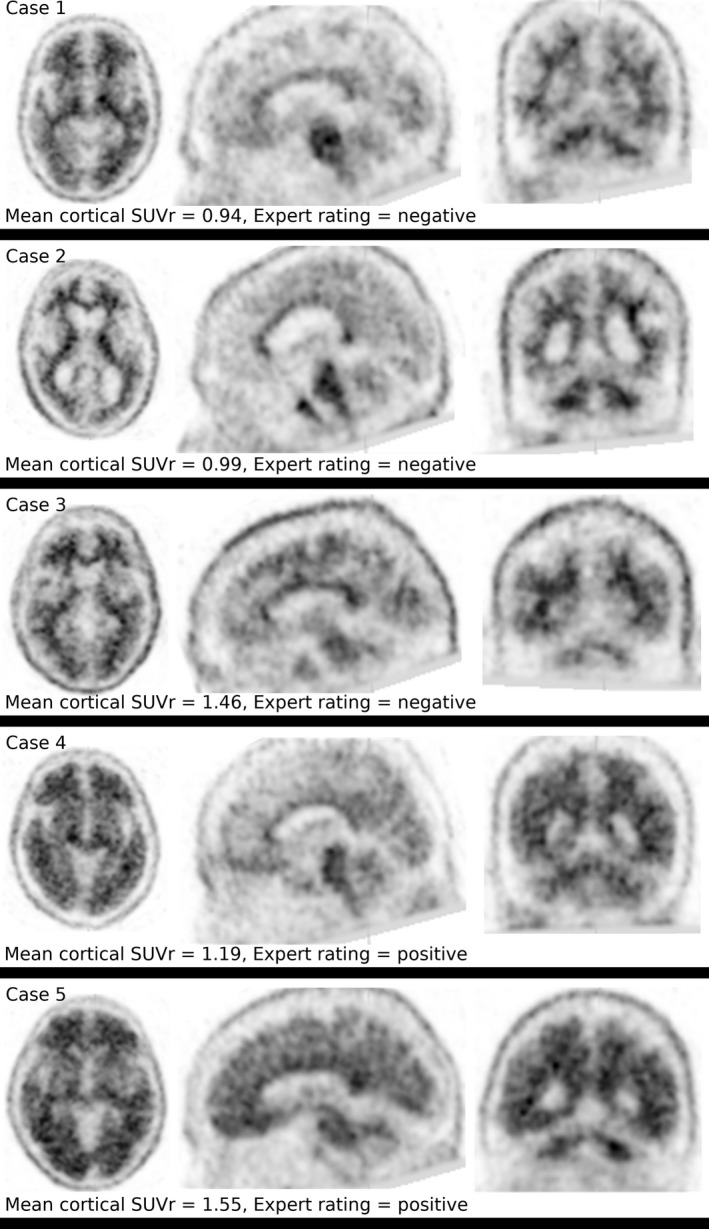
Florbetapir PET imaging of Aβ amyloid in 5 patients with PDD. Representative axial (left), sagittal (center), and coronal (right) views of florbetapir PET images are shown. Dark regions indicate increased florbetapir tracer retention. Scans from cases 1, 2, and 3 were interpreted as negative by expert readers and scans from cases 4 and 5 were positive (see Table 1 for details).
A diagnostic cut‐off value for cortical/cerebellar SUVr for florbetapir of 1.10 appears to be useful for identifying premortem AD.22 This cutoff was consistent with the expert rating for 4 of the 5 participants (Table 1). In case 3, the global cortical/cerebellar SUVr value (1.46) was inconsistent with the unanimous negative expert rating of the florbetapir PET imaging (Fig. 1; Table 1). Upon further analysis, the scan in this case demonstrated unusually low cerebellar uptake that elevated SUVr calculations (data not shown). For this reason, we analyzed raw SUV values for each of six cortical regions (frontal cortex, parietal cortex, temporal cortex, precuneus, anterior cingulate gyrus, and posterior cingulate gyrus), in addition to the average cortical SUV (Fig. 2). Cases with positive florbetapir scans (cases 4 and 5) has significantly higher mean cortical SUV values as compared to cases with negative scans (cases 1, 2, and 3) by a linear mixed‐effects model (β = 227(64.7); z = 3.5; P < 0.0001), whereas individual cortical region had no effect (β = 0.934(4.44); z = 0.21; P = 0.833). Because raw SUV can vary with factors unrelated to PET tracer binding,23 we also calculated a global cortical SUVr using the centrum semiovale as an alternate reference region.8 These SUVrs were consistent with expert rating of all 5 participants (Fig. 2; Table 1).
Figure 2.
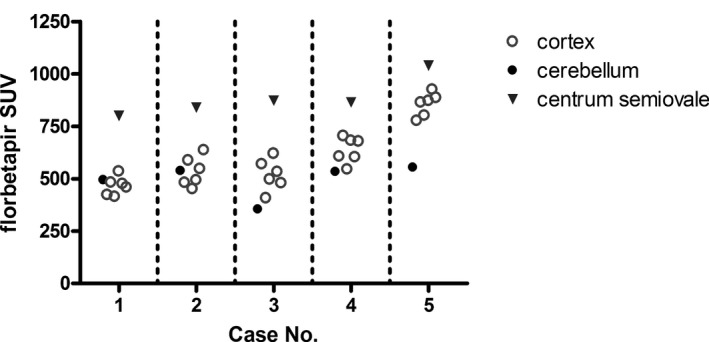
Individual SUVs for cortical brain regions. Florbetapir SUV measures for each of six brain regions (frontal cortex, parietal cortex, temporal cortex, precuneus, anterior cingulate gyrus, and posterior cingulate gyrus) are depicted in open circles for each participant. For comparison, SUVs for centrum semiovale and entire cerebellum are shown (triangles and closed circles, respectively). Mean cortical/cerebellar SUVr measures for participants 4 and 5 were significantly elevated as compared to the other participants (see Table 1 for details). The discordant global cortical/cerebellar SUVr value for case 3 (Table 1) was explained by an abnormally low whole‐cerebellar raw uptake value.
Neuropathological Examination
All five participants met neuropathological criteria for PD24 and had moderate‐to‐severe Lewy body (LB) pathology in the brainstem and limbic regions (Table 1). Three participants (cases 1, 4, and 5) also had significant LB pathology in cortical regions and severe depigmentation of the SN on gross examination. The remaining participants had LB pathology of the limbic transitional (case 2) or brainstem‐predominant (case 3) type.
Three participants (cases 2, 4, and 5) had detectable Aβ amyloid neuropathology (Table 2). Case 2 had low‐to‐moderate density of Aβ amyloid pathology in the angular gyrus, medial frontal lobe, and, to a lesser degree, the superior temporal lobe (Thal phase 1; A1) and only rare neuritic plaques in the frontal and angular cortex (Consortium to Establish a Registry for Alzheimer's Disease [CERAD] C0). There were mild neurofibrillary tau tangles and neuritic thread pathology in CA1 and limbic regions (Braak stage II; B1; Table 2). There was moderate intracranial artery atherosclerosis on gross examination. These findings were consistent with low probability of AD by National Institute on Aging (NIA)‐Reagan criteria25 and low level of AD neuropathological change by the new NIA/Alzheimer's Association criteria.26 Florbetapir Aβ imaging did not detect these low levels of Aβ neuropathological change in this participant (Fig. 1).
Table 2.
Semiquantitative neuropathological Aβ amyloid and tau burden
| Case | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Aβ | Tau | Aβ | Tau | Aβ | Tau | Aβ | Tau | Aβ | Tau | |
| Medial frontal lobe | 0 | 1+ | 1+ | Rare | 0 | 0 | 3+ | 1+ | 3+ | 0 |
| Superior/middle temporal lobe | 0 | 1+ | Rare | Rare | 0 | 0 | 3+ | 1+ | 3+ | Rare |
| Angular gyrus | 0 | 1+ | 2+ | 0 | 0 | 0 | 3+ | 0 | 3+ | Rare |
| Entorhinal cortex | 0 | 1+ | 0 | 1+ | 0 | 3+ | 2+ | 3+ | 3+ | 3+ |
| Anterior/posterior cingulate gyrus | 0 | 1+ | Rare | Rare | 0 | 1+ | 3+ | 2+ | 3+ | 1+ |
| CA1/subiculum | 0 | 1+ | 0 | 1+ | 0 | 2+ | 2+ | 2+ | 1+ | 2+ |
Case 4 had moderate neurofibrillary tangles in limbic regions and mild tangles in brainstem and neocortex (Braak III–IV; B2), as well as moderate neuritic plaques in neocortex and limbic regions (CERAD C2; Table 2). There was moderate intracranial artery atherosclerosis. Case 5 had tau‐positive neurofibrillary tangles in limbic regions and rarely in neocortical areas (Braak IV; B2), accompanied by a high burden of neocortical neuritic plaques (CERAD C3) and diffuse plaques in cortical, limbic, subcortical, and brainstem regions (Thal stage 4; A3; Table 2). This individual also had moderate Aβ amyloid angiopathy in the cerebellum and cortical and limbic regions, although intracranial artery atherosclerosis was mild on gross examination. These findings were consistent with intermediate probability (for case 4) and high probability (for case 5) of AD by NIA‐Reagan criteria.25 Both cases 4 and 5 met criteria for intermediate AD neuropathological change by NIA/Alzheimer's Association criteria26 and had positive Aβ PET imaging by florbetapir (Fig. 1).
The remaining 2 cases (nos. 1 and 3) did not have detectable Aβ amyloid neuropathology (Thal phase 0 and CERAD C0; Table 2; Fig. 3). Case 1 had mild‐to‐moderate neurofibrillary tangles in entorhinal and neocortex (Braak V–VI; B3) and a predominance of tau pathology in the amygdala, medulla, and cerebellum (Table 2). There were tau‐positive globose neurofibrillary tangles, rare coiled bodies, and frequent tufted astrocytes in both gray and white matter. Grumose degeneration was noted in the cerebellar dentate nuclei. Tangles were present to a lesser extent in both the STN and globus pallidus. There was severe gross intracranial artery atherosclerosis. Given the lack of Aβ plaques and the morphology and distribution of tau deposits, the neuropathological examination was most consistent with PSP27 in combination with LB disease (diffuse neocortical subtype).
Figure 3.
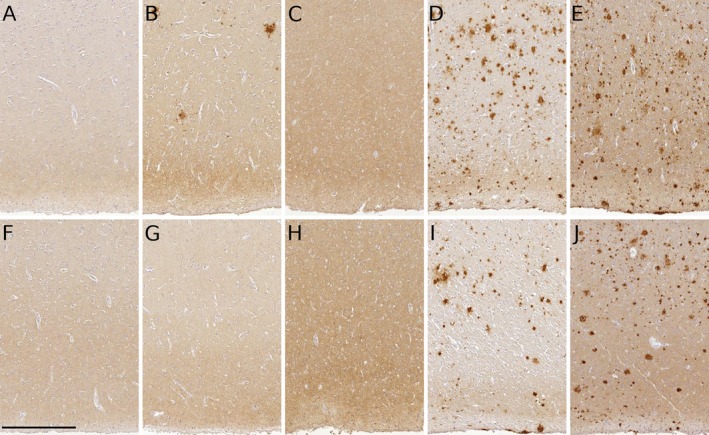
Representative Aβ neuropathology in 5 PDD patients. Medial frontal lobe (A–E) and temporal lobe (F–J) were examined for Aβ amyloid neuropathology using the NAB228 antibody.20 Cases 4 and 5 had significant Aβ amyloid neuropathology in medial frontal lobe (D and E) and temporal lobe (I and J), respectively, consistent with florbetapir Aβ amyloid imaging in these cases. No Aβ amyloid neuropathology was detectable in medial frontal lobe from cases 1 (A) and 3 (C) or in temporal lobe from cases 1 (F), 2 (G), and 3 (H). Low numbers of Aβ amyloid plaques were observed in medial frontal lobe from case 2 (B). Scale bar = 50 μm.
Case 3 had a dense burden of tau‐positive grain pathology in the amygdala and hippocampus, associated with granular intracellular “pretangle” inclusions in the hippocampus (Table 2). There were rare coiled bodies and diffuse white matter tau pathology in the entorhinal cortex. There were no intracellular neurofibrillary tangles (Braak 0; B0), consistent with argyrophilic grain disease (AGD)28 in addition to LB disease (brainstem‐predominant subtype). There was moderate gross intracranial artery atherosclerosis. Both participants 1 and 3 had negative florbetapir Aβ imaging (Fig. 1).
All 5 participants had moderate‐to‐severe neuronal loss in the SN, basal ganglia, and cortex. Gross depigmentation in locus ceruleus was only observed in 2 participants (cases 1 and 5). Mild‐to‐moderate Aβ amyloid angiopathy was only observed in case 5 and affected multiple cortical regions. There were no cerebral infarctions or hemorrhages and no microvascular disease in any case. No significant TDP‐43 or ubiquitin‐positive pathology was observed in any participant.
Discussion
In this study, we examined 5 patients with PDD using florbetapir PET imaging and correlated these findings with Aβ amyloid neuropathology at autopsy. As expected, all 5 cases had LB disease and nigral neuronal loss consistent with their clinical diagnosis. Two cases had significant Aβ amyloid neuropathology and abnormal florbetapir imaging before death. The remaining 3 participants either had no Aβ neuropathology or a low level of AD neuropathological change, and all 3 had normal Aβ amyloid imaging. Of the florbetapir‐negative cases, 2 had additional neuropathological findings—1 with PSP and 1 with AGD—as additional substrates to explain their dementia. Our studies suggest that florbetapir PET Aβ amyloid imaging may reliably identify Aβ‐positive neuritic plaque pathology when present in patients with clinical PDD, including among those with other neuropathological diagnoses.
There is an emerging literature on the utility of Aβ amyloid imaging in PD patients, both for diagnostic purposes and for prognosis. Efforts to use PiB imaging to improve clinical diagnostic accuracy in a mix of LB diseases have found inconsistent results. Using established clinical diagnostic criteria for PD, PDD, and DLB,16, 29 a multicenter study of PiB uptake in a mix of these disorders showed that only the DLB group had significantly elevated PIB uptake in cortical regions, as compared to age‐matched controls.30 A smaller study using florbetapir imaging showed higher average cortical retention levels in AD and DLB patients, as compared to healthy controls or PD patients without cognitive impairment.31 In contrast, other studies have found that both PDD and DLB patients have abnormal PiB scans, yet the quantitative PiB retention in PDD patients was either lower than in DLB patients32, 33 or similar to DLB patients.34 These differences may reflect the variable extent of cortical Aβ pathology at diagnosis, which could contribute to the timing of cognitive impairment with respect to clinical parkinsonism and thereby influence the clinical categorization of PDD versus DLB.29 All 5 cases in our study met clinical criteria for PDD and not DLB,29 and our results suggest that abnormal Aβ amyloid imaging in PDD patients accurately mirrors underlying AD neuropathological change.
To our knowledge, only one study has investigated the value of Aβ amyloid imaging in predicting cognitive decline in PD.35 In a prospective study of nondemented PD patients, abnormal PiB scans at baseline associated with decline in executive function and conversion to greater degrees of clinical cognitive impairment.35 Similar studies with florbetapir Aβ amyloid imaging have not been performed and are needed to determine whether this imaging modality offers prognostic value in PD, noting that the severity of cognitive decline in PD is likely related to both AD and cortical LB pathology.
Our study is the first to show that Aβ amyloid imaging by florbetapir correlates with Aβ neuropathology in PD patients. Only two other autopsy studies with Aβ amyloid imaging (both with PiB) in patients with LB diseases have been published.13, 14 In the first, 2 of 3 PDD patients had both abnormal PiB imaging and high burdens of cortical Aβ diffuse plaque with lower levels of fibrillar Aβ neuritic plaque.13 The second study did not quantify diffuse plaques. However, 1 of 3 DLB patients had high PiB retention with sparse neuritic plaques, whereas the remaining 2 had borderline PiB retention and only sparse or moderate neuritic plaques.14 These studies suggest that the presence of diffuse plaques is sufficient for detection by antemortem PiB imaging. In our study, cases 4 and 5 had both diffuse and neuritic plaques, and therefore we cannot draw a conclusion regarding florbetapir specificity in recognizing these two forms of Aβ amyloid, which likely exist on a continuum.36
Our work supports previous clinicopathological studies from our group3 and others4 that suggest that multiple underlying neuropathologies contribute to dementia in PD, including LB pathology.37 Identification of these neuropathologies before death might identify individuals who would benefit from therapies designed specifically for Aβ and tau. We found 2 cases with significant Aβ neuropathology, 2 with significant, non‐AD tauopathy (PSP and AGD), and 1 with no discernible cause of dementia, all in addition to brainstem and subcortical LB disease. Our study was not designed to compare the contribution of LB pathology versus Aβ or tau pathology to dementia, given that all of these cases had clinical PDD and were expected to have LB pathology at autopsy. However, although this sample size is small, our findings suggest that florbetapir PET imaging is reliable in identifying Aβ amyloid pathology irrespective of other pathologies, such as tauopathy and synucleinopathy. Previously, concomitant AGD was not found to influence florbetapir binding when discovered in an autopsied AD patient cohort.38 Vascular disease is also frequently present in PD patients with clinical dementia.3, 4 Although all 5 cases had some degree of arterial atherosclerosis on gross examination, we did not observe significant microvascular disease in any case.
The standard assessment method for florbetapir PET imaging remains the qualitative binary interpretation by trained expert raters, which has excellent inter‐rater reliability.31 Case 3 had an elevated SUVr measure that was discordant with both the expert rating and the autopsy findings. The discrepancy was attributable to an abnormally low cerebellar SUV value, a region typically used for normalization between subjects (Fig. 2). We could not identify a structural or technical reason why this particular region's SUV was low in this case. Brain MRI in this case did not show significant cerebellar atrophy. However, normalization to the centrum semiovale did not show the same limitation (Fig. 2). This finding highlights the importance of the trained expert reader analysis as the quantitative analysis methods for florbetapir PET imaging continue to mature. However, by their nature qualitative visual inspections may overlook subtle differences in regional uptake. More quantitative measures, including regional raw SUV and SUVr, could better reflect underlying regional Aβ amyloid neuropathology, but this hypothesis should be tested in a larger cohort of PD patients. According to our semiquantitative rating system, cases 4 and 5 met the highest level of pathology (3+) in many brain regions, producing a ceiling effect that precluded a precise correlative analysis between raw SUV and immunohistochemistry.
Our study had several limitations. First, the scan–death interval was fairly long in some cases. As a result, the florbetapir PET imaging results might underestimate the degree of Aβ neuropathology observed at autopsy given the longer interval for cases 2, 3, and 5. Case 2 met neuropathological criteria for low probability of AD by NIA‐Reagan criteria25 and low level of AD neuropathological change by the new NIA/Alzheimer's Association criteria,26 yet his florbetapir PET scan was negative. It is possible that this Aβ amyloid pathology was not present at the time his florbetapir PET scan was obtained, and we cannot exclude the possibility that had the scan been performed closer to the time of autopsy, the PET scan would have been positive. However, the low amount of Aβ amyloid pathology observed in case 2 may be undetectable by florbetapir PET irrespective of the scan to autopsy interval. In an autopsy study of normal controls and individuals with MCI, clinical AD, or other non‐AD dementias, florbetapir was 92% sensitive for identifying cases with moderate‐to‐frequent density of neuritic plaques at autopsy, but did not result in abnormal PET scans in cases with no or sparse plaques.11 By recent consensus criteria, an intermediate level of pathology, usually involving moderate‐to‐frequent plaques as detected by florbetapir, and a Braak stage ≥3 is considered sufficient to account for cognitive impairment.26 Additional research is needed to determine whether low neuritic plaque levels (i.e., sparse plaques, as observed in case 2), have any implications for the presence or future development of cognitive impairment. Future radiological correlative studies in PD would also benefit from measures of Aβ amyloid imaging close in time to death to allow a comparison between imaging and pathology findings. In addition, each participant had their florbetapir PET during the middle‐to‐later periods of their PD disease course, as evidenced by the generally low total DRS‐2 and high modified H & Y scores. This limitation is somewhat unavoidable given the source study cohort from where these participants were derived. Other large study cohorts, including the Parkinson Progression Marker Initiative, have included Aβ amyloid imaging closer to disease onset in their multimodal biomarker collections. These cohorts are invaluable in determining both the contribution of Aβ PET imaging to a prediction of cognitive decline as well as making strong radiological correlative studies should these participants be followed to autopsy.
Our results suggest that florbetapir Aβ amyloid imaging may allow premortem identification of Aβ amyloid neuropathology in PD patients. These findings may have implications in refining the characterization of cognitive impairment in PD, assessing the risk for future dementia in nondemented PD patients, and designing clinical trials to mitigate the accumulation of Aβ aggregates.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution, D. Obtained Funding; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
R.S.A.: 1A, 1C, 1D, 2B, 3A
S.X.X.: 2A, 2C, 3B
L.B.: 1C, 3B
M.J.P.: 1C, 3B
H.I.H.: 1B, 1D, 3B
J.Q.T.: 1C, 1D, 3B
D.W.: 1A, 1D, 3B
A.D.S.: 1A, 1B, 1C, 1D, 3B
Disclosures
Funding Sources and Conflicts of Interest: This study was funded by the National Institutes of Health (NIH; P50‐NS053488 and P30‐AG010124) and by Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company. Dr. Pontecorvo and Dr. Siderowf are employees of Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company. Dr. Trojanowski may accrue revenue in the future on patents submitted by the University of Pennsylvania wherein he is coinventor and he received revenue from the sale of Avid Radiopharmaceuticals to Eli Lilly and Company as coinventor on imaging‐related patents submitted by the University of Pennsylvania. The authors report no conflicts of interest.
Financial Disclosures for previous 12 months: Dr. Akhtar is supported by the NIH (K08‐NS093127) and by a Clinical Research Training Fellowship from the American Brain Foundation/American Academy of Neurology. Dr. Xie is supported by the NIH (P30‐AG010124, P50‐NS053488, P01‐AG032953, and P01‐AG017586). Dr. Hurtig is supported by the NIH (P50‐NS053488) and receives royalties from UpToDate, an online educational website for health care providers. Dr. Trojanowski is supported by the NIH (P30‐AG010124, P50‐NS053488, and P01‐AG017586) and by funding from GlaxoSmithKline, Janssen, and several nonprofit organizations. Dr. Weintraub is supported by the Michael J. Fox Foundation for Parkinson's Research, NIH, Novartis Pharmaceuticals, Department of Veterans Affairs, and Alzheimer's Disease Cooperative Study; received honoraria from Biotie, Teva Pharmaceuticals, Lundbeck Inc., Acadia, UCB, Clintrex LLC, Medivation, CHDI Foundation, and the Weston Foundation; license fee payments from the University of Pennsylvania for the QUIP and QUIP‐RS; royalties from Wolters Kluweland; and fees for testifying in two court cases related to impulse controls disorders in Parkinson's disease (March 2013–April 2014, payments by Eversheds and Roach, Brown, McCarthy & Gruber, P.C.)
Acknowledgments
The authors thank Dr. Stacy S. Horn for patient recruitment and Terry Shuck and John L. Robinson for technical help.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord 2008;23:837–844. [DOI] [PubMed] [Google Scholar]
- 2. Todorova A, Jenner P. Ray Chaudhuri K. Non‐motor Parkinson's: integral to motor Parkinson's, yet often neglected. Pract Neurol 2014;14:310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kotzbauer PT, Cairns NJ, Campbell MC, et al. Pathologic accumulation of alpha‐synuclein and Abeta in Parkinson disease patients with dementia. Arch Neurol 2012;69:1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy‐ and Alzheimer‐type pathologies in Parkinson's disease dementia: which is more important? Brain 2011;134:1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord 2009;23:295–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galvin JE, Pollack J, Morris JC. Clinical phenotype of Parkinson disease dementia. Neurology 2006;67:1605–1611. [DOI] [PubMed] [Google Scholar]
- 8. Wong DF, Rosenberg PB, Zhou Y, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F‐AV‐45 (florbetapir [corrected] F 18). J Nucl Med 2010;51:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newberg AB, Arnold SE, Wintering N, Rovner BW, Alavi A. Initial clinical comparison of 18F‐florbetapir and 18F‐FDG PET in patients with Alzheimer disease and controls. J Nucl Med 2012;53:902–907. [DOI] [PubMed] [Google Scholar]
- 10. Choi SR, Schneider JA, Bennett DA, et al. Correlation of amyloid PET ligand florbetapir F 18 binding with Abeta aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord 2012;26:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐beta plaques: a prospective cohort study. Lancet Neurol 2012;11:669–678. [DOI] [PubMed] [Google Scholar]
- 12. Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir‐PET for imaging beta‐amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burack MA, Hartlein J, Flores HP, Taylor‐Reinwald L, Perlmutter JS, Cairns NJ. In vivo amyloid imaging in autopsy‐confirmed Parkinson disease with dementia. Neurology 2010;74:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging 2012;33:2091–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 2007;22:1689–1707. [DOI] [PubMed] [Google Scholar]
- 17. Litvan I, Goldman JG, Troster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov Disord 2012;27:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stebbins GT, Goetz CG, Burn DJ, Jankovic J, Khoo TK, Tilley BC. How to identify tremor dominant and postural instability/gait difficulty groups with the Movement Disorder Society Unified Parkinson's Disease Rating Scale: comparison with the Unified Parkinson's Disease Rating Scale. Mov Disord 2013;28:668–670. [DOI] [PubMed] [Google Scholar]
- 19. Amyvid (Florbetapir F18 Injection) for Intravenous Use: Full Prescribing Information. Indianapolis, IN: Eli Lilly and Company; 2013. [Google Scholar]
- 20. Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 2014;10:477–484.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laird NM, Ware JH. Random‐effects models for longitudinal data. Biometrics 1982;38:963–974. [PubMed] [Google Scholar]
- 22. Landau SM, Breault C, Joshi AD, et al. Amyloid‐beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013;54:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Basu S, Zaidi H, Houseni M, et al. Novel quantitative techniques for assessing regional and global function and structure based on modern imaging modalities: implications for normal variation, aging and diseased states. Semin Nucl Med 2007;37:223–239. [DOI] [PubMed] [Google Scholar]
- 24. Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 2009;8:1150–1157. [DOI] [PubMed] [Google Scholar]
- 25. Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997;56:1095–1097. [DOI] [PubMed] [Google Scholar]
- 26. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy). Neurology 1994;44:2015–2019. [DOI] [PubMed] [Google Scholar]
- 28. Braak H, Braak E. Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 1989;15:13–26. [DOI] [PubMed] [Google Scholar]
- 29. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 30. Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338. [DOI] [PubMed] [Google Scholar]
- 31. Siderowf A, Pontecorvo MJ, Shill HA, et al. PET imaging of amyloid with Florbetapir F 18 and PET imaging of dopamine degeneration with 18F‐AV‐133 (florbenazine) in patients with Alzheimer's disease and Lewy body disorders. BMC Neurol 2014;14:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gomperts SN, Locascio JJ, Marquie M, et al. Brain amyloid and cognition in Lewy body diseases. Mov Disord 2012;27:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foster ER, Campbell MC, Burack MA, et al. Amyloid imaging of Lewy body‐associated disorders. Mov Disord 2010;25:2516–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gomperts SN, Locascio JJ, Rentz D, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 2013;80:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thal DR, Capetillo‐Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ 2006;2006:re1. [DOI] [PubMed] [Google Scholar]
- 37. Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha‐synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology 2000;54:1916–1921. [DOI] [PubMed] [Google Scholar]
- 38. Dugger BN, Clark CM, Serrano G, et al. Neuropathologic heterogeneity does not impair florbetapir‐positron emission tomography postmortem correlates. J Neuropathol Exp Neurol 2014;73:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]