

Abstract
Background
The Lundehund is a highly specialized breed characterized by a unique flexibility of the joints and polydactyly in all four limbs. The extremely small population size and high inbreeding has promoted a high frequency of diseased dogs affected by the Lundehund syndrome (LS), a severe gastro-enteropathic disease.
Results
Comprehensive analysis of bead chip and whole-genome sequencing data for LS in the Lundehund resulted in a genome-wide association signal on CFA 34 and LS-specific runs of homozygosity (ROH) in this region. Filtering analysis for variants with predicted high or moderate effects revealed a missense mutation in LEPREL1 1.2 Mb proximal to the region of the genome-wide association, which was shown to be significantly associated with LS. LS-affected Lundehund harbored the mutant LEPREL1:g.139212C>G genotype A/A whereas all controls of other breeds showed the C/C wild type.
In addition, ROH analysis for the Lundehund indicated a high enrichment of genes in potential signatures of selection affecting protein activation and immunoregulatory processes like NOD1 potentially involved in LS breed disposition.
Conclusions
Sequencing results for Lundehund specific traits reveal a potential causative mutation for LS in the neuropeptide operating gene LEPREL1 and suggests it as a precursor of the inflammatory process. Analyses of ROH regions give an insight into the genetic background of characteristic traits in the Lundehund that remain to be elucidated in the future.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2844-6) contains supplementary material, which is available to authorized users.
Keywords: LEPREL1, Lundehund syndrome, Runs of homozygosity
Background
The Norwegian Lundehund represents a specific but small group in the variety of dog breeds which have evolved since domestication [1]. It stands out by exceptional characteristics like double dewclaws and extreme flexibility in shoulder and neck, which represent traits fixed in the Lundehund population [2]. In addition, the Lundehund harbors a breed disposition for a syndrome comprising particular features of protein-losing enteropathy (PLE), intestinal lymphangiectasia, gastrointestinal disturbance, inflammatory bowel disease and malabsorption designated as Lundehund syndrome (LS) [3, 4]. Clinical signs are diarrhea, vomiting, weight loss, edema and apathy often accompanied with decreased concentrations of albumin and globulin in blood profile [2]. A similar condition has been described in the Soft Coated Wheaten Terrier affecting immune system, gut and kidney [5, 6]. It was supposed that one or more genes involved in these complex systems might be responsible for a breed disposition for PLE. An increased occurrence of PLE could also be found in Rottweilers and Yorkshire terriers [7–9]. In Basenji and German Shepherd, hypoalbuminemia and hypoglobulinemia was found in dogs with severe lymphocytic-plasmacytic enteritis characterized by excessive infiltrates of mononuclear inflammatory cells [10, 11]. It was suggested that the inflammatory bowel disease in German Shepherd was the result of a complex etiology with the involvement of different variant effects similar to Crohn’s disease, the chronic inflammatory bowel disease in human [12–15].
In the Lundehund, a signature of selection for LS was suggested to be located on CFA 9 in the region of Caspase Recruitment Domain Family, Member 9 (CARD9), which is known to be associated with Crohn’s disease and ulcerative colitis in human [16, 17]. In addition, extended haplotype homozygosity (EHH) tests suggested further breed specific characteristics like polydactyly, body size or flexibility of the joints as targets of specific selection [17]. Genotyping the limb development membrane protein 1 (LMBR1) mutation DC-2, which was shown to be associated with preaxial polydactyly in western dogs, revealed the mutant genotype (A/A) in all tested Lundehund [18, 19]. In general, the frequency of long stretches of homozygous genotypes on basis of bead chip data was shown to be high which was suggested to be a result of an extremely low genetic variability in the Lundehund breed [1, 17, 20].
In this study whole-genome sequencing was performed in two Lundehund pools comprising LS-affected and LS-unaffected individuals. Sequence data were investigated for runs of homozygosity (ROHs) and variants predicted to have high or moderate effects in order to elucidate the genetic background of LS.
Results
Phenotype
Samples of six Lundehund dogs with typical characteristics of a puffin hunter phenotype including an additional toe and joint flexibility (Fig. 1) were chosen for whole-genome sequencing in two pools. One pool enclosed three healthy dogs whereas the other pool comprised three Lundehund showing severe signs of gastroenteropathy known as LS. In addition, 12 LS-affected Lundehund with clinical signs and low blood protein parameters, 6 Lundehund suspected to be LS-affected due to recurrent diarrhea and vomiting and 12 Lundehund without clinical signs of LS classified as LS-unaffected were available for further analysis. Characteristic clinical signs were diarrhea, vomiting, weight loss and apathy (Fig. 2). The onset of first signs varied widely from the age of 2.5 to 10.5 years (Additional file 1). Some cases also showed ascites and edema in the limbs. The results of blood screening revealed hypoalbuminemia in all analyzed cases, sometimes accompanied with hypoglobulinemia, reduced levels of fructosamines and vitamin B, hypo- or hypercalcemia, as well as increased concentrations of serum folate and alanine aminotransferase.
Fig. 1.
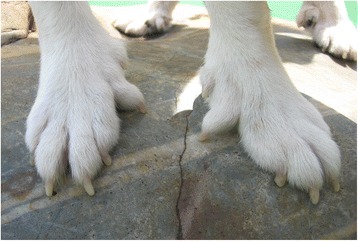
Polydactyly in the Lundehund. Polydactyly with six toes at all four limbs is a characteristic trait in the Lundehund
Fig. 2.
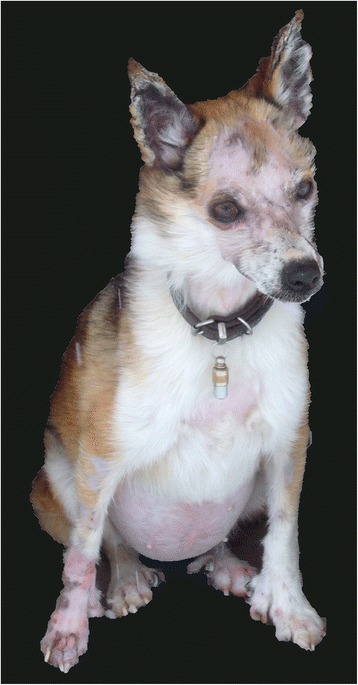
Clinical picture of Lundehund syndrome (LS). A five year old Lundehund with severe signs of LS is shown. Persistent diarrhea, vomiting and ascites resulted in marked weight and hair loss. The dog had to be euthanized due to poor prognosis and unresponsive therapy
Pedigree and association analysis
Inspection of the pedigree data of all Lundehund under study revealed close relationships among affected dogs (Additional file 2). In total 24 female and 16 male LS-affected Lundehund were included in this pedigree data. A complex segregation analysis revealed a recessive major gene model as the most likely mode of inheritance with the lowest −2 log-likelihood at 52.68 (Additional file 3).
Genome-wide association analysis for LS in 17 LS-affected and 8 LS-unaffected Lundehund showed a highly significant peak on CFA 34 at 26,384,304-27,498,705 bp (CanFam 2.0) corresponding to 23,373,982-24,488,983 bp in CanFam 3.1 assembly (Fig. 3) in the region of fibroblast growth factor 12 (FGF12; ENSCAFG00000031187) and mab-21 domain containing 2 (MB21D2; ENSCAFG00000014075). The highest -log10P value (−log10P = 7.7) could be shown for one SNP at 24,152,349 bp whereas further 30 SNPs in the associated region reached -log10P-values of 5.6-5.9. Expected versus observed -log10P-values (quantile-quantile-plot) showed that inflation due to stratification effects had not increased -log10P-values. The distribution of genotypes suggested a recessive effect as proposed by segregation analysis.
Fig. 3.
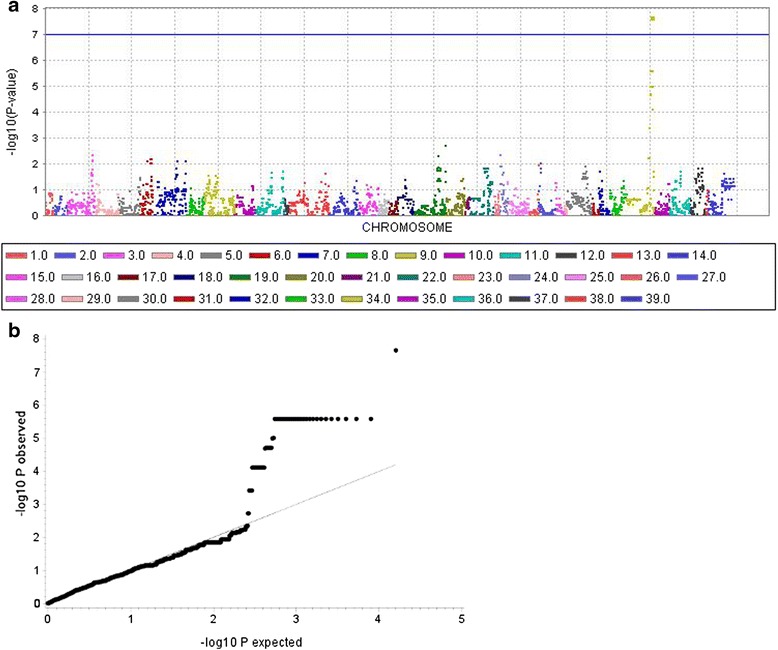
Genome-wide association analysis (GWAS) for Lundehund syndrome (LS). a The Manhattan-plot of the -log10 P-values shows a highly significant peak on canine chromosome (CFA) 34 in the region of 23,373,982-24,488,983 bp (CanFam 3.0). b Observed versus expected -log10 P-values (Q-Q plot) reveal 31 SNPs with highly significant -log10 P-values
Sequencing and variant detection
Whole-genome sequencing of two Lundehund pools with three dogs each resulted in a mean coverage of 15.56X for Lundehund pool 1 (LS-affected) and 14.48X Lundehund pool 2 (LS-unaffected). In total 10,584,482 SNPs and 3,223,567 insertions/deletions (INDEL) could be detected for pool 1 as well as 10,530,165 SNPs and 3,207,163 INDEL for pool 2 in raw data after variant calling. Stringent filters for read depth and quality of each variant led to a set of 8,815,395 SNPs (pool 1) as well as 8,799,793 SNPs (pool 2) for ROH analysis. For these SNPs the mean heterozygosity per site was 0.11 in both pools.
LS-specific ROH detection
A total of 1713 ROHs specific for LS-affected dogs were detected. Three ROHs could be found directly in the region of association on CFA 34 at 23,223,356-24,490,050 bp harboring the genes Coiled-Coil Domain Containing 50 (CCDC50), FGF12 and MB21D2 (Additional file 4). In private ROHs analysis for homozygous regions which could be exclusively found in LS-affected as well as LS-unaffected Lundehund, 472 ROH regions could be detected for pool 1 (LS-affected, Additional file 5) and 408 ROH regions for pool 2 (LS-unaffected, Additional file 6). The largest private ROH region for pool 1 could also be found on CFA 15 in a wider area of 7,812,609-13,628,020 bp including 51 genes.
Breed specific ROH
Consensus ROH detection was performed to identify potential signatures of selection for unique breed specific characteristics whose intense human selection might be collaterally associated with the high incidence of LS in the Lundehund population.
In total 660 Lundehund specific consensuses ROH regions could be detected for 500-SNP windows (Additional file 7). A notably high number of large stretches of consecutive homozygous genotypes was found on CFA 12 in the region of 32,968,627-49,633,816 bp and CFA 7 in the region of 28,049,903-54,984,739 bp. These regions harbored a large number of genes including PRRX1 (paired related homeobox 1) and GREM2 (gremlin 2, DAN family BMP antagonist) on CFA 7. Furthermore, we detected a ROH region on CFA 14 at 42,891,687-43,618,177 bp harboring Nucleotide-Binding Oligomerization Domain Containing 1 (NOD1). In contrast, no ROH region could be detected harboring the candidate gene LMBR1 (limb development membrane protein 1) for canine polydactyl mutations [19]. Further genotyping of the intronic LMBR1 SNP DC-2 revealed the mutant DC-2 allele homozygous in all analyzed Lundehund but also in the Norwegian Buhund, Bernese Mountain Dogs, Briard and Irish Wolfhound (Additional file 8).
Variants in ROH regions
Detection of SNPs or INDEL predicted to have high or moderate effects in ROH regions was performed to identify LS-specific or Lundehund-specific variants. No variant with predicted high or moderate effects could be detected within FGF12 and MB21D2 located in the region of ROHs found in the LS-affected (pool 1) which matched the region of genome-wide association. In total 2036 intron variants with a homozygous mutant genotype in LS-affected Lundehund, one 3′UTR and 13 5′UTR variants could be found in FGF12 with predicted low or modifier effects only. In MB21D2 no LS-associated variant could be detected.
Analysis of private LS-specific ROH regions resulted in one variant with a homozygous mutant genotype in LS-affected Lundehund, a heterozygous genotype in LS-unaffected Lundehund and a wildtype genotype in controls. This missense variant COL28A1:g.159951T>A on ECA14 was predicted to be deleterious (SIFT) as well as probably damaging (PolyPhen).
In ROH regions detected in all Lundehund (pool 1 and 2) seven variants with predicted tolerated or deleterious (low confidence) effects could be found. The candidate genes PRRX1 and GREM2 did not harbor variants with predicted high or moderate effects. Nevertheless, ten intronic variants could be detected in PRRX1 with mutant homozygous genotypes exclusively found in the Lundehund pools.
Functional annotation of ROH regions
Functional gene classification of human orthologues detected in ROH regions showed a particularly high involvement in biological processes affecting cellular processes (GO:0009987), localization (GO:0051179), response to stimulus (GO:0050896) and metabolic processes (GO:0008152) in all data sets (Additional file 9). PANTHER overrepresentation test in ROH regions in LS-affected Lundehund revealed an enrichment of genes involved in cellular process (GO:0009987, GO:0006464), metabolic process (GO:0008152, GO:0019538, GO:0044238, GO:0019219) and transcription (GO:0006351). Private ROHs detected in LS-affected dogs revealed a >5 fold enrichment of genes known to play a role in negative regulation of complement activation (GO:0045916), protein activation cascade (GO:2000258), humoral immune response (GO:0002921) and also in the perception of taste (GO:0001580, GO:0050913, GO:0050912, GO:0050909). In consensus Lundehund ROH regions genes affecting single-organism (GO:0044763) as well as cellular (GO:0009987) and biological (GO:0008150) processes were predicted to be overrepresented (Table 1).
Table 1.
Statistical overrepresentation test for biological processes. All human orthologues derived from ROH detection were analyzed for an enrichment of genes involved in specific processes using PANTHER tools. The number of detected genes in ROH regions in 500-SNP windows and Gene Ontology (GO) terms are shown for Bonferroni corrected significant P-values <0.05
| Number of reference genes (Homo sapiens) | ROH regions in LS-affected: expected | Bonferroni corrected P-value for multiple testing | Private ROH regions in LS-affected: expected | Bonferroni corrected P-value for multiple testing | Private ROH regions in LS-unaffected: expected | Bonferroni corrected P-value for multiple testing | Lundehund consensus ROH regions: expected | Bonferroni corrected P-value for multiple testing | |
|---|---|---|---|---|---|---|---|---|---|
| Single-organism cellular process (GO:0044763) | 11415 | ─ | ─ | ─ | ─ | ─ | ─ | 480.42 | 1.03E-06 |
| Single-organism process (GO:0044699) | 12755 | ─ | ─ | ─ | ─ | ─ | ─ | 536.82 | 5.36E-06 |
| Cellular process (GO:0009987) | 14147 | 1714.55 | 3.37E-05 | ─ | ─ | ─ | ─ | 595.41 | 4.70E-07 |
| Biological process (GO:0008150) | 16542 | ─ | ─ | ─ | ─ | ─ | ─ | 696.2 | 1.02E-04 |
| Generation of neurons (GO:0048699) | 1551 | ─ | ─ | ─ | ─ | 61.18 | 1.61E-02 | ─ | ─ |
| Neurogenesis (GO:0022008) | 1628 | ─ | ─ | ─ | ─ | 64.22 | 2.46E-02 | ─ | ─ |
| Cellular component organization or biogenesis (GO:0071840) | 5188 | ─ | ─ | ─ | ─ | 204.64 | 1.47E-02 | ─ | ─ |
| Cellular protein modification process (GO:0006464) | 1317 | 336.62 | 1.15E-03 | ─ | ─ | ─ | ─ | ─ | ─ |
| Protein metabolic process (GO:0019538) | 2692 | 688.07 | 2.95E-04 | ─ | ─ | ─ | ─ | ─ | ─ |
| Metabolic process (GO:0008152) | 8247 | 2107.91 | 7.09E-04 | ─ | ─ | ─ | ─ | ─ | ─ |
| Primary metabolic process (GO:0044238) | 6825 | 1744.45 | 1.39E-02 | ─ | ─ | ─ | ─ | ─ | ─ |
| Transcription, DNA-dependent (GO:0006351) | 1941 | 496.11 | 4.71E-03 | ─ | ─ | ─ | ─ | ─ | ─ |
| Regulation of nucleobase-containing compound metabolic process (GO:0019219) | 1700 | 434.52 | 1.03E-02 | ─ | ─ | ─ | ─ | ─ | ─ |
Filtering for mutations with predicted high or moderate effects
Specific filtering for variants with predicted high or moderate effects in the region of genome-wide association for LS revealed no variant directly in this region but one missense mutation 1.2 Mb proximal to the peak of association in the candidate gene LEPREL1 (Table 2). It was predicted to result in a substitution glutamic acid to glutamine.
Table 2.
Filtered variants from whole-genome analysis. Variants with predicted high or moderate effects and a homozygous mutant genotype exclusively found in the LS-affected pool as well as a heterozygous or homozygous wild-type genotype in the LS-unaffected pool are shown. All five reference dogs of four different breeds do not show the mutant genotype. Potential functional effects of these variants were predicted using SIFT and PolyPhen
| CFA | Position | Base change | Amino acid change | Consequence | Genotype (LS-affected Lunde-hund pool) | Genotype (LS-unaffected Lunde-hund pool) | Genotype (5 reference dogs) | Gene (transcript) | SIFT | PolyPhen-2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 13398018 | T>G | T>P/ S>R | missense variant | 1/1 | 0/0 | 0/0 | ENSCAFG00000031329 (novel gene; (ENSCAFT00000043665) and ENSCAFG00000030129 (novel gene; ENSCAFT00000048679) | tolerated (0.07)/ deleterious (0.02) | benign (0.074)/- |
| 1 | 111903572 | A>G | T>A | missense variant | 1/1 | 0/1 | 0/0 | CEACAM1 (ENSCAFT00000007749/ ENSCAFT00000046087/ ENSCAFT00000022623/ ENSCAFT00000047331/ ENSCAFT00000049292) | tolerated (0.08/ 0.13/ 0.2/ 0.21/ 0.2) | benign (0.00)/ benign (0.310)/ possibly damaging (0.659)/ possibly damaging (0.605)/ benign (0.250) |
| 3 | 56483857 | G>T | P>T | missense variant | 1/1 | 0/1 | 0/0 | IL16 (ENSCAFT00000021964) | tolerated (0.59) | benign (0.310) |
| 3 | 62265062 | A>T | F>I | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000017475 (novel gene; ENSCAFT00000027691) | deleterious (0.01) | possibly damaging (0.614) |
| 5 | 16233038 | G>T | L>M | missense variant | 1/1 | 0/1 | 0/0 | CEP164 (ENSCAFT00000020686) | deleterious (0.02) | probably damaging (0.996) |
| 6 | 67204999 | C>T | R>Q | non coding transcript variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000005648 (novel gene; ENSCAFT00000009086) | - | - |
| 6 | 67205308 | A>C | Y>* (stop codon) | non coding transcript variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000005648 (novel gene; ENSCAFT00000009086) | - | - |
| 6 | 67205754 | G>A | R>* (stop codon) | non coding transcript variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000005648 (novel gene; ENSCAFT00000009086) | - | - |
| 6 | 67205959 | A>G | L>P | non coding transcript variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000005648 (novel gene; ENSCAFT00000009086) | - | - |
| 6 | 67205966 | T>G | T>P | non coding transcript variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000005648 (novel gene; ENSCAFT00000009086) | - | - |
| 7 | 2590167 | G>A | S>N | missense variant | 1/1 | 0/1 | 0/0 | KIF14 (ENSCAFT00000017720) | tolerated (0.21) | benign (0.002) |
| 7 | 4169250 | A>T | F>Y | missense variant | 1/1 | 0/1 | 0/0 | PTPRC (ENSCAFT00000017964/ ENSCAFT00000017955) | tolerated (0.3/ 0.31) | benign (0.023/ 0.347 ) |
| 8 | 22616300 | C>T | P>L | missense variant | 1/1 | 0/1 | 0/0 | FANCM (ENSCAFT00000046644/ ENSCAFT00000022327/ ENSCAFT00000048988) | tolerated (0.42/ 0.34/ 0.42) | benign (0.009/ 0.009/ 0.004 ) |
| 8 | 47544408 | G>A | G>R | missense variant | 1/1 | 0/1 | 0/0 | VRTN (ENSCAFT00000026784) | tolerated (0.56) | possibly damaging (0.560) |
| 8 | 73685892 | C>G | Q>H | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000029996 (novel gene; ENSCAFT00000049952) | deleterious (0) | benign (0.152) |
| 8 | 73685923 | C>G | W>S | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000029996 (novel gene; ENSCAFT00000049952) | deleterious (0) | probably damaging (1.000) |
| 8 | 73685927 | T>G | S>R | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000029996 (novel gene; ENSCAFT00000049952) | tolerated (0.21) | probably damaging (0.963) |
| 9 | 37739213 | C>T | R>W | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000032731 (novel gene; ENSCAFT00000047013) | tolerated (0.07) | probably damaging (0.998) |
| 9 | 51227945 | T>C | H>R | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000019863 (novel gene; ENSCAFT00000031603) | deleterious (0) | possibly damaging (0.898) |
| 10 | 36330685 | C>T | A>V | missense variant | 1/1 | 0/1 | 0/0 | FAM32A (ENSCAFT00000003279) | tolerated (0.16) | benign (0.328) |
| 11 | 67454670 | A>C | S>R | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000028946 (novel gene; ENSCAFT00000045538) |
tolerated (0.33) | - |
| 12 | 40430108-40430109 | A>AGG | R47fs | frameshift variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000030790 (novel gene; ENSCAFT00000045206) | - | - |
| 12 | 52354031 | A>G | F>S | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000030881 (novel gene; ENSCAFT00000046435) |
deleterious (0.01) | - |
| 12 | 52354041 | G>A | L>F | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000030881 (novel gene; ENSCAFT00000046435) |
deleterious (0.02) | - |
| 14 | 16187782 | T>G | I>M | missense variant | 1/1 | 0/1 | 0/0 | CFAP69 (ENSCAFT00000002973) | tolerated (0.23) | possibly damaging (0.648) |
| 14 | 22993351 | A>T | C>S | missense variant | 1/1 | 0/1 | 0/0 | COL28A1 (ENSCAFT00000044570) | deleterious (0) | probably damaging (1.000) |
| 18 | 39577830 | C>T | A>V | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000030998 (novel gene; ENSCAFT00000047197) | tolerated (1) | benign (0.000) |
| 20 | 54722514 rs22884799 |
T>C | I>M | missense variant | 1/1 | 0/1 | 0/0 | KDM4B (ENSCAFT00000030040) | tolerated (0.7) | possibly damaging (0.728) |
| 27 | 2322565 | C>T | R>C | missense variant | 1/1 | 0/1 | 0/0 | KRT3 (ENSCAFT00000011634) | deleterious (0.03) | possibly damaging (0.942) |
| 28 | 31706107 | A>C | N>T | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000012412 (novel gene; (ENSCAFT00000019700) | tolerated (0.78) | benign (0.000) |
| 32 | 21555092 | C>T | D>N | missense variant | 1/1 | 0/1 | 0/0 | TRMT10A (ENSCAFT00000016732) | tolerated (0.31) | probably damaging (0.997) |
| 34 | 22046092 | C>G | E>Q | missense variant | 1/1 | 0/1 | 0/0 | LEPREL1 (ENSCAFT00000022188) | tolerated (0.43) | possibly damaging (0.945) |
| X | 96702059 rs24643372 |
A>G | R>G | missense variant | 1/1 | 0/1 | 0/0 | ENSCAFG00000029138 (novel gene; ENSCAFT00000044717) | tolerated (0.41) | benign (0.001) |
* is the official HGVS-symbol for a stop codon
In addition, specific filtering was done for further candidate variants which could potentially be involved in disease development of LS and might have been missed in the specified ROH detection windows or association analysis. We extracted genetic variants putatively associated with LS with predicted high or moderate effects which showed a homozygous mutant genotype only in LS-affected and a heterozygous mutant or wildtype genotype in LS-unaffected dogs. In total 32 SNPs and one INDEL resulted from filtering analysis.
Based on these results we chose the missense mutation LEPREL1:g.139212C>G near the region of genome-wide association and further six single nucleotide variants (SNV) in other genomic regions which were predicted to be deleterious (SIFT [21]) as well as possibly or probably damaging (PolyPhen [22]) for validation in all Lundehund samples and across several dog breeds. Three of these SNVs located in regions of an extreme high density of mutations, probably a result of inaccurate gene annotation, were assumed to be false positive results and therefore omitted from analysis. Genotyping of the remaining four non-synonymous SNVs in the region of the candidate genes leprecan-like 1 (LEPREL1), centrosomal protein 164 kDa (CEP164), collagen, type XXVII, alpha 1 (COL28A1) and keratin 3 (KRT3) in 36 Lundehund revealed a significant association of CEP164:g.57380G>T and LEPREL1:g.139212C>G with LS (Table 3).
Table 3.
Genotypic distribution of candidate SNPs for LS in Lundehund dogs. Chi-square test results are shown for the four candidate SNPs chosen from filtering analysis for mutations predicted to have high or moderate effects. Genotypes are assigned to Lundehund without signs of LS (0), LS-affected Lundehund (1) and LS-suspicious dogs (1 susp.) due to clinical signs
| CFA | Gene | Polymorphism | Chi-Square genotype (Probability) | Chi-Square allele (Probability) | Chi-Square trend (Probability) | LS status affected (1) or unaffected (0) | Genotype 0/0 (number and frequency) |
Genotype 0/1 (number and frequency) |
Genotype 1/1 (number and frequency) |
|---|---|---|---|---|---|---|---|---|---|
| 34 | LEPREL1 | LEPREL1:g.139212C>G | 6.807 (P = 0.033) | 4.959 (P = 0.026) | 4.293 (P = 0.038) | 0 | 1 (2.78) | 7 (19.44) | 7 (19.44) |
| 1 | 0 (0.00) | 0 (0.00) | 15 (41.70) | ||||||
| 1 (susp.) | 1 (2.78) | 2 (5.56) | 3 (8.34) | ||||||
| 5 | CEP164 | CEP164:g.57380G>T | 6.264 (P = 0.044) | 4.538 (P = 0.033) | 4.412 (P = 0.036) | 0 | 4 (11.43) | 6 (17.14) | 5 (14.29) |
| 1 | 0 (0.00) | 7 (19.46) | 7 (19.46) | ||||||
| 1 (susp.) | 0 (0.00) | 2 (5.56) | 4 (11.12) | ||||||
| 14 | COL28A1 | COL28A1:g.159951T>A | 0.614 (P = 0.736) | 0.019 (P = 0.889) | 0.021 (P = 0.883) | 0 | 3 (8.57) | 7 (20.00) | 5 (14.29) |
| 1 | 2 (5.72) | 8 (22.80) | 4 (11.44) | ||||||
| 1 (susp.) | 1 (2.86) | 4 (11.44) | 1 (2.86) | ||||||
| 27 | KRT3 | KRT3:g.2584C>T | 3.379 (P = 0.184) | 2.483 (P = 0.115) | 2.146 (P = 0.143) | 0 | 3 (8.33) | 8 (22.22) | 4 (11.11) |
| 1 | 3 (8.33) | 4 (11.12) | 8 (22.22) | ||||||
| 1 (susp.) | 0 (0.00) | 2 (5.56) | 4 (11.12) |
Further genotyping of 186 dogs of 17 breeds with no signs of gastroenteropathic disease showed only LEPREL1:g.139212C>G and COL28A1:g.159951T>A to be breed specific mutations for the Lundehund (Additional file 10) and revealed no significant P-value for COL28A1:g.159951T>A, but a significant P-value of 1.503E-29 for LEPREL1:g.139212C>G (Table 4). A closer look at the LS-phenotypes showed that all 17 LS-affected Lundhund harbored the homozygous mutant LEPREL1:g.139212C>G genotype whereas three of the six dogs which were estimated to be LS-suspicious due to clinical signs, did not show the homozygous mutant genotype.
Table 4.
Chi-square test for Lundehund dogs and seventeen different breeds. Other breeds are used as controls for Lundehund-specific LS. The test results are shown for the four candidate SNPs chosen from filtering analysis for mutations predicted to have high or moderate effects
| CFA | Gene | Polymorphism | Chi-Square genotype (Probability) | Chi-Square allele (Probability) | Chi-Square trend (Probability) |
|---|---|---|---|---|---|
| 34 | LEPREL1 | LEPREL1:g.139212C>G | 132.735 (P = 1.503E-29) | 238.686 (P = 7.607E-54) | 130.861 (P = 2.655E-30) |
| 5 | CEP164 | CEP164:g.57380G>T | 83.884 (P = 6.092E-19) | 120.165 (P = 5.822E-28) | 83.716 (P = 5.712E-20) |
| 14 | COL28A1 | COL28A1:g.159951T>A | 101.034 (P = 1.150E-22) | 116.566 (P = 3.573E-27) | 79.531 (P = 4.748E-19) |
| 27 | KRT3 | KRT3:g.2584C>T | 3.379 (P = 0.184) | 2.483 (P = 0.115) | 2.146 (P = 0.173) |
Candidate gene sequencing
Sequence analysis of LEPREL1 cDNA confirmed the missense mutation in exon 13 in LS-affected Lundehund dogs. No further SNVs could be found in LEPREL1 to be associated with LS (Additional file 11). Sequencing the complementary DNA (cDNA) of MB21D2 in an affected Lundehund and a reference German Shepherd dog revealed three mutations. Nevertheless, none of these mutations could be exclusively found in the Lundehund. The predicted gene FGF12 cDNA could not be amplified at all.
Discussion
Whole-genome sequencing analysis in two pools of Lundehund dogs revealed a candidate gene for LS-disposition and gave evidence for potential LS-related signatures of selection. The genome-wide association analysis resulted in a significant peak on CFA 34 which could also be shown to be located in a ROH region for LS-affected dogs. Screening of whole-genome data did not reveal any LS-associated mutations with predicted high impacts on protein function but the missense mutation LEPREL1:g.139212C>G 1.2 Mb proximal of the region of significant genome-wide association. We assume that the shift of the peak of LS-association and LS-associated ROH region might be a result of the low bead chip marker density that could be found in the region of LEPREL1. Furthermore, it could be shown that the detection of ROHs is strongly dependent on the window size and marker density [23] so that not all homozygous stretches can be detected in 500-SNP windows. We assume that smaller windows could probably have enabled the detection of a ROH region comprising LEPREL1 but might have increased the number of false positive results, too. According to the suggested recessive mode of inheritance the missense mutation LEPREL1:g.139212C>G could be shown to be homozygous in all analyzed dogs in NGS data. LS-affected dogs showed the mutant genotype G/G whereas LS-unaffected controls of other breeds did harbor the homozygous wild type C/C. In addition, seven Lundehund with no clinical signs for LS also showed the mutant genotype G/G. Due to the variable onset of the disease it can be proposed that these dogs will develop signs of the disease later in their life. We suggest this SNV as a potential causative mutation for a genetic disposition for LS.
LEPREL1 was shown to be expressed in a subpopulation of neuroendocrine cells of the intestinal mucosa and suggested to be important for processing and secretion of neuropeptides [24]. It was proposed that the enteric nervous system could trigger the occurrence of inflammatory bowel disease through neuropeptide secretion [25]. Neuropeptides were proposed to play a key role in inflammatory bowel diseases [25, 26]. Blockades for these molecules were considered for therapeutic approaches. We assume mutant LEPREL1 might be an essential precursor for LS in the Lundehund. Lundehund without clinical signs for LS who harbor this mutant RNA could possibly be subclinically affected or fall ill later in their life by specific trigger mechanisms. It was shown that not a few Lundehund who appeared healthy often had abnormal findings in histopathologic examinations [2]. The onset of the disease could be influenced by external factors triggering phenotypic expression. In the Soft Coated Wheaten Terrier it was proposed that food allergies could play a role in the development of PLE [5, 27]. In addition to external factors, further genes might be involved in triggering LS as well as influence the severity of symptoms. In human Crohn’s disease various mutations have been shown to be associated with disease development in different populations [13, 28–33]. It was suggested that complex regulatory processes driven by various genes could affect the extent of defects and result in different severities.
We assume that further genes might be involved in LS-development and could potentially be related to targets of selection for breed specific traits. This could probably explain the high incidence of LS in the population. In general, ROH analysis for the Lundehund showed a high frequency of long stretches of homozygous genotypes as expected from the extremely low genetic variability of this breed [1]. A high enrichment of genes in these ROH regions affecting protein activation, complement activation and humoral immune response suggested that protein-protein interactions and immunoregulatory processes might play an important role in the development of LS in the Lundehund as it was previously proposed for PLE in the Soft Coated Wheaten Terrier [5].
Variants in the candidate gene NOD1 (CARD4), which could be found to harbor a Lundehund specific ROH region, have been shown to be highly susceptible to inflammatory bowel disease [30]. It was proposed that NOD1 plays an important role in colonic epithelial protection against intracellular organisms similar to mucin genes. In Lundehund affected with atrophic gastritis an abnormal presence of mucous neck cells could be detected in the gastric mucosa [34]. We suppose that this gene, potentially under targeted selection in the Lundehund, might predispose this breed in general for gastrointestinal problems.
Further potential signatures of selection could be detected in the region of genes that might be involved in breed specific conformational traits like joint flexibility or polydactyly. According to Lundehund breed standard, the Lundehund was reported to have at least six toes at fore and hind limps [2, 18, 20]. Mapping analysis for hind limp specific canine preaxial polydactyly revealed a potentially causative mutation in LMBR1 (DC-2) in western dog breeds [19]. The homozygous mutant DC-2 genotype was also found in the Lundehund [18] and could be confirmed in all 36 Lundehund in our study. Nevertheless, it was still unclear if this mutation could explain the characteristic phenotype in the Lundehund showing polydactyly at both fore and hind limps in contrast to the usually unchanged forelimb in dogs with polydactyly [19]. We found evidence in Lundehund consensus ROH regions for potential modifier genes that might play a role in limb development. GREM2 was shown to be expressed in osteoblasts during in vivo skeletogenesis and involved in the regulation of bone formation genes [35, 36]. It was suggested that GREM2 was bidirectionally regulated by bone morphogenetic protein 2 (BMP2) together with GREM1, a gene which was shown to be expressed along with sonic hedgehog (SHH) and play an important role in limb bud development [36, 37]. A similar effect was suggested for PRRX1 (PRX1). Loss-of-function mutant PRRX1/PRRX2 mice resulted in shortened zeugopods of the forelimbs and hindlimbs as well as postaxial polydactyly in the forelimb [38]. EHH analysis in the Lundehund revealed PRRX2 in long-range homozygous haplotypes suggesting this gene as a candidate for Lundehund-polydactyly [17]. We assume that these genes under potential targeted selection could explain the specific phenotypic characteristic for a four-limb polydactyly in the Lundehund.
Conclusions
In conclusion, our study of whole-genome sequencing data of Lundehund dogs suggests LEPREL1:g.139212C>G as a potential causative mutation for LS as well as NOD1 as a potential precursor gene which might play a role in LS breed predisposition. In addition, we identified potential signatures of selection for characteristic breed specific traits whose targeted selection might have increased genetic risk factors for disease development.
The results of our analysis represent a significant step to identify the genetic background of Lundehund specific traits which still remain to be further investigated for proper understanding of the underlying complex genetic mechanisms.
Methods
Animals
Genomic DNA was obtained from blood samples of 36 Lundehund and 186 dogs of different dog breeds including Norwegian Buhund and Norrbottenspets using a standard saline precipitation method [39]. Lundehund specific phenotypes and affection-status were obtained on basis a questionnaire filled in by dog owners and veterinarians. Information about the pedigree, date of birth, date of euthanasia and signs of LS like diarrhea, vomiting and cachexia was recorded using this form. Further blood parameters were added from clinically examined dogs.
Association analysis
In total 25 Lundehund (17 LS-affected and 8 LS-unaffected) were genotyped on the canine Illumina high density bead chip (Illumina) according to manufacturer’s protocols. Quality of genotyping data was controlled using a minor allele frequency (MAF) >0.05 and a genotyping rate per SNP >0.90. Filtering resulted in 93,882 SNPs with a mean genotyping rate of 99 %. Genome-wide association analysis was run using TASSEL version 3.0 [40]. Generalized linear model (GLM) with sex, inbreeding coefficients and three PCA’s was run to test the model. Quantile-quantile (Q-Q) plots for observed versus expected -log10P-values were calculated to control for population stratification using SAS/Genetics.
Pedigree analysis
Pedigree analysis was performed using regressive logistic models and the procedure SEGREG of S.A.G.E. (Statistical Analysis for Genetic Epidemiology, Release 6.3: http://darwin.cwru.edu, 2012) to test the most likely mode of inheritance for LS. We tested the hypothesis for the μ model without any genetic component, monogenic inheritance, polygenic inheritance and mixed major gene inheritance with a polygenic component and an independently segregating major gene locus with two alleles.
Whole-genome sequencing
Whole genome sequencing of Lundehund samples was performed on the Illumina MiSeq (Illumina, San Diego, CA) in paired-end mode. Libraries were prepared for one DNA-pool of three LS-affected Lundehund and for one DNA-pool of three LS-unaffected Lundehund using the Illumina Nextera DNA Sample Prep Kit according to standard protocols. We performed four runs for each pool with v2 Reagent Kits (Illumina) on a single lane flow cell (2 × 250 bp reads). Fastq-files were quality controlled using fastqc 0.11.3 [41]. Mapping to the reference genome CanFam 3.1.78 was performed using BWA 0.7.12 [42] followed by conversion into binary format using SAMtools 1.2 [43] and PCR duplicate marking using Picard tools (http://broadinstitute.github.io/picard/, version 1.130). Furthermore, we locally realigned reads and performed quality score recalibration and SNP calling using GATK [44]. In addition to the Lundehund fastq files we added five whole-genome sequences of one Korean Jindo Dog (DRR001566), one Afghan Hound (SRR1061643), two German Shepherd dogs (SRR1130247 and SRR1124304) and one Border Collie (SRR654728) from Sequence Read Archive (NCBI) to analysis. Quality of the data was controlled filtering variants for a read depth >2 and <1000 and quality values >20 (qual). Variant effects were predicted using the genetic variant annotation and effect prediction toolbox SNPEff version 4.1 b (2015-02-13) [45].
ROH detection
ROHs were detected using a stringently quality-controlled autosomal dataset of 8,585,517 SNPs showing a minimum read depth of 3, a maximum read depth of 60, a minimum mean read depth of 12 and minor allele frequency 0.01 for all samples. Sliding windows of 500 SNPs were chosen for ROH analysis. Homozygous regions of >150 kb were detected as ROHs using PLINK, version 1.07 (http://pngu.mgh.harvard.edu/purcell/plink/). Based on the genome size covered with SNPs divided by the number of SNPs we estimated the minimum distance of SNPs as 0.3. A maximum of three SNPs with missing genotypes and three heterozygous SNPs were admitted in each window. ROHs were filtered for LS-specific and Lundehund specific ROH regions based on hom.summary data outputs from ROH analysis for each dog using SAS/Genetics, version 9.4 (Statistical Analysis System, Cary, NC). The homozygosity information of each SNP (1 = homozygosity, 0 = no homozygosity) was detected and compared to the other individuals in order to identify overlapping and private regions, the number of SNPs and length of the regions. These specific ROH regions were searched for variants predicted to have high or moderate effects (SNPEff).
Functional annotation
Chromosomal positions of ROH regions were merged with Ensembl gene predictions (ensGene) using Galaxy intersection tool (https://usegalaxy.org/) [46, 47]. The obtained gene lists were converted into human orthologous genes using g:Profiler [48, 49] and tested for statistical overrepresentation in biological processes (PANTHER Overrepresentation Test; release 20150430, version 10.0) [50].
Filtering analysis
Analysis of sequencing data was performed identifying variants with SNPEff-predicted high or moderate effects with a homozygous mutant genotype in the LS-affected pool, a heterozygous or homozygous wild-type genotype in LS-unaffected pool and a homozygous wild-type genotype in the other five dogs of different breeds using SAS/Genetics 9.4. Variants flagged with warnings about possible annotation accuracy problems were omitted from analysis. The candidate SNP in LEPREL1 and further 23 SNPs were investigated for functional effects using the Variant Effect Predictor [51] for SIFT [21] predictions and PolyPhen [22]. Regions of SNPs that were predicted to be deleterious (SIFT) and possibly or probably damaging (PolyPhen) had been further investigated for a high density of mutations around the candidate SNP that could reference to annotation problems.
Genotyping
Genotyping of the candidate SNPs LEPREL1:g.139212C>G, KRT3:g.2584C>T as well as the polydactyly-associated polymorphism in LMBR1 (DC-2) was performed using restriction fragment length polymorphisms according to standard protocols [52]. DC-2 primers were obtained from previous study [18]. In addition the missense variants CEP164:g.57380G>T and COL28A1:g.159951T>A were validated using Kompetitive Allele Specific PCR (KASP) genotyping assays (LGC, Middlesex, UK) [53]. After the KASP standard thermal cycling touchdown protocol was run on a thermocycler TProfessional 96 (Biometra, Göttingen, Germany) using an annealing temperature of 61 °C (Additional file 12), allelic discrimination was performed on the ABI7300 sequence detection system (Applied Biosystems, Waltham, Massachusetts, USA).
Sequencing of candidate genes
Sanger sequencing of the cDNA of MB21D2 and LEPREL1 was performed in one LS-affected Lundehund and one Great Dane as control dog. RNA was obtained from hair roots in stabilized RNALater reagent (Quiagen, Maryland, USA), transcribed into cDNA and used for PCR amplification according to a standardized protocol [54] (Additional file 13). Alignment and variant detection was performed using the analysis software Sequencher 4.8 (Genes Codes, Ann Arbor, MI, USA). Despite the use of Q-solution (Quiagen) as enhancer reagent and Gradient PCR for optimizing reaction conditions, the coding region of FGF12 could not be amplified from these hair root samples at all.
Abbreviations
CARD9, Caspase Recruitment Domain Family, Member 9; CCDC50, Coiled-Coil Domain Containing 50 ; cDNA, complementary DNA; CEP164, centrosomal protein 164 kDa; COL28A1, collagen, type XXVII, alpha 1; EHH, extended haplotype homozygosity; FGF12, fibroblast growth factor 12; GLM, generalized linear model; GREM2, gremlin 2, DAN family BMP antagonist; GWAS, genome-wide association analysis; KRT3, keratin 3; LEPREL1, leprecan-like 1; LMBR1, limb development membrane protein 1; LS, Lundehund syndrome; MB21D2, mab-21 domain containing 2; NOD1, Nucleotide-Binding Oligomerization Domain Containing 1; PLE, protein-losing enteropathy; PRRX1, paired related homeobox 1; ROH, runs of homozygosity; SHH, sonic hedgehog; SNV, single nucleotide variant
Acknowledgements
The authors thank all dog owners and veterinarians for their support and for their donation of data. We also thank J. Wrede for his help in data analysis and M. Drabert for his support in DNA isolation.
Funding
No funding was obtained for this study.
Availability of data and material
All fastq files analysis can be obtained by NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra), BioProject PRJNA309755 (SRX1546460, SRX1546461).
Authors’ contributions
JM, OD designed the study, carried out the experiments and data analysis, drafted and finalized the manuscript. SP performed data collection and sample preparation for bead chip analysis. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the dog owners to use their samples from the bio-bank for Lundehund research and in addition to use individual images from their dogs for further investigations and publication.
Ethics approval and consent to participate
All animal work has been conducted according to the national and international guidelines for animal welfare. EDTA-blood samples were derived from the bio-bank for diagnostic purposes and did therefore not require an animal welfare committee approval according to the German Animal Welfare Law.
Additional files
Clinical data of 21 Lundehund with signs of LS. Blood parameters of total protein (TP), albumin (ALB), globulin (GLOB), fructosamine (F), alkaline phosphatase (ALKP), alanine aminotransferase (ALT), vitamin B12 (B12) and folic acid (FA) are shown. (DOCX 18 kb)
Pedigree of LS-affected Lundehund. In total, samples of twenty-one LS-affected and fifteen LS-unaffected Lundehund were available for analysis. A common pedigree for all these dogs shows a close relationship in-between all Lundehund dogs. Whole genome sequencing was performed in three affected and three LS-unaffected dogs (numbers in boxes) whereas genotyping on the Illumina high density bead chip was done for seventeen LS-affected and eight LS-unaffected Lundehund (numbers S1-S25). (TIF 2248 kb)
Results of complex segregation analysis using regressive logistic models for LS in the Lundehund. (DOCX 12 kb)
Runs of homozygosity for LS-affected Lundehund in 500-SNP windows. The chromosomal position of ROH regions, number of SNPs in these regions (n), size in base pairs (size_bp), canine genes (gene) and human orthologues (human gene) are shown. (XLSX 319 kb)
Private runs of homozygosity for LS-affected Lundehund in 500-SNP windows. The chromosomal position of ROH regions, number of SNPs in these regions (n), size in base pairs (size_bp), canine genes (gene) and human orthologues (human gene) are shown. (XLSX 60 kb)
Private runs of homozygosity for LS-unaffected Lundehund in 500-SNP windows. The chromosomal position of ROH regions, number of SNPs in these regions (n), size in base pairs (size_bp), canine genes (gene) and human orthologues (human gene) are shown. (XLSX 65 kb)
Runs of homozygosity for Lundehund specific regions in 500-SNP windows. The chromosomal position of ROH regions, number of SNPs in these regions (n), size in base pairs (size_bp), canine genes (gene) and human orthologues (human gene) are shown. (XLSX 88 kb)
Genotyping results for DC-2 mutation. The intronic LMBR1 SNP which is known to be associated with polydactyly (Park, 2008) was genotyped for all Lundehund and further 13 different dog breeds. (DOCX 13 kb)
Functional annotations for runs of homozygosity (ROH) in the Lundehund. PANTHER gene list analysis shows the proportion of gene hits against total number of process hits for genes detected in Lundehund consensus as well as LS-specific and private ROHs for LS-affected and LS-unaffected dogs. Detected ROHs were investigated for genes and their involvement in specific biologic processes. (DOCX 16 kb)
Allele frequencies of candidate SNPs. In total 186 samples of 17 breeds were genotyped for the LS-candidate SNPs LEPREL1:g.139212C>G, KRT3:g.2584C>T, COL28A1:g.159951T>A and CEP164:g.57380G>T. In KRT3:g.2584C>T and CEP164:g.57380G>T the mutant allele could be detected in other breeds whereas LEPREL1:g.139212C>G and COL28A1:g.159951T>A could not be found in any other dog breed than the Lundehund. (DOCX 14 kb)
Mutations detected in sequencing analysis of MB21D2 and LEPREL1. None of the synonymous as well as 3 prime UTR mutations could be shown to be exclusively found in the Lundehund except LEPREL1:c.1849C>G. (DOCX 13 kb)
Primer sequences used for genotyping of candidate SNPs. The two variants located in KRT3 and LEPREL1 were genotyped by the use of restriction fragment length polymorphisms (RFLP). KASP-primers were used for genotyping CEP164 and COL28A1 variants. Primer pairs, amplicon size (AS) in base pairs (bp), annealing (AT), restriction enzyme and incubation temperature (IT) are shown. The polymorphism DC-2 in LMBR1 was detected using primer pairs previously published (Kropatsch, 2015). (DOCX 14 kb)
Sequences of primers used for the investigation of MB21D2 and LEPREL1 complementary DNA. The gene region, product sizes in base pairs (bp) and annealing temperatures are shown. (DOCX 13 kb)
Contributor Information
Julia Metzger, Email: julia.metzger@tiho-hannover.de.
Sophia Pfahler, Email: sophia.pfahler@web.de.
Ottmar Distl, Email: ottmar.distl@tiho-hannover.de.
References
- 1.Melis C, Borg ÅA, Espelien IS, Jensen H. Low neutral genetic variability in a specialist puffin hunter: the Norwegian Lundehund. Anim Genet. 2013;44(3):348–351. doi: 10.1111/age.12000. [DOI] [PubMed] [Google Scholar]
- 2.Berghoff N, Ruaux CG, Steiner JM, Williams DA. Gastroenteropathy in Norwegian Lundehunds. America. 2007;1:4–7. [PubMed] [Google Scholar]
- 3.FlesjÅ K, Yri T. Protein-losing enteropathy in the Lundehund. J Small Anim Pract. 1977;18(1):11–23. doi: 10.1111/j.1748-5827.1977.tb05819.x. [DOI] [PubMed] [Google Scholar]
- 4.Kolbjørnsen Ø, Press CM, Landsverk T. Gastropathies in the Lundehund. APMIS. 1994;102(7–12):647–661. doi: 10.1111/j.1699-0463.1994.tb05216.x. [DOI] [PubMed] [Google Scholar]
- 5.Littman MP, Dambach DM, Vaden SL, Giger U. Familial protein‐losing enteropathy and protein‐losing nephropathy in Soft Coated Wheaten Terriers: 222 cases (1983–1997) J Vet Intern Med. 2000;14(1):68–80. doi: 10.1892/0891-6640(2000)014<0068:fpleap>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 6.Littman M, Giger U. Familial protein-losing enteropathy (PLE) and/or protein-losing nephropathy (PLN) in Soft-coated Wheaten Terriers (SCWT) J Vet Intern Med. 1990;4:133. doi: 10.1892/0891-6640(2000)014<0068:fpleap>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 7.Dijkstra M, Kraus JS, Bosje JT, Den Hertog E. Protein-losing enteropathy in Rottweilers. Tijdschr Diergeneeskd. 2010;135(10):406–412. [PubMed] [Google Scholar]
- 8.Lecoindre P, Chevallier M, Guerret S. Protein-losing enteropathy of non neoplastic origin in the dog: a retrospective study of 34 cases. Schweiz Arch Tierheilkd. 2010;152(3):141–146. doi: 10.1024/0036-7281/a000033. [DOI] [PubMed] [Google Scholar]
- 9.Kimmel SE, Waddell LS, Michel KE. Hypomagnesemia and hypocalcemia associated with protein-losing enteropathy in Yorkshire terriers: five cases (1992–1998) J Am Vet Med Assoc. 2000;217(5):703–706. doi: 10.2460/javma.2000.217.703. [DOI] [PubMed] [Google Scholar]
- 10.MacLachlan N, Breitschwerdt E, Chambers J, Argenzio R, De Buysscher E. Gastroenteritis of Basenji dogs. Veterinary Pathology Online. 1988;25(1):36–41. doi: 10.1177/030098588802500105. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs G, Collins-Kelly L, Lappin M, Tyler D. Lymphocytic-plasmacytic enteritis in 24 dogs. J Vet Intern Med. 1990;4(2):45–53. doi: 10.1111/j.1939-1676.1990.tb00875.x. [DOI] [PubMed] [Google Scholar]
- 12.Kathrani A, Lee H, White C, Catchpole B, Murphy A, German A, Werling D, Allenspach K. Association between nucleotide oligomerisation domain two (Nod2) gene polymorphisms and canine inflammatory bowel disease. Vet Immunol Immunopathol. 2014;161(1):32–41. doi: 10.1016/j.vetimm.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cézard J-P, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 14.Vignes S, Bellanger J. Primary intestinal lymphangiectasia (Waldmann’s disease) Orphanet J Rare Dis. 2008;3(5):b1. doi: 10.1186/1750-1172-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169(1):345–350. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC, Monsuur AJ, Bevova M, Nijmeijer RM, van ’t Slot R, Heijmans R, Boezen HM, van Heel DA, van Bodegraven AA, Stokkers PC, Wijmenga C, Crusius JB, Weersma RK. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet. 2008;82(5):1202–1210. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pfahler S, Distl O. Effective population size, extended linkage disequilibrium and signatures of selection in the rare dog breed Lundehund. Plos One. 2015;10(4):e0122680. doi: 10.1371/journal.pone.0122680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kropatsch R, Melis C, Stronen AV, Jensen H, Epplen JT: Molecular Genetics of Sex Identification, Breed Ancestry and Polydactyly in the Norwegian Lundehund Breed. J Hered. 2015;106.4:403-406. [DOI] [PubMed]
- 19.Park K, Kang J, Subedi KP, Ha J-H, Park C. Canine polydactyl mutations with heterogeneous origin in the conserved intronic sequence of LMBR1. Genetics. 2008;179(4):2163–2172. doi: 10.1534/genetics.108.087114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfahler S, Distl O. A massive reduction of the genetic diversity in the Lundehund. Anim Genet. 2014;45(1):154. doi: 10.1111/age.12084. [DOI] [PubMed] [Google Scholar]
- 21.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metzger J, Karwath M, Tonda R, Beltran S, Agueda L, Gut M, Gut IG, Distl O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genomics. 2015;16(1):764. doi: 10.1186/s12864-015-1977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Järnum S, Kjellman C, Darabi A, Nilsson I, Edvardsen K, Åman P. LEPREL1, a novel ER and Golgi resident member of the Leprecan family. Biochem Biophys Res Commun. 2004;317(2):342–351. doi: 10.1016/j.bbrc.2004.03.060. [DOI] [PubMed] [Google Scholar]
- 25.Gross KJ, Pothoulakis C. Role of neuropeptides in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(7):918–932. doi: 10.1002/ibd.20129. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto H, Morise K, Kusugami K, Furusawa A, Konagaya T, Nishio Y, Kaneko H, Uchida K, Nagai H, Mitsuma T, Nagura H. Abnormal neuropeptide concentration in rectal mucosa of patients with inflammatory bowel disease. J Gastroenterol. 1996;31(4):525–532. doi: 10.1007/BF02355052. [DOI] [PubMed] [Google Scholar]
- 27.Vaden SL, Hammerberg B, Davenport DJ, Orton SM, Trogdon MM, Melgarejo LT, VanCamp SD, Williams DA. Food hypersensitivity reactions in Soft Coated Wheaten Terriers with protein-losing enteropathy or protein-losing nephropathy or both: gastroscopic food sensitivity testing, dietary provocation, and fecal immunoglobulin E. J Vet Intern Med. 2000;14(1):60–67. doi: 10.1111/j.1939-1676.2000.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 28.Franke A, Hampe J, Rosenstiel P, Becker C, Wagner F, Hasler R, Little RD, Huse K, Ruether A, Balschun T. Systematic association mapping identifies NELL1 as a novel IBD disease gene. PLoS One. 2007;2(1):e691. doi: 10.1371/journal.pone.0000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoll M, Corneliussen B, Costello CM, Waetzig GH, Mellgard B, Koch WA, Rosenstiel P, Albrecht M, Croucher PJ, Seegert D. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36(5):476–480. doi: 10.1038/ng1345. [DOI] [PubMed] [Google Scholar]
- 30.McGovern DP, Hysi P, Ahmad T, van Heel DA, Moffatt MF, Carey A, Cookson WO, Jewell DP. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet. 2005;14(10):1245–1250. doi: 10.1093/hmg/ddi135. [DOI] [PubMed] [Google Scholar]
- 31.Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, Cardon L, Takazoe M, Tanaka T, Ichimori T. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14(22):3499–3506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 32.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 33.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolbjornsen O, Press CM, Landsverk T. Gastropathies in the Lundehund. II. A study of mucin profiles. APMIS. 1994;102(11):801–809. doi: 10.1111/j.1699-0463.1994.tb05238.x. [DOI] [PubMed] [Google Scholar]
- 35.Ideno H, Takanabe R, Shimada A, Imaizumi K, Araki R, Abe M, Nifuji A. Protein related to DAN and cerberus (PRDC) inhibits osteoblastic differentiation and its suppression promotes osteogenesis in vitro. Exp Cell Res. 2009;315(3):474–484. doi: 10.1016/j.yexcr.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki D, Yamada A, Aizawa R, Funato S, Matsumoto T, Suzuki W, Takami M, Miyamoto Y, Suzawa T, Yamamoto M, Baba K, Kamijo R. BMP2 differentially regulates the expression of Gremlin1 and Gremlin2, the negative regulators of BMP function, during osteoblast differentiation. Calcif Tissue Int. 2012;91(1):88–96. doi: 10.1007/s00223-012-9614-5. [DOI] [PubMed] [Google Scholar]
- 37.Zeller R, Zuniga A. Shh and Gremlin1 chromosomal landscapes in development and disease. Curr Opin Genet Dev. 2007;17(5):428–434. doi: 10.1016/j.gde.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 38.ten Berge D, Brouwer A, Korving J, Martin JF, Meijlink F. Prx1 and Prx2 in skeletogenesis: roles in the craniofacial region, inner ear and limbs. Development. 1998;125(19):3831–3842. doi: 10.1242/dev.125.19.3831. [DOI] [PubMed] [Google Scholar]
- 39.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 41.Andrews S: FastQC: A quality control tool for high throughput sequence data. In: Reference Source. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; 2010.
- 42.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome project data processing s: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blankenberg D, Kuster GV, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;89:19. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g: Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35(suppl 2):W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reimand J, Arak T, Vilo J. g:Profiler--a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39(Web Server issue):W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(D1):D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Metzger J, Schrimpf R, Philipp U, Distl O. Expression Levels of LCORL Are Associated with Body Size in Horses. PLoS One. 2013;8(2):e56497. doi: 10.1371/journal.pone.0056497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He C, Holme J, Anthony J: SNP genotyping: the KASP assay. In: Crop Breeding. New York: Springer; 2014: 75–86. [DOI] [PubMed]
- 54.Metzger J, Wohlke A, Mischke R, Hoffmann A, Hewicker-Trautwein M, Kuch EM, Naim HY, Distl O. A Novel SLC27A4 splice acceptor site mutation in great Danes with ichthyosis. Plos One. 2015;10(10):e0141514. doi: 10.1371/journal.pone.0141514. [DOI] [PMC free article] [PubMed] [Google Scholar]