

Abstract
Silver nanoparticles (AgNPs) have been extensively used as antibacterial component in numerous healthcare, biomedical, and consumer products. Therefore, their adverse effects to biological systems have become a major concern. AgNPs have been shown to be absorbed into circulation and redistributed into various organs. It is thus of great importance to understand how these nanoparticles affect vascular permeability and uncover the underlying molecular mechanisms. A negatively charged mecaptoundeonic acid capped silver nanoparticle (MUA@AgNP) was investigated in this work. Ex-vivo experiments in mouse plasma revealed that MUA@AgNPs caused plasma prekallikrein cleavage, while positively charged or neutral AgNPs, as well as Ag ions had no effect. In-vitro tests revealed that MUA@AgNPs activated the plasma kallikrein-kinin system (KKS) by triggering Hageman factor autoactivation. By using specific inhibitors aprotinin and HOE 140, we demonstrated that KKS activation caused the release of bradykinin, which activated B2 receptors and induced the shedding of adherens junction protein, VE-cadherin. These biological perturbations eventually resulted in endothelial paracellular permeability in mouse retina after intravitreal injection of MUA@AgNPs. The findings from this work provided key insights for toxicity modulation and biomedical applications of AgNPs.
Keywords: Silver NP, kallikrein-kinin system, retinal endothelial cell, vascular permeability, cell junction
1. Introduction
Silver nanoparticles (AgNPs), as a common antibacterial agent, have been extensively used in a variety of products such as household appliances, healthcare supplies, and water disinfection agents (Wilson center, 2014). However, concerns have recently been raised regarding the potential biological toxicities of this agent (Ahamed, 2010; Geranio, 2009; Nel, 2006). Human exposure to AgNPs normally occurs via dermal, digestive and respiratory pathways. Once internalized, AgNPs are transported through blood circulation to multiple organs and cause the corresponding deleterious effects (Ahamed, 2010; Takenaka, 2001). Although some possible mechanisms have been proposed on basis of direct interactions between AgNPs and vascular barriers such as small particle transit across barriers via transcytosis (Trickler, 2010) and AgNPs induced oxidative damage to the barriers (Baruwati, 2013), AgNPs mediated biological processes in blood circulation and penetration of vascular barriers are not fully understood. It is thus of great importance to investigate how AgNPs interact with plasma components and potentially cause vascular permeability.
The contact system, also known as the kallikrein-kinin system (KKS), is involved in the regulation of vascular permeability, inflammation and coagulation (Björkqvist, 2013; Colman, 1997; Feener, 2013). The KKS consists of several plasma serine proteases including Hagemen factor (FXII), plasma prekallikrein (PPK), and high molecular weight kininogen (HK) (Björkqvist, 2013). On contact with negatively charged artificial or biologic surfaces, the zymogen, FXII, will undergo autoactivation, leading to the formation of FXIIa and PPK activation (contact activation). Activated plasma kallikrein (PK) acts in two ways that reciprocally activate FXII to produce more FXIIa and cleave HK to release a biological active peptide, bradykinin (BK). The cascade activation was shown in Figure S1. As an inflammatory mediator, BK may mediate retinal vascular permeability elevation, potentially inducing macular edema and even causing vision loss in some clinical disease cases like diabetes (Kempen, 2004; Williams, 2004). Since the KKS is central to vascular permeability, and activated by interactions with negatively charged surfaces, it is of particular interest to characterize the potential effects of nanoparticles on the KKS and the resultant effects on physiological systems and organs.
Up to now, very few studies explored the effects of engineered nanomaterials on the KKS or the related zymogens. Superparamagnetic iron oxide nanoparticles were reported to induce contact activation of KKS both in vitro and in vivo (Simberg, 2009). Multiple-walled carbon nanotubes were found to accelerate thrombosis by activating the intrinsic coagulation cascade (Burke, 2011) which cross-talks with the KKS via FXII activation. However, key questions, such as how nanoparticles interact with plasma zymogens and the KKS pathway and how nanoparticles break the vascular barrier remain unanswered. Furthermore, widely used AgNPs that provide better aqueous solubility, biocompatibility and shelf stability often have a negative surface charge (Badawy, 2010; Levard, 2012). Thus, since AgNPs have a high propensity to activate the contact cascades due to its negatively charged surface, this interaction will be the primary focus for this investigation.
In this report, we describe the first mechanistic study of contact system activation by mecaptoundecanoic acid capped silver nanoparticles (MUA@AgNPs). MUA@AgNPs activate the KKS cascade, cause the shedding of adherens junction protein, VE-cadherin, and enhance retinal vascular permeability after intravitreal injection in mice. These results provide key insights into the mechanism by which AgNPs increase vascular permeability and further guide its usage in potential medical applications and toxicity modulation.
2. Methods
2.1 Preparation and Characterization of AgNPs
Firstly, citrate capped silver nanoparticles (Cit@AgNPs, 10 nm) were synthesized with a chemical reduction reaction (details shown in supplementary information). Secondly, MUA@AgNPs with the same size was prepared by the ligand exchange with 11-mecaptoundecanoic acid (MUA, Sigma-Aldrich). The modification was conducted by incubating 264 μg/mL 11-mecaptoundecanoic acid with 20 μg/mL Cit@AgNPs in 10 mM PBS buffer (pH 7.4) at room temperature overnight. The other two nanoparticles of BEPI@AgNPs (nanoComposix) and PVP@AgNPs (Shanghai Zhuiguang Technology Co., LTD,) were commercially purchased.
The morphology of AgNPs was observed with transmission electronic microscopy (2100f, JEOL; H-7500, Hitachi). The hydrated size and zeta potential were characterized on a particle size analyzer (Mastersizer 2000, Malvern). The X-ray photoelectron energy spectroscopy (AXIS UltraDLD, Kratos) and UV-Visible absorption (DU-800, Beckman) were used to demonstrate the surface information of MUA@AgNPs. The release of Ag+ was evaluated by the ratio of silver content in 3 kDa ultrafiltrated supernatant to that in MUA@AgNPs suspension quantified with ICP-MS (Agilent 7500ce). The properties and the dissolution over time (0, 1, 2 and 6 h) of MUA@AgNPs in mouse plasma were evaluated as well to understand their fate and responses in blood.
2.2 Ex-Vivo Experiments
Treatment for plasma contact activation
3 μL of 4 mg/mL kaolin (Sinopharm) suspension, 40 μg/mL diverse AgNPs suspensions (MUA@AgNPs, PVP@AgNPs and BEPI@AgNPs), 40 μg/mL AgNO3 solution and 40 μg/mL MUA were mixed with 27 μL of mouse plasma (C57BL/6, Vital River Laboratory Animal Technology Co. Ltd.), respectively, and incubated at 37 °C for 1 h. The control plasma sample was prepared by using 3 μL of distilled water instead. The tested concentrations of MUA@AgNPs for dose-response study were set as 0.5, 5, 10, 40, 200 μg/mL and the incubation time was 1 h. As for the time-course experiment, different incubation durations of 0, 5, 15, 30 and 60 min were controlled when MUA@AgNPs (40 μg/mL) was introduced into the plasma. Ex-vivo contact activation in human plasma (healthy volunteers from the lab) was performed in the similar way by using MUA@AgNPs (40 μg/mL, 400 μg/mL) as the activator.
Western blot assay
The ex-vivo contact activation was stopped by the addition of 4× sample buffer (Laemmli buffer, containing 4% β-mecaptoethanol) and the mixtures (AgNPs activated plasma samples) were subsequently submitted for Western blot assay. It mainly included gel separation (4–20% gradient, Bio-Rad), membrane transferring (cellulose acetate, 0.45 μm, EMD millipore), immunoblotting with the primary and secondary antibodies and development with Thermo ECL reagents and Kodak film. The primary antibodies included mouse PK antibody (1:1000, R&D), and those for human proteins, PK (1:1000, Abcam), FXII (1:1000, Cedarlane Laboratories), HK (1:2000, R&D). The secondary antibodies were peroxidase-conjugated rabbit anti-goat and goat anti-rabbit IgG (1:500, ZSGB-BIO).
PK Activity Assay
27 μL of fresh mouse plasma samples were incubated with 3 μL of H2O, human PK (80 μg/mL, Enzyme Research Laboratories), Kaolin (4 mg/mL) and MUA@AgNPs (4 mg/mL) for 1 h, respectively. After centrifugation, 4 μL of supernatants were mixed with 95 μL of 10 mM phosphate buffer (pH7.4) containing 50 μM ZnSO4 in Corning® 96-well black plate. After 1 μL of 20 mM fluorogenic kallikrein substrate (H-D-Val-Leu-Arg-AFC·2HCl, Calbiochem, EMD Millipore) was added, the fluorescence emission was instantly monitored by the multifunction microplate reader (Thermo Scientific, VARIOSKAN FLASH) at 400 nm/505 nm (λex/λem) for continuous 30 min at an interval of 30 s.
2.3 In-Vitro Experiments
Purified human protein including FXII, FXIIa, PPK, PK and HK obtained from Enzyme Research Laboratories were used for the in-vitro experiments.
Native PAGE assay
In total 25 μL of 10 mM PBS buffer system (pH 7.4, 50 μM ZnSO4), 0.2 μg FXII was incubated with suitable amount of MUA@AgNPs at 37 °C for 1 h. The molar ratios of MUA@AgNPs/FXII were 0, 0.1, 0.2, 0.5, 0.8, 1, 1.5, 2, 5, and 8. The mixture was separated on 12% native PAGE gel. Using Western blot, the amount of unbound FXII was visualized and the binding ratio of the complex was confirmed based on the densitometric measurement of the unbound FXII in the separation gel.
CD determination
As for the conformational change analysis, FXII (44 μg/mL) was incubated with MUA@AgNPs (40 μg/mL) in 10 mM phosphate buffer (pH7.4) containing 50 μM ZnSO4 at 37 °C for 1 h. Together with the relative protein controls (FXII, FXIIa, 44 μg/mL), the samples were characterized using CD measurement. The CD spectra were acquired from 195 to 300 nm at room temperature on a spectropolarimeter (Jasco-815, Jasco). The samples were under nitrogen protection when the data were recorded by using a 0.05% (w/v) polybrene (Sigma-Aldrich) pretreated quartz cell with a scan rate of 200 nm min−1. Each spectrum was accumulated twice, and the results were expressed as CD milli-degrees. The data were further analyzed with CDpro software package (Sreerama, 2004).
In-vitro KKS activation
The in-vitro KKS zymogen activation was performed by the incubation of 400 μg/mL MUA@NPs with 0.5 μg FXII, 1 μg PPK, 1.6 μg HK and the combination in 30 μL of 10 mM PBS buffer system (pH 7.4, 50 μM ZnSO4) at 37 °C for 1 h. The samples were subsequently submitted to Western blot assays according to the protocols used for ex-vivo contact activation in human plasma.
2.4 In-Vivo Experiments
Animals
Animals for in-vivo studies were 10-week old male C57BL/6 mouse with an average weight of 22 g and they were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing). All the animal experiments were approved by the Animal Care and Use Committee of the RCEES, Chinese Academy of Sciences. All the materials used on the mice were sterile and the surgeries were performed under the disinfection condition. Before the surgeries, the mice were anaesthetized with 50 mg/kg pentobarbital sodium (Sigma-Aldrich) by peritoneal injection. All in-vivo treatments were performed on three independent mice, unless otherwise specified.
In-vivo PPK activation
The anaesthetized mice were slowly infused 2.5 mg/kg MUA@AgNPs or PBS into the jugular vein through the catheter (MRE-025, Braintree Scientific, Inc.). At circulation time of 0, 30 and 60 min, about 50 μL of mouse blood samples were collected through the catheter with the syringe pre-filled with the anticoagulant (3.6% trisodium citrate solution). The blood was subsequently centrifuged at 2500 g (15 min, 4 °C) for the plasma preparation. These plasma samples (2 μL) were diluted with sample buffer (1x, Laemmli buffer) by 20 folds and further processed for PK Western blot assay with the same condition as ex-vivo PPK activation in mouse plasma.
FITC-dextran infiltration assay
On the anaesthetized mice, intravitreal injection of MUA@AgNPs (500 μg/mL, 1 μL), BK (10 μM, 1 μL) or PBS (1 μL) were performed using Hamilton syringe with 30G needle. Fifteen minutes later, the probe solution (FITC-dextran in PBS, 70kDa, Sigma,) was infused into the blood circulation system of the mice through the jugular vein at the dose of 200 mg/kg and allowed to circulate for 2 h. The mice were subsequently sacrificed with CO2. Both eyes in each mouse were harvested and fixed separately in 10% formalin for 1 h. The retina samples were carefully dissected and mounted on glass slides with anti-fade mounting medium (Vector Labs). The fluorescent images were photographed using a confocal fluorescent microscope (Leica TCS SP5).
EB assay leakage assay
The anaesthetized mice were performed with intravitreal injections of PBS (1 μL), BK (10 μM, 1 μL), or MUA@AgNPs (50 μg/mL, 500 μg/mL, 1 μL). More than 5 mice were tested for each group and the detailed animal numbers were specified in Figure 5d. Fifteen minutes later, Evans blue dye (EB, 90 mg/mL, 2 μL/g mouse) was infused through jugular catheter into the mouse and it was allowed to circulate for 1 h. The mouse was subsequently sacrificed and around 300 μL of blood was collected from the postcava to prepare the plasma sample. The cardiac perfusion was successively performed on the mice with PBS (5 mL) and 10% formalin (5mL) at a physiological pressure through a 0.22 μm syringe filter to remove the residue blood in the whole vasculature. The retina samples were then harvested and 2 retinas from the same mouse were combined as 1 sample. After vacuum dried for 4 h and weighed, each retina sample was separately extracted by 130 μL of formamide (Sigma) at 72°C for 18 h. The supernatants containing the dye leaked into the retina tissues were collected by the centrifugation and 50 μL of the samples were loaded onto the 96-well plate. Together with the formamide diluted plasma (1000 folds dilution, 50 μL), the absorbance of all the samples were measured at 620 nm (Ab for Evans blue) and 740 nm (background) on the multifunctional plate reader (Thermo Scientific, VARIOSKAN FLASH), respectively. All the values were calibrated by Ab[620nm] - Ab[740nm] and the EB leakage in each retina sample was calculated according to the following equation.
Figure 5. MUA@AgNPs activated plasma increased the paracellular permeability of human retina endothelial cells (HRECs) through regulating adherens junction protein, VE-cadherin.

(a) Monolayer HREC permeability changes upon MUA@ANPs-activated mouse plasma stimulation for 6 h. (b) VE-cadherin expression in HRECs: 1) PBS, 2) 1% (V/V) mouse plasma, 3) 2 μg/mL MUA@AgNPs, 4) 10 μM BK, 5) L-p@AgNPs (0.2 μg/mL MUA@AgNPs activated mouse plasma, 1%, V/V), 6) H-p@AgNPs (2 μg/mL MUA@AgNPs activated mouse plasma, 1%, V/V), 7) 10 μM aprotinin + H-p@AgNPs (2 μg/mL MUA@AgNPs activated mouse plasma, 1%, V/V with 10 μM aprotinin pretreatment), and 8) 2 μM HOE 140 + H-p@AgNPs (HRECs were pretreated with 2 μM HOE 140, then stimulated with 2 μg/mL MUA@AgNPs activated mouse plasma, 1%, V/V). (c) The statistical analysis of the Western blots for VE-cadherin expression; (d) Immunostaining of VE-cadherin on HREC cells treated as indicated. (e) The statistical result of the continuity grade for VE-cadherin expression on HRECs. All results were from three independent experiments, *P < 0.05 and **P < 0.01 by ANOVA.
Retinal PPK Leakage assay
PPK levels, as one of the representative plasma component, were analyzed in the moue retina tissues by Western blot for the evaluation of retinal vascular permeability upon intravitreal treatments of PBS (1 μL), BK (10 μM, 1 μL) or MUA@AgNPs (500 μg/mL, 1 μL) in anaesthetized mice. One hour later after intravitreal injection, the mice were sacrificed and cardiac perfused with 10 mL of PBS. The retina samples were dissected and snap frozen in liquid nitrogen. Each retina sample was sonicated in 100 μL of lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100 and 1% cocktail solution) on ice. The lysates were submitted to protein quantification using BCA assay kit (Pierce, Thermo) and subsequent PPK Western blot assay using the similar condition for mouse plasma PPK immunoblot. β-actin in the retina tissues were analyzed as the loading control (rabbit anti-beta-actin antibody, 1:2000, Beijing Biosynthesis biotechnology). The quantitative analysis was performed by scanning the grey density of the protein bands using Quantity One software (http://www.bio-rad.com/en-us/product/quantity-one-1-d-analysis-software).
2.5 Cell Experiments
Cell cultures
The HREC (ScienCell Research Laboratories) was cultured in the 100-mm culture dish pre-coated with 0.1% gelatin (Gibico) and incubated at 37 °C under 5% CO2. The culture medium was ECM (Sciencell) containing 5% FBS (Gibico), 1% endothelial cell growth supplement (ScienCell Research Laboratories) and 1% antibiotic reagents (Gibico), and was changed every day. As for the following cell studies, triplicate experiments were performed independently.
Cell stimulation
All the cell stimulations were performed in ECM medium containing 1% BSA (Sigma-Aldrich). The exposure groups included negative controls (PBS, 1% mouse plasma and 2 μg/mL MUA@AgNPs), positive control (10 μM BK), 1% mouse plasma activated by MUA@AgNPs (0.2 μg/mL for L-p@AgNPs, 2 μg/mL for H-p@AgNPs), 10 μM aprotinin with H-p@AgNPs and 2 μM HOE 140 with H-p@AgNPs. The stimulus of L-p@AgNPs and H-p@AgNPs were freshly prepared by adding 2 μL of MUA@AgNPs (0.2, 2 mg/mL) in 18 μL of mouse plasma, respectively and incubating at 37°C for 10 min before cell stimulation. As for the stimuli of aprotinin with H-p@AgNPs, 1 mM aprotinin (Sigma-Aldrich) was pre-mixed with mouse plasma and it was subsequently processed with the protocol for H-p@AgNPs preparation. In view of 2 μM HOE 140 with H-p@AgNPs, HRECs were pre-incubated with 2 μM HOE 140 (Sigma-Aldrich) for 30 min, then treated with H-p@AgNPs. The stimulations were performed by directly adding the stimulus described above to the cell exposure medium at the ratio of 1:100 (V/V). The specific details were given in the respective assays. BK stimulation was lasted for 30 min, while the exposure duration for the other stimulus was 2 h or 6 h.
Alamar blue assay
HRECs were seeded in 96-well pates with the density of 10 000 cells/well. After 24-h incubation, the cell medium was replaced by 100 μL of ECM medium containing 1% BSA and the cells were starved for 3 h. The treatments with the stimulus described in section of cell stimulation were performed for 2 h and 6 h, respectively. Three parallel wells were set for each condition. 10 μL of 100 μM Alamar Blue® reagent (Sigma Aldrich) was added and the plates were incubated at 37°C with 5% CO2 for another 2 h. The fluorescence at 590 nm/530 nm (λem/λex) was measured for the evaluation of cell viability.
Permeability assay for monolayer HRECs
HRECs were seeded on 0.1% gelatin pre-coated polyester filter of a transwell device (24-well format, 0.4 μm pore size, Millipore) at the density of 50 000 cells/cm2 and cultured at 37 °C under 5% CO2. The trans-endothelial electrical resistance was monitored for 7 days by Millicell® ERS-2 voltohmmeter (Merck Millipore) until a monolayer of HRECs was formed in each well to simulate iBRB in vitro. The culture medium was replaced by 100 μL and 600 μL of ECM medium containing 1% BSA (Sigma-Aldrich) in upper and bottom wells, respectively, and the cells were starved for 3 h before stimulation. The respective stimuli described in section of cell stimulation together with the fluorescent probe of RITC-dextran (70kDa, Sigma, 5 mg/mL) were added into the upper compartment. After 6-h exposure, 50 μL aliquot of the media in each bottom chamber was collected for the fluorescence measurement (λex/em 543 nm/584 nm). The fluorescent signal of the probe penetrating across the cell monolayer in each group was used for the evaluation of the changes in the permeability of simulated iBRB.
VE-Cadherin expression in HRECs
HRECs were seeded in 0.1% gelatin coated 6-well plates. When the confluency increased to 80–90 %, the cells were processed with 3 h starving and 2 h stimulation (as shown in section of cell stimulation). After exposure, the cells were washed with DPBS containing Ca2+/Mg2+ twice and lyzed in 50 μL of lysis buffer containing 1 mM NaF, 1 mM Na3VO4, 1% cocktail and 0.6 mM H2O2 on ice for 1 h. After centrifugation, the lysates were submitted to Western blot for VE-cadherin expression (anti-VE-cadherin antibody, 1:100, Invitrogen)) and β-actin was detected as the loading control with the similar protocol for mouse retina samples.
Immunostaining of VE-cadherin in HRECs
HRECs were grown in 0.1% gelatin pre-coated glass bottom cell culture dishes (35 mm, NEST) for 12 h. The confluent cells were starved and submitted to cell stimulation as described above. At the end of stimulation (2 h), the cells were gently washed three times with DPBS containing Ca2+/Mg2+ and fixed with ice-cold methanol (Sinopharm) for 5 min. After the cells were rinsed three times, DPBS containing 5% BSA was added to block the non-specific binding sites at room temperature for 30 min. Following triple rinses (5 min for each), the cells were blotted with human VE-cadherein antibody (1:150, Abcam) in DPBS buffer containing 2% BSA overnight at 4 °C. After triple washes with DPBS (5 min for each), the cells were incubated with dylight549 conjugated anti-rabbit antibody (1:200, Abbkine) in DPBS containing 2% BSA at room temperature for 1 h. The cells were then washed with DPBS for three times (5 min for each). The samples were imaged for VE-cadherin distribution under confocal fluorescence microscope. The integrity of AJs was evaluated by scoring the continuity grade of stained VE-cadherin (Murakami, 2009). Briefly, grade 1 was for the cell borders with < 25% continuous staining; grade 2 for ~25% continuous staining; grade 3 for ~50% continuous staining; grade 4 for almost continuous staining with slight breaks; and grade 5 for border completely stained. Thirty cells in each group were randomly selected for the integrity scoring.
2.6 Statistical Analysis
All experiments were independently performed by three times or more and the results were the mean value ± standard deviation. The statistical analysis was conducted with ANOVA and the significant difference was expressed by P value less than 0.05 or 0.01.
3. Results
3.1 Characterization of AgNPs
As an in-vivo imaging agent, MUA@AgNPs have robust stability and favorable biocompatibility (Lee, 2007). In this study, it was used as AgNPs model to investigate the potential interaction with the KKS in plasma. The MUA@AgNPs depicted in Figure 1a were prepared by adding mecaptoundenic acid molecule to replace the citrate on the citrate-coated silver nanoparticles (Cit@AgNPs) (Lee, 1982). The transmission electron microscopic (TEM) image (Figure 1b) showed that the prepared MUA@AgNPs were monodispersive spheric particles and the statistical size distribution was 9.7 ± 0.9 nm (Figure 1b, inset). The C1s spectra of MUA@AgNPs (Figure 1c) was composed of four components of C-C at 284.9 eV, C-S at 286.3 eV, Ag-coordinated O-C=O at 288.5 eV, and free O-C=O at 289.3 eV (Janardhanan, 2009; Yang, 2012), and the signal of coordinated O-C=O from the molecule of citrate ion almost disappeared. Together with the decreased doublet electronic-binding energy of S2p at 161.3 eV and 162.4 eV (Figure 1d), MUA@AgNPs were demonstrated to be well passivated by the mecaptoundenic acid molecules through the Ag-S coordination bond. The hydrated size and zeta potential of MUA@AgNPs was 16.0 ± 2.0 nm and −38.3 ± 0.8 mV, respectively. The typical measurement curves were shown in Figure 1e and 1f. As the released Ag+ is commonly believed to be vital for the biological toxicity of nanosilver (Lapresta-Fernández, 2012; Liu, 2010), the fraction of Ag+ in the MUA@AgNPs suspension was determined after 7-day storage in water at room temperature (25 °C). As a result, the low amount of 1.5% Ag+ release indicate that Ag+ is unlikely to cause the resulting biological effect from MUA@AgNPs suspension. The morphorage of MUA@AgNPs in mouse plasma depicted in Figure 1g was similar to those in distilled water (Figure 1b). The nanoparticles remained monodisperse in plasma and the partilce size was 13.2 ± 0.8 nm under TEM examination. The hydrate size and zeta potential in plasma were 42.4 ± 0.7 nm and −38.4 ± 1.8 mV, repspectively. The UV-Vis absorbance at 402 nm confirmed the spectrometric characteristic of AgNPs in plasma. The dynamic dissolution over time showed silver ion release was less than 0.3% during 6 h incubation in plasma (Figure S2), confirming the negligible silver ion release during the short-term exposure experiments (≤ 6 h) in this study. Moreover, two other silver nanoparticles, the branched polyethyleneimine coated silver nanoparticles (BPEI@AgNPs) and polyvinylpyrrolidone coated silver nanoparticles (PVP@AgNPs) were also used to study the effect of surface charge on ex-vivo activation of plasma KKS. These two AgNPs had uniform dispersity with the equivalent size around 25 nm (Figure S3). The zeta potentials were + 18.9 mV for BPEI@AgNPs and −2.6 mV for PVP@AgNPs, respectively.
Figure 1. Characterization of MUA@AgNPs.

(a) Cartoon depiction of the negatively charged MUA@AgNPs. (b) Transmission electron microscopic (TEM) image and the size distribution of MUA@AgNPs in water. (c and d) High-resolution XPS spectra of C1s and S2p at room temperature. (e and f) Hydrated size and zeta potential of MUA@AgNPs in distilled water and mouse plasma as indicated. (g) TEM image and the size distribution of MUA@AgNPs in mouse plasma.
3.2 AgNPs Induced PPK Activation in Mouse Plasma
With reference to KKS activation by the non-physiological activator of kaolin (Maas, 2011) which is characterized by PPK activation with the disappearance of PPK band (~75 kDa) and the occurrence of PK band (~52 kDa) (Figure 2a), the effect of AgNPs on ex-vivo KKS activation was explored in mouse plasma. As shown in Figure 2b, similar to kaolin, the negatively charged MUA@AgNPs (lane 4) efficiently cleaved PPK to produce its active form, PK. In contrast, neither the positively charged BPEI@AgNPs nor neutral PVP@AgNPs caused the activation of PPK zymogen. This indicated that only the particle with a negatively charged surface triggered this biological process. The released Ag+ from AgNPs is always considered to play important roles in AgNPs induced biological effects through denaturing enzyme or depolarizing cell (Ahamed, 2010; Lapresta-Fernández, 2012). Incubating equivalent amount of Ag+ with mouse plasma showed that Ag+ induced a negligible amount of PPK cleavage in contrast to MUA@AgNPs. This suggested that the activation of PPK in plasma caused by AgNPs was particle-specific and independent of released Ag+. Likewise, the free ligand of 11-mecaptoundecanoic acid molecule which was used as the coating material for MUA@AgNPs preparation had no effect on PPK activation itself (Figure 2c), showing that the AgNPs core was critical for this biological process. Time-course studies indicated that MUA@AgNPs activated PPK within 5 minutes (Figure 2d) and the contact activation was dose-dependent in the range of 0.5 – 200 μg/mL of MUA@AgNPs (Figure 2e). PK activity was also tested to further confirm the potential effect of MUA@AgNPs on contact activation in mouse plasma. As shown in Figure 2f, the fresh mouse plasma with 10% sodium citrate anticoagulant itself showed no capability to cleave the fluorogenic kallikrein substrate, while the plasma with exogenous PK efficiently cleave the substrate and generate a fluorescent signal. As a positive control, kaolin also significantly elevated the fluorescence emission by inducing the endogenous PK generation in the fresh mouse plasma through contact activation. Similarly, MUA@AgNPs treated plasma exhibited increased kallikrein activity, suggesting that MUA@AgNPs are equally potent to trigger plasma contact activation like kaolin. Therefore, it was confirmed that ex-vivo PPK activation through contact activation was induced by the negatively charged MUA@AgNPs in mouse plasma.
Figure 2. Negatively charged silver nanoparticles cause ex-vivo activation of PPK in mouse plasma (MUA@AgNPs abbreviated as AgNPs in all figures).

(a) Western blot for kaolin-mediated PPK activation in mouse plasma: 1) plasma control, 2) kaolin-treated plasma (positive control, 400 μg/mL). (b) The effect of AgNPs and silver ion on PPK activation in mouse plasma: 1) plasma control, 2) 40 μg/mL BPEI@AgNPs, 3) 40 μg/mL PVP@AgNPs, 4) 40 μg/mL MUA@AgNPs, 5) 40 μg/mL Ag+. (c) The effect of mecaptoundecanoic acid molecule on PPK activation in mouse plasma: 1) plasma control, 2) 40 μg/mL 11-mecaptoundecanoic acid. (d) Time course for PPK activation induced by 40 μg/mL MUA@AgNPs: 1)-6) 0, 5, 15, 30 and 60 min. (e) Dose-dependent PPK activation: 1)-6) 0, 0.5, 5, 10, 40 and 200 μg/mL. (f) PK activity induction in plasma upon PK (1 μg/mL), Kaolin (400 μg/mL) and MUA@AgNPs (400 μg/mL) treatment.
3.3 AgNPs Triggered KKS Cascade Activation In Vitro and Ex Vivo
To investigate the waterfall cascade activation triggered by MUA@AgNPs, purified human plasma zymogens in the KKS were used for in-vitro tests. As stated previously, contact activation is triggered by the conformational change and autocatalysis of FXII and it is initially mediated through the binding of FXII to a negatively charged surface (Konings, 2011). A native gel separation coupled with FXII Western blot was employed to investigate the binding of FXII with MUA@NPs, since the formation of FXII complex would restrain its migration in non-denaturing PAGE due to the conformational changes and size increase (Citarella, 1996). The result in Figure 3a showed that the free FXII protein decreased with the increasing molar ratio of MUA@AgNPs to FXII in the reaction system. By densitometric measurement of the unbound protein, the binding molar ratio for MUA@AgNPs/FXII was about 1.5:1. The conformational change of FXII on the negatively charged surface of MUA@AgNPs was further examined by circular dichroism (CD) after incubating FXII with MUA@AgNPs at 37°C for 1 h. As shown in Figure 3b, the CD signals in UV region ranging from 195 to 260 nm indicated that FXII was characterized with 14.9% of β-sheet, 33.8% of βturn, and 51.3% of random coil in structure. Upon the interaction with MUA@AgNPs, the secondary structure of FXII changed dramatically. The ratio of β-sheet was increased to 34.8%, the β-turn was decreased to 11.5%, while the random coil was unchanged. This conformational change showed that MUA@AgNPs treated FXII exhibited a high similarity to its active form, α-FXIIa (blue curve in Figure 3b), thus suggesting that MUA@AgNPs are potential contact activators of FXII.
Figure 3. Activation of KKS cascade upon MUA@AgNPs treatment.
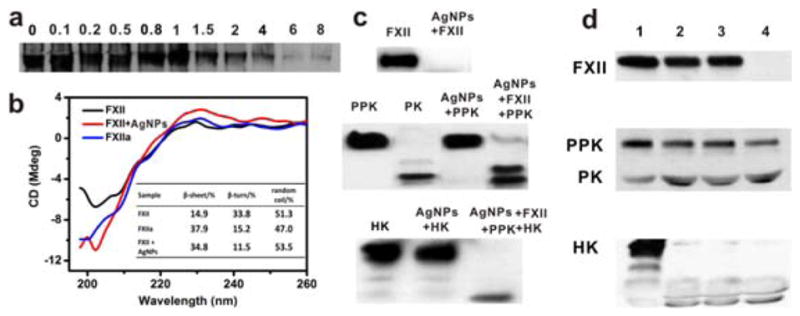
(a) Non-denaturing PAGE analysis for the binding of FXII with MUA@AgNPs. The FXII/AgNPs molar ratios were set as indicated. (b) The CD spectra and the related structure parameters (inset) of FXII (black), FXIIa (blue), and FXII (red) incubated with MUA@AgNPs at 37°C for 1 h, respectively. (c) Western blots for KKS cascade activation in vitro based on the incubation of MUA@AgNPs with the purified zymogens including FXII), PPK and HK. (d) Ex-vivo KKS cascade activation in human plasma (37°C, 1 h): 1) control, 2) Kaolin, 3) 40 μg/mL MUA@AgNPs and 4) 400 μg/mL MUA@AgNPs.
The interaction of the non-physiologic activators with FXII may cause subsequent autocleavage at amino acid residues 353–354 to yield the two-chain enzyme active factor XII (i.e. α-FXIIa) (Colman, 1997). On SDS-PAGE, the disulfide bond of α-FXIIa was dissociated, causing the complete loss of the zymogen band (~80 kDa). As evidenced by the Western blot in Figure 3c, the pre-incubation of FXII with MUA@AgNPs (37°C, 1 h) caused the complete loss of FXII zymogen in the reaction system. The formation of α-FXIIa converts PPK into PK and PK subsequently cleaves its main plasma substrate, HK to liberate the bioactive peptide BK. These biological processes were also confirmed by the cleavage of zymogens PPK and HK and the formation of PK through Western blot analysis (Figure 3c). In addition, MUA@AgNPs itself had no effect on PPK or HK activation, suggesting that the autoactivation of FXII on negatively charged nanoparticle surfaces was crucial for triggering the contact system activation.
In comparison to in-vitro system, KKS activation in plasma may be modulated by negative feedback control through endogenous serine protease inhibitors that can regulate KKS activity (van der Graaf, 1983). It was thus questioned whether MUA@AgNPs could induce KKS activation in plasma when endogenous inhibitors for FXIIa or PK, such as C1 esterase inhibitor (C1INH) and α2-macroglobulin, were abundant (Maas, 2011; Harpel, 1985). Therefore, KKS activation in plasma was also performed using human plasma with or without MUA@AgNPs treatment. Similar to the in-vitro results, MUA@AgNP-induced cleavages of FXII, PPK and HK in a dose-dependent manner were observed in plasma (Figure 3d). Taken together, both in-vitro and ex-vivo experiments confirmed that MUA@AgNPs triggered waterfall cascade activation of the KKS through FXII autoactivation.
3.4 AgNPs Caused Retinal Vascular Permeability by Activating the KKS In Vivo
By using a C57BL/6 mouse model, we subsequently tested MUA@AgNP-induced KKS activation in vivo. MUA@AgNPs were administered through a jugular catheter and allowed to circulate for 1 h. Plasma samples were collected at 0 min, 30 min and 1 h and analyzed by Western blot for PPK activation. The zymogen PPK level in Figure 4a exhibited a continuous decrease throughout 1 h, while the active PK formation increased from 0 to 30 min, then decreased during continued circulation (Figure 4b), in contrast to the time course for ex-vivo PK formation (Figure 2c). These findings indicated that MUA@AgNPs were able to induce in-vivo KKS activation in the presence of intricate KKS regulatory mechanisms in mice.
Figure 4. MUA@AgNPs stimulated in-vivo KKS activation and induced increased retinal vascular permeability in mice (n=3).

(a, b) The jugular infusion of MUA@AgNPs stimulated a time-dependent KKS activation in vivo, which was characterized by PPK loss (a) and PK formation (b). Inset: Western blots for PPK and PK in mouse plasma. (c) FITC-dextran (70 kDa) penetration assay for retinal vascular permeability in mice following intravitreal injection of PBS (1 μL), MUA@AgNPs (500 μg/mL, 1 μL) and BK (10 μM, 1 μL) as indicated per panel. (d) Evans blue leakage for retinal following intravitreal injection of PBS, MUA@AgNPs (50 μg/mL for L@AgNPs, 500 μg/mL for H@AgNPs) and BK (10 μM), respectively. Data are shown per mouse (two retina = one sample) and the groups are averaged for each treatment (e) Quantification and Western blots for PPK leakage in mouse retina with intravitreal injection of MUA@AgNPs (500 μg/mL) and BK (10 μM). *P < 0.05 and **P < 0.01 by ANOVA.
KKS activation in plasma increases the release of the biologically active peptide, BK, which is a potent inducer of vascular permeability via binding with B2 receptor on endothelial cells (Taylor, 2013). The retinal endothelium is very responsive to vasoactive agents such as the enhanced generation of BK which would lead to hypervasopermeability (Warboys, 2009). Whether MUA@AgNPs induced KKS activation increases retinal vascular permeability is an important question regarding eye drops of colloidal silver can be applied directly to heal eye infections (TSE, 2015). Therefore, retinal vascular permeability was evaluated in mice by intravitreal injections of MUA@AgNPs followed by jugular infusion of fluorescent probe of FITC-dextran. Intravitreal injections of PBS and BK were used as the negative and positive controls, respectively. Confocal images in Figure 4c showed the intact and compact capillary blood vessels highlighted by the green fluorescence were well distributed in the retinas of mice with PBS treatment. Enhanced retinal vascular permeability was obviously visualized by the infiltration of fluorescent probe from the damaged vasculature in MUA@AgNPs treated mice into the extravascular space (marked with yellow circles) which was similar to the effect caused by BK.
Evans blue (EB) is a dye that specifically binds with serum albumin and commonly used to quantitatively estimate vascular leakage (Xu, 2001). EB assay was thus used for the quantification of retinal vascular permeability induced by MUA@AgNPs in mice. The results in Figure 4d showed, similar to the effect of BK, that intravitreal injection of MUA@AgNPs significantly increased the EB leakage into retina in a dose-dependent manner. Likewise, Western blot analysis of retinal PPK in Figure 4e showed that PPK levels were elevated in the MUA@AgNPs and BK treated groups which indicated that blood ingredients passed through retinal vessels via increased retinal vascular permeability.
3.5 AgNPs Increased Paracellular Permeability Through Modulating VE-Cadherin
Normally, retinal vascular permeability is tightly regulated by inner blood retinal barrier (iBRB) which is formed by the compact alignment of retinal endothelial cells inside the blood vessel wall, blocking the plasma proteins and circulating cells from leaking out of the blood vessels (Cunha-Vaz, 2011; Cunha-Vaz, 1976). Herein, an artificial iBRB model was simulated by growing a tight monolayer of human retina endothelial cell (HRECs) on the transwell insert to test nanoparticle-induced perturbations. As shown in the schematic diagram for the transwell experiments (Figure S4), paracellular flux, an important index of barrier function, was measured by fluorescent probe (RITC-dextran) that penetrates through HRECs monolayer across the inserts to the bottom chambers. The results (Figure 5a) showed that in contrast to PBS control, mouse plasma and MUA@AgNPs alone, BK (10 μM) induced an increase in paracellular flux, suggesting BK mediated endothelial paracellular permeability. Similarly, MUA@AgNPs activated plasma significantly elevated the endothelial permeability in a dose-dependent manner (0.2, 2 μg/mL). Aprotinin is able to efficiently inhibit the activities of proteases of FXIIa and PK (Sherman, 1968), while HOE 140 can specifically bind with the B2 receptor and block the activity of BK (Wirth, 1991). When the mouse plasma was pre-mixed with aprotinin and subsequent treated with MUA@AgNPs before cell stimulation, the paracellular flux significantly decreased compared to the cells treated with MUA@AgNPs activated plasma (Figure 5a). Likewise, the pretreatment of HRECs with HOE 140 inhibited the effect of MUA@AgNPs activated plasma on endothelial cell barrier permeability. In addition, cell viability of HRECs by Alamar Blue assay (Figure S5) confirmed that the increased endothelial paracellular permeability induced by MUA@AgNPs activated plasma was irrelevant to non-specific cytotoxic effects.
The integrity of tight junctions (TJs) and adherens junctions (AJs) on endothelial cells dictates the performance of iBRB (Dejana, 2004; Dejana, 2009). More specifically, the AJ exhibits a key role in the formation and maintenance of the junction structure, and it also affects the TJ organization, promoting the homophilic adhesions between the vascular endothelial cells and sustaining the integrity of the blood barriers (Behrens, 1985; Ohsugi, 1997; Taddei, 2008). The AJ impairment might directly lead to the dysfunction of barrier rigidity (Song, 2006). Recently, it has been reported that the inflammatory mediators, such as BK and histamine can bind and activate the receptors on endothelial cells and induce the shedding of VE-cadherin, a main component of AJ proteins, thus increasing vascular permeability (Orsenigo, 2012). To further elucidate the molecular mechanism for the enhanced endothelial paracellular permeability induced by MUA@AgNPs activated plasma, we investigated VE-cadherin expression in HRECs upon the stimulations similar to the transwell experiments described above. Similar to BK, MUA@AgNPs activated plasma decreased VE-cadherin expression in a dose-dependent manner (Figure 5b and 5c). The treatments with plasma or MUA@AgNPs alone did not change VE-cadherin expression in HRECs. The application of aprotinin and HOE 140 recovered VE-cadherin expression by 36% and 33%, respectively, when compared to that of MUA@AgNP activated plasma group.
The localization of VE-cadherin in HRECs was further visualized by immunocytostaining with the corresponding antibodies conjugated with Dylight549 probe. The results in Figure 5d showed VE-cadherin stained by green fluorescent probe was continuously distributed on the borders of HRECs and constituted the intact cell AJs in the control group. Neither plasma nor MUA@AgNPs alone affected VE-cadherin distribution in HRECs. However, the fluorescence on cellular border lost its continuity and obvious breaks were observed after BK treatment (marked with yellow arrows in Figure 5d), indicating that BK decreased the expression of VE-cadherin on the junctions. When HRECs were treated with MUA@AgNP activated plasma, the integrity of VE-cadherin distribution on AJs was damaged (indicated by the yellow arrows in Figure 5d) similar to the effect induced by BK. The inhibitors of aprotinin and HOE 140 blocked the destructive effects on VE-cadherin expression induced by MUA@AgNPs activated plasma. The integrity level of AJs was further quantified by scoring the continuity grade of stained VE-cadherin on HRECs (Murakami, 2009). The statistical results in Figure 5e demonstrated that the continuity grades of HRECs after the treatments with PBS, plasma or MUA@AgNPs were 4.31, 4.32 and 4.20, respectively, and the scores of HRECs exposed to BK and MUA@AgNPs activated plasma reduced to 2.46 and 2.35, separately. Aprotinin and HOE 140 in combination with MUA@AgNPs activated plasma were scored 3.62 and 3.40, respectively, showing their substantial protective effects. The results obtained from the transwell experiments, VE-cadherin Western blots and immunocytostaining assays collectively suggested that MUA@AgNP activated plasma increased iBRB permeability through the regulation of B2 receptor.
In summary, activation of the KKS cascade triggered by negatively charged AgNPs liberates the biological active peptide, BK, which binds with B2 receptor and results in the shedding of VE-cadherin in retinal endothelial cells. The loss of VE-cadherin impairs AJs integrity and causes increased retinal vascular permeability. The proposed mechanism is illustrated in Figure 6.
Figure 6.

Schematic diagram depicting the mechanism for the elevated retinal vascular permeability induced by negatively charged AgNPs through contact activation.
4. Discussion
In view of the potential biological hazards of nanosilver, massive toxicological data has been obtained so far to demonstrate its diverse effects including pulmonary inflammation, hepatic and renal damage and cerebral edema (Stebounova, 2011; Sung, 2008; Yin, 2013). However, the biological process for AgNPs infiltration through vascular barrier and the underlying mechanism remain ambiguous. The KKS in plasma regulates vascular permeability and its hyper-activation may cause life-threatening clinical disorders like hereditary angioedema (Kaplan, 2010). The investigation on the interaction of AgNPs with the KKS would be helpful to address the suspending question mentioned above. In this work, the elevated retinal vascular permeability was found by the intravitreal injection of AgNPs in vivo, and the molecular mechanism for KKS activation by the negatively charged AgNPs and the disruption of adherens junction was further uncovered.
As the initial zymogen in the KKS, FXII autoactivates by contact with a variety of artificial or biologic negatively charged surfaces (contact activation), resulting in the waterfall cascade activation of KKS (Schmaier, 2008). Activated FXIIa cleaves zymogen PPK at the single peptide bond at Arg371-Ile372 to form the active PK protein with two chains (Colman, 1997). In this study, the heavy chain of PK with a molecular weight of approximate 52 kDa was detected by resolving the mouse plasma treated with kaolin, a well-known FXII activator on the reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the cleavage of PPK caused loss of the band at ~75 kDa meanwhile (Figure 2a), which was used as a key marker to probe contact activation in our study of AgNPs.
Ex-vivo experiments on three species of AgNPs with different surface charges indicated that only the particles with the negatively charged surface (MUA@AgNPs) triggered this biological process (Figure 2b). When the toxicity of nanomaterials with respect to surface charge is compared, it is commonly believed that the positive one has greater toxicity due to its higher ability to direct penetrate the cell membrane with negative surface charges in most instances (Mecke, 2005; Mecke, 2004). Nevertheless, this study brought a new distinct insight for the influence of surface charges on the biological effects of AgNPs. The next important question into the biological effects of AgNPs is whether the particles or the released ions are the active species as the controversial debates are occurrent on this topic (Ahamed, 2010; Lapresta-Fernández, 2012). The result in Figure 2b shows Ag+ itself doesn’t induce ex-vivo PPK activation. Moreover, the free capping MUA molecules are reactionless (Figure 2c). Taken together, it can be concluded that the negative surface charge and AgNPs particulate core are essential for triggering KKS activation, which was consistent with the extant theory of FXII autoactivation on negatively charged, hydrophilic surfaces. The binding assay and the conformational characterization of FXII zymogen confirmed its autoactivation upon MUA@AgNPs treatment (Figure 3a, b), which trigged the waterfall cascade activation of the KKS. As evidenced by Western blots for in-vitro and ex-vivo experiments (Figure 3c, d and Figure 4a, b), the consecutive cleavages of zymogens including FXII, PPK and HK, proved that MUA@AgNPs were capable of inducing contact activation. In-vivo KKS activation characterized by PPK cleavage showed the distinctive time-course for PK formation (Figure 4b) occurred in contrast to ex vivo results (Figure 2d) because multiple endogenous plasma protease inhibitors are constantly recruited to bind and degrade activated FXII and PK upon contact activation thus balancing the homeostasis in blood circulation (Kaplan, 1985; Schmaier, 2007).
Previous scientific studies on the evaluation of potential risks related to the short- and long-term toxicities (Ahamed et al, 2010) of AgNPs cover a wide range of dosages on cell cultures (from several to hundreds μg/mL) and in animal tests (from several to hundreds mg/kg body weight). Human bodies may also be exposed to AgNPs directly due to their medical usages including dental instruments, coating contact lenses, bandages, endodontic filling materials, medical catheters, cardiovascular implants, wound dressing, bone cement and other implants (Ge et al, 2014), which brings the opportunities for AgNPs to enter into blood circulation system. In medical products, the concentration of AgNPs varies according to the specific purposes. For example, there is an alternative-medicine scene that recommends a daily intake of colloidal silver at the concentration of less than 10 μg/mL to prevent sickness (Nanoinitiative Bayern GmbH, 2015). For immune support, one half of a teaspoon of colloidal silver (1–10 nm, 500 μg/mL) is suggested to be administered for most adults for the duration of 10–14 days (NPSW, 2015). According to ex-vivo experiments in our study, AgNPs can trigger plasma KKS activation when the concentration is above 5 μg/mL. In view of the real situation for AgNPs usage and the accurate evaluation for the biological effects of AgNPs, the highest concentration of 50 μg/mL (1 μL of 500 μg/mL AgNPs into approximate 10 μL of vitreous body) was accordingly used for in-vivo retinal vascular permeability study. Moreover, considering the short lifetime of the released BK and B2 receptor activation in vivo (Gousseva et al, 2008), the relevant effects on vascular permeability were evaluated based on acute stimulations in both animal studies (≤ 2 h) and cell experiments (≤ 6 h).
The inner blood-retinal barrier (iBRB) consisted of tightly joined retinal endothelial cells is vulnerable to KKS activation. Dysfunction of the retinal endothelial cells due to the imbalance of local KKS homeostasis disrupts barrier function which leads to pathological conditions like retinal hemorrhage and diabetic retinopathy (Cunha-Vaz, 2011; Klaassen, 2013). In-vivo experiments using FITC-dextran and EB assay showed that intravitreal injection of MUA@AgNPs caused increased retinal vascular leakage (Figure 4c and d) potentially through the induction of local endogenous KKS activation. The tests on the HREC monolayer using MUA@AgNPs activated plasma confirmed the crucial role of KKS activation in the mediation paracellular permeability of the simulated iBRB (Figure 5a). The antagonist tests using aprotinin and HOE140 verified KKS activation and BK availability were essential for iBRB breakdown upon MUA@AgNPs stimulation.
The AJs impairment may directly lead to the dysfunction of barrier rigidity. The trans-membrane adhesive protein, VE-cadherin is the key protein involved in the formation of endothelial AJs. BK induces increased vascular permeability by activating B2 receptor, which results in phosphorylation, internalization and destruction of the VE-cadherin (Bouillet, 2011). Western blots and imunocytostainning of VE-cadherin (Figure 5b and 5d) demonstrated the generation of FXIIa, PK and BK in MUA@AgNPs treated plasma played critical roles for VE-cadherin loss in HRECs, which provided the underlying molecular mechanism for the increased paracellular permeability in HRECs monolayer simulated iBRB.
5. Conclusions
This study elucidated that the enhanced iBRB permeability by the negatively charged AgNPs was mediated through the plasma contact activation, resulting in the shedding of VE-cadherin on the borders of retinal endothelial cells, as illustrated in Figure 6. Different from neutral or positive charged nanoparticles or silver ion, negatively charged AgNPs triggered the waterfall cascade activation of KKS in the plasma through the binding to FXII and causing its autoactivation. This contact activation caused the liberation of BK that bound to B2 receptor on endothelial cells and disturbed the paracellular AJ protein, VE-cadherin, thus resulting in retinal vascular permeability. The findings obtained from this work provide key insights for nanoparticle synthesis in order to regulate vascular permeability besides the understanding of the potential biological hazards of AgNPs.
Supplementary Material
Footnotes
This material included Cit@AgNPs preparation, KKS activation scheme, results for characterization of BEPI@AgNPs and PVP@AgNPs, the dynamic dissolution of Ag+ from MUA@AgNPs in plasma, schematic diagram of transwell experiment and cell viability of HRECs upon various stimulus treatments including mouse plasma, MUA@AgNPs, MUA@AgNPs activated plasma, BK, aprotinin and HOE140.
Declaration of Interest Section
This work was jointly supported by the National Basic Research Program of China (2011CB936001), National Natural Science Foundation of China (21137002, 21477153, 21307151) and the China Postdoctoral Science Foundation (2013M541055). The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ahamed M, Alsalhi MS, Siddiqui MK. Silver nanoparticle applications and human health. Clin Chim Acta. 2010;411:1841–1848. doi: 10.1016/j.cca.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Badawy AME, Luxton TP, Silva RG, Scheckel KG, Suidan MT, Tolaymat TM. Impact of environmental conditions (pH, ionic strength, and electrolyte type) on the surface charge and aggregation of silver nanoparticles suspensions. Environ Sci Technol. 2010;44:1260–1266. doi: 10.1021/es902240k. [DOI] [PubMed] [Google Scholar]
- Baruwati B, Simmons SO, Varma RS, Veronesi B. “Green” synthesized and coated nanosilver alters the membrane permeability of barrier (intestinal, brain endothelial) cells and stimulates oxidative stress pathways in neurons. ACS Sustain Chem Eng. 2013;1:753–759. [Google Scholar]
- Behrens J, Birchmeier W, Goodman SL, Imhof BA. Dissociation of Madin-Darby canine kidney epithelial cells by the monoclonal antibody anti-arc-1: mechanistic aspects and identification of the antigen as a component related to uvomorulin. J Cell Biol. 1985;101:1307–1315. doi: 10.1083/jcb.101.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkqvist J, Jämsä A, Renné T. Plasma kallikrein: the bradykinin-producing enzyme. Thromb Haemost. 2013;110:399–407. doi: 10.1160/TH13-03-0258. [DOI] [PubMed] [Google Scholar]
- Bouillet L, Mannic T, Arboleas M, Subileau M, Massot C, Drouet C, Huber P, Vilgrain I. Hereditary angioedema: Key role for kallikrein and bradykinin in vascular endothelial-cadherin cleavage and edema formation. J Allergy Clin Immunol. 2011;128:232–234. doi: 10.1016/j.jaci.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Burke AR, Singh RN, Carroll DL, Owen JD, Kock ND, D’Agostino R, Jr, Torti FM, Torti SV. Determinants of the thrombogenic potential of multiwalled carbon nanotubes. Biomaterials. 2011;32:5970–5978. doi: 10.1016/j.biomaterials.2011.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citarella F, Ravon DM, Pascucci B, Felici A, Fantoni A, Hack CE. Structure/function analysis of human factor XII using recombinant deletion mutants. Eur J Biochem. 1996;238:240–249. doi: 10.1111/j.1432-1033.1996.0240q.x. [DOI] [PubMed] [Google Scholar]
- Colman RW, Schmaier AH. Contact system: A vascular biology modulator with anticoagulant, Profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–3843. [PubMed] [Google Scholar]
- Cunha-Vaz J, Bernardes R, Lobo C. The blood-retinal barrier. Eur J Ophthalmol. 2011;21:3–9. doi: 10.5301/EJO.2010.6049. [DOI] [PubMed] [Google Scholar]
- Cunha-Vaz JG. The blood-retinal barriers. Doc Ophthalmol. 1976;41:287–327. doi: 10.1007/BF00146764. [DOI] [PubMed] [Google Scholar]
- Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Feener EP, Zhou Q, Fickweiler W. Role of plasma kallikrein in diabetes and metabolism. Thromb Haemost. 2013;110:434–441. doi: 10.1160/TH13-02-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Li Q, Wang M, Ouyang J, Li X, Xing MMQ. Nanosilver particles in medical applications: synthesis, performance, and toxicity. Int J Nanomed. 2014;9:2399–2407. doi: 10.2147/IJN.S55015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geranio L, Heuberger M, Nowack B. The behavior of silver nanotextiles during washing. Environ Sci Technol. 2009;43:8113–8118. doi: 10.1021/es9018332. [DOI] [PubMed] [Google Scholar]
- Gousseva V, Simaan M, Laporte SA, Swain PS. Inferring the lifetime of endosomal protein complexes by fluorescence recovery after photobleaching. Biophys J. 2008;94:679–687. doi: 10.1529/biophysj.107.115188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpel PC, Lewin MF, Kaplan AP. Distribution of plasma kallikrein between C-1 inactivator and alpha 2-macroglobulin in plasma utilizing a new assay for alpha 2-macroglobulin-kallikrein complexes. J Bio Chem. 1985;260:4257–4263. [PubMed] [Google Scholar]
- Janardhanan R, Karuppaiah M, Hebalkar N, Rao TN. Synthesis and surface chemistry of nano silver particles. Polyhedron. 2009;28:2522–2530. [Google Scholar]
- Kaplan AP, Joseph K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104:193–204. doi: 10.1016/j.anai.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Kaplan AP, Gruber B, Harpel PC. Assessment of hageman factor activation in human plasma: quantification of activated hageman factor-C1 inactivator complexes by an enzyme- linked differential antibody immunosorbent assay. Blood. 1985;66:636–641. [PubMed] [Google Scholar]
- Kempen JH, O’Colmain BJ, Leske MC, Haffner SM, Klein R, Moss SE, Taylor HR, Hamman RF. THe prevalence of diabetic retinopathy among adults in the united states. Arch Ophthalmol. 2004;122:552–563. doi: 10.1001/archopht.122.4.552. [DOI] [PubMed] [Google Scholar]
- Klaassen I, Van Noorden CJF, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res. 2013;34:19–48. doi: 10.1016/j.preteyeres.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Konings J, Govers-Riemslag JWP, Philippou H, Mutch NJ, Borissoff JI, Allan P, Mohan S, Tans G, ten Cate H, Ariëns RAS. Factor XIIa regulates the structure of the fibrin clot independently of thrombin generation through direct interaction with fibrin. Blood. 2011;118:3942–3951. doi: 10.1182/blood-2011-03-339572. [DOI] [PubMed] [Google Scholar]
- Lapresta-Fernández A, Fernández A, Blasco J. Nanoecotoxicity effects of engineered silver and gold nanoparticles in aquatic organisms. TrAC, Trends Anal Chem. 2012;32:40–59. [Google Scholar]
- Lee KJ, Nallathamby PD, Browning LM, Osgood CJ, Xu X-HN. In vivo imaging of transport and biocompatibility of single silver nanoparticles in early development of zebrafish embryos. ACS Nano. 2007;1:133–143. doi: 10.1021/nn700048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PC, Meisel D. Adsorption and surface-enhanced raman of dyes on silver and gold sols. J Phys Chem. 1982;86:3391–3395. [Google Scholar]
- Levard C, Hotze EM, Lowry GV, Brown GE., Jr Environmental transformations of silver nanoparticles: impact on stability and toxicity. Environ Sci Technol. 2012;46:6900–6914. doi: 10.1021/es2037405. [DOI] [PubMed] [Google Scholar]
- Liu J, Sonshine DA, Shervani S, Hurt RH. Controlled release of biologically active silver from nanosilver surfaces. ACS Nano. 2010;4:6903–6913. doi: 10.1021/nn102272n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas C, Oschatz C, Renné T. The plasma contact system 2.0. Semin Thromb Hemost. 2011;37:375–381. doi: 10.1055/s-0031-1276586. [DOI] [PubMed] [Google Scholar]
- Mecke A, Majoros IJ, Patri AK, Baker JR, Banaszak Holl MM, Orr BG. Lipid Bilayer Disruption by Polycationic Polymers: The roles of size and chemical functional group. Langmuir. 2005;21:10348–10354. doi: 10.1021/la050629l. [DOI] [PubMed] [Google Scholar]
- Mecke A, Uppuluri S, Sassanella TM, Lee D-K, Ramamoorthy A, Baker JR, Jr, Orr BG, Banaszak Holl MM. Direct observation of lipid bilayer disruption by poly(amidoamine) dendrimers. Chem Phys Lipids. 2004;132:3–14. doi: 10.1016/j.chemphyslip.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Bio Chem. 2009;284:21036–21046. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanoinitiative Bayern GmbH. Application of nano silver in medicine. 2015 Available at: http://www.nanosilber.de/en/nano-silver/examples-of-application/
- Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- NPSW (Natural Path Silver Wings) 2015 Available at: http://www.npswsilver.com/shop/COLLOIDAL%20SILVER%20500ppm/cat/11956.
- Ohsugi M, Larue L, Schwarz H, Kemler R. Cell-Junctional and cytoskeletal organization in mouse blastocysts lacking E-cadherin. Dev Biol. 1997;185:261–271. doi: 10.1006/dbio.1997.8560. [DOI] [PubMed] [Google Scholar]
- Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Young Koh G, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M, Dejana E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaier AH. The elusive physiologic role of factor XII. J Clin Invest. 2008;118:3006–3009. doi: 10.1172/JCI36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaier AH, McCrae KR. The plasma kallikrein–kinin system: its evolution from contact activation. J Thromb Haemost. 2007;5:2323–2329. doi: 10.1111/j.1538-7836.2007.02770.x. [DOI] [PubMed] [Google Scholar]
- Sherman MP, Kassell B. The tyrosine residues of the basic trypsin inhibitor of bovine pancreas. Spectrophotometric titration and iodination. Biochemistry. 1968;7:3634–3641. doi: 10.1021/bi00850a042. [DOI] [PubMed] [Google Scholar]
- Simberg D, Zhang W-M, Merkulov S, McCrae K, Park J-H, Sailor MJ, Ruoslahti E. Contact activation of kallikrein-kinin system by superparamagnetic iron oxide nanoparticles in vitro and in vivo. J Control Release. 2009;140:301–305. doi: 10.1016/j.jconrel.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2006;109:1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N, Woody RW. Computation and analysis of protein circular dichroism spectra. Meth Enzymol. 2004;383:318–351. doi: 10.1016/S0076-6879(04)83013-1. [DOI] [PubMed] [Google Scholar]
- Stebounova LV, Adamcakova-Dodd A, Kim JS, Park H, O’Shaughnessy PT, Grassian VH, Thorne PS. Nanosilver induces minimal lung toxicity or inflammation in a subacute murine inhalation model. Part Fibre Toxicol. 2011;8:1–5. doi: 10.1186/1743-8977-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Ji JH, Yoon JU, Kim DS, Song MY, Jeong J, Han BS, Han JH, Chung YH, Kim J, Kim TS, Chang HK, Lee EJ, Lee JH, Yu IJ. Lung function changes in Sprague-Dawley rats after prolonged inhalation exposure to silver nanoparticles. Inhal Toxicol. 2008;20:567–574. doi: 10.1080/08958370701874671. [DOI] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- Takenaka S, Karg E, Roth C, Schulz H, Heinzmann AZU, Schramel P, Heyder aJ. Pulmonary and systemic distribution of inhaled ultrafine silver particles in rats. Environ Health Persp. 2001;109:547–551. doi: 10.1289/ehp.01109s4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SL, Wahl-Jensen V, Copeland AM, Jahrling PB, Schmaljohn CS. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLos Pathog. 2013;9:e1003470. doi: 10.1371/journal.ppat.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trickler WJ, Lantz SM, Murdock RC, Schrand AM, Robinson BL, Newport GD, Schlager JJ, Oldenburg SJ, Paule MG, Slikker W, Jr, Hussain SM, Ali SF. Silver nanoparticle induced blood-brain barrier inflammation and increased permeability in primary rat brain microvessel endothelial cells. Toxicol Sci. 2010;118:160–170. doi: 10.1093/toxsci/kfq244. [DOI] [PubMed] [Google Scholar]
- TSE (The Silver Edge) Using colloidal silver to heal eye infections. 2015 Available at: http://www.thesilveredge.com/eye_infections.shtml#.VZOchTsVjmQ.
- van der Graaf F, Koedam JA, Bouma BN. Inactivation of kallikrein in human plasma. J Clin Invest. 1983;71:149–158. doi: 10.1172/JCI110743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warboys CM, Toh H-B, Fraser PA. Role of NADPH Oxidase in retinal microvascular permeability increase by RAGE activation. Invest Ophth Vis Sci. 2009;50:1319–1328. doi: 10.1167/iovs.08-2730. [DOI] [PubMed] [Google Scholar]
- Williams R, Airey M, Baxter H, Forrester J, Kennedy-Martin T, Girach A. Epidemiology of diabetic retinopathy and macular oedema: a systematic review. Eye. 2004;18:963–983. doi: 10.1038/sj.eye.6701476. [DOI] [PubMed] [Google Scholar]
- Wilson center. Project on emerging nanotechnologies. Consumer products inventory. 2014 http://www.nanotechproject.org/cpi.
- Wirth K, Hock Fj, Albus U, WL, Alpermann HG, Anagnostopoulos H, Henk S, Breipohl G, Konig W, Knolle J, Schölkens BA. Hoe 140 a new potent and long acting bradykinin-antagonist: in vivo studies. Br J Pharmacol. 1991;102:774–777. doi: 10.1111/j.1476-5381.1991.tb12249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Qaum T, Adamis AP. Sensitive blood–retinal barrier breakdown quantitation using evans blue. Invest Ophth Vis Sci. 2001;42:789–794. [PubMed] [Google Scholar]
- Yang H, Liu Y, Shen Q, Chen L, You W, Wang X, Sheng J. Mesoporous silica microcapsule-supported Ag nanoparticles fabricated via nano-assembly and its antibacterial properties. J Mater Chem. 2012;22:24132–24138. [Google Scholar]
- Yin N, Liu Q, Liu J, He B, Cui L, Li Z, Yun Z, Qu G, Liu S, Zhou Q, Jiang G. Silver nanoparticle exposure attenuates the viability of rat cerebellum granule cells through apoptosis coupled to oxidative stress. Small. 2013;9:1831–1841. doi: 10.1002/smll.201202732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.