

Abstract
Metformin-induced activation of the 5′-AMP-activated protein kinase (AMPK) has been associated with enhanced glucose uptake in skeletal muscle, but so far no direct causality has been examined. We hypothesized that an effect of in vivo metformin treatment on glucose uptake in mouse skeletal muscles is dependent on AMPK signaling. Oral doses of metformin or saline treatment were given to muscle-specific kinase dead (KD) AMPKα2 mice and wild-type (WT) littermates either once or chronically for 2 wk. Soleus and extensor digitorum longus muscles were used for measurements of glucose transport and Western blot analyses. Chronic treatment with metformin enhanced insulin-stimulated glucose uptake in soleus muscles of WT (∼45%, P < 0.01) but not of AMPK KD mice. Insulin signaling at the level of Akt protein expression or Thr308 and Ser473 phosphorylation was not changed by metformin treatment. Insulin signaling at the level of Akt and TBC1D4 protein expression as well as Akt Thr308/Ser473 and TBC1D4 Thr642/Ser711 phosphorylation were not changed by metformin treatment. Also, protein expressions of Rab4, GLUT4, and hexokinase II were unaltered after treatment. The acute metformin treatment did not affect glucose uptake in muscle of either of the genotypes. In conclusion, we provide novel evidence for a role of AMPK in potentiating the effect of insulin on glucose uptake in soleus muscle in response to chronic metformin treatment.
Keywords: metformin, AMPK, skeletal muscle, glucose uptake, insulin
metformin is worldwide the most prescribed drug used to treat insulin resistance in type 2 diabetic patients and is also utilized in other patient groups with decreased insulin sensitivity. Most attention on the beneficial effects of metformin in regard to amelioration of insulin sensitivity has been in the liver. Both acute and chronic metformin treatments reduce hepatic glucose production by reducing gluconeogenesis and glucose output (73). Several in vitro (9, 30, 60, 77) and in vivo (6, 42, 58) studies in rodents have documented that metformin both acutely (77) and chronically (6, 10, 42) are also able to stimulate peripheral glucose disposal by increasing glucose uptake in skeletal muscle. The clinical relevance of these latter findings are supported by human clamp studies showing increased peripheral glucose disposal after metformin treatment (45, 52, 57), although this is not a totally consistent finding (15).
The molecular mechanisms underlying the effects of metformin are still not fully described, but inhibition of complex I in the mitochondria respiration chain has been reported (19, 54). Metformin treatment also induces activation of the 5′-AMP-activated protein kinase (AMPK) in muscle cells (9, 23, 40, 60), intact rodent muscles (60, 66, 77), and human diabetic skeletal muscles (45, 52). AMPK is activated under conditions of cellular stress, e.g., exercise, muscle contractions, ischemia, and hypoxia (31, 46, 75), where it catalyzes changes in the cellular metabolism to reduce ATP consumption as well as raising ATP production by an inhibitory effect on the anabolic and a stimulatory effect on the catabolic processes in the cell (69). Hence, AMPK activation by metformin has been suggested to be linked to complex I inhibition, which may lead to disturbance of the cell energy balance and thus AMPK activation (29).
Genetic evidence supports a role for AMPK in processes involved in glucose uptake and GLUT4 regulation in relation to acute and chronic treatment with the pharmacological activator AICAR (33, 34). Although metformin treatment has been coupled to both AMPK activation and upregulation of glucose uptake in skeletal muscle, only a few studies have measured both AMPK activation and glucose uptake (42, 45, 52, 60, 77). In fact, to our knowledge, no studies have investigated any causality between metformin, AMPK activation, and glucose uptake in skeletal muscle.
We hypothesized that potential effects of metformin treatment in vivo on glucose uptake in skeletal muscles would be dependent on an intact AMPK signaling system. Therefore, we investigated the role of AMPK in the acute and chronic effects of metformin to stimulate glucose transport in skeletal muscle by using muscle-specific kinase dead (KD) AMPKα2 mice (50).
METHODS
Animals
Experiments were approved by the Danish Animal Experiments Inspectorate and complied with the European convention for protection of vertebrate animals used for scientific purposes. The mice used in these experiments were muscle-specific kinase dead (KD) AMPKα2 mice, described previously (50). Female mice 22–30-wk old on a C57BL/6J background were studied and wild-type (WT) littermates were used as controls.
Treatment Models
All animals were kept on a 10:14-h light-dark cycle with unlimited access to standard rodent chow food and water.
Acute metformin time course experiment.
WT mice were given one dose of metformin (150 mg/kg 1,1-dimethylbiguanide HCl; Sigma-Aldrich, Germany) or saline by gavage. Muscles were removed from anesthetized animals 2, 4, 6, 8, and 10 h after treatment and immediately frozen in liquid N2.
Acute (6-h) metformin treatment experiment.
WT and KD mice were divided into two groups each, one treated with metformin and the other given saline. Metformin (150 mg/kg) and saline solutions were administered once by gavage 6 h before dissection and isolation of soleus and EDL muscles for incubations ex vivo (see Muscle Incubation Procedure).
Chronic metformin treatment experiment.
WT and KD mice were divided into two groups each, one treated with metformin and the other given saline. Metformin (150 mg/kg) and saline solutions were administered by gavage twice daily in the morning (around 8–10 AM) and afternoon (around 4–6 PM) for 2 wk. The last dose of metformin/saline was given the afternoon before the experimental day.
Acute (16-h) metformin control experiment.
A control experiment was conducted to differentiate the outcome in the chronic metformin treatment from possible effects related to the last dose of metformin administered the day before. WT mice were treated once with metformin (150 mg/kg) or saline by gavage the afternoon before the experimental day, i.e., around 16–18 h before isolation of muscles.
Muscle Incubation Procedure
Fed mice were anesthetized by intraperitoneal injection of pentobarbital sodium (0.1 mg/g body wt). Soleus and extensor digitorum longus (EDL) muscles were removed from the animal and suspended at resting tension in incubation chambers (Multi Myograph System; Danish Myo-Technology) with basal buffer (Krebs-Henseleit buffer, pH 7.4: 117 mM NaCl, 4.7 mM KCl, 2.5 mM Ca2Cl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 24.6 mM NaHCO3, 5 mM HEPES, 0.1% BSA, 8 mM mannitol and 2 mM sodium pyruvate) at 30°C oxygenated with 95% O2 and 5% CO2 gas. For maximal insulin stimulation, insulin (10,000 μU/ml Actrapid; Novo Nordisk, Denmark) was added to the basal buffer. Basal and insulin-stimulated conditions were paired muscles from each leg of the same mice. All muscles were incubated for 40 min. For the assessment of glucose uptake, radioactively labeled tracers were added to the medium during the last 10 min of incubation (see next section). Following the 40-min incubation muscles were harvested and washed in ice-cold buffer, dried on filter paper and quickly frozen in liquid nitrogen. Muscles used for Western blot analyzes (see next section) were treated in the same way but without measuring glucose uptake during the last 10-min incubation. Muscles were stored at −80°C.
Muscle Preparation Used for Glucose Uptake
Glucose uptake was measured as accumulation of 2-deoxy[3H]glucose (1 mM; Perkin Elmer). [14C]mannitol (8 mM) (PerkinElmer) was used as a marker of the extracellular space. For measurement of the accumulated 2-deoxy[3H]glucose and [14C]mannitol, muscles were heated and shaken at 80°C for 10 min in 1 M NaOH, neutralized with 1 M HCl, and centrifuged for 2 min at 13,000 g. Subsequently, radioactivity in the supernatants was measured by liquid scintillation counting (Ultima gold LSC-cocktail and Liquid Scintillation Analyzer Tri-Carb 2910 TR, PerkinElmer) and related to the specific activity of the incubation buffer.
Muscle Lysate Preparation Used for Western Blot Analyzes
Whole muscles were homogenized in ice-cold buffer (10% glycerol, 20 mM sodium pyrophosphate, 150 mM NaCl, 50 mM HEPES, 1% NP-40, 20 mM β-glycerophosphate, 10 mM NaF, 2 mM PMSF, 1 mM EDTA, 1 mM EGTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 2 mM Na3VO4, 3 mM benzamidine, pH 7.5) by a tissue lyser for 2 × 1 min at 30 Hz (Tissue Lyser II; Qiagen Retch, Germany). Muscle homogenates were rotated end over end at 4°C for 1 h, after which they were centrifuged for 20 min at 15,000 g. The supernatants were harvested as the muscle lysate and stored at −80°C. Total protein content in supernatants was determined by the bicinchoninic acid method (Pierce Biotechnology).
SDS-Page and Western Blot Analyses
Lysate proteins were separated by SDS-PAGE on self-cast Tris·HCl (7–12%) PAGE gels and by semidry blotting transferred to a PVDF membrane (Immobilon Transfer Membranes; Millipore, Denmark). The membrane was blocked in a washing buffer (10 mM Tris base, 150 mM NaCl, and 0.25% Tween 20, pH 7.4) containing low-fat milk protein (2–5%) or BSA (3%) solution and afterward probed with primary antibodies and appropriate secondary antibodies (see next section). Protein bands were visualized using a Kodak Image Station (2000 MM; Kodak, Rochester, NY) after probing with enhanced chemiluminescence (ECL, Millipore). Bands were quantified using Kodak 1D 3.6 software (Kodak). Membranes used for detecting phosphorylations of AMPK pThr172, Akt pSer473 and TBC1D1/D4 pThr642 were stripped (buffer containing 100 mM 2-mercaptoethanol, 2% SDS, and 62.5 mM Tris·HCl). After checking for successful removal of the primary antibody, the membrane were reprobed with the corresponding total protein antibody. The phosphorylation results are not related to the total protein blots.
Antibodies Used for Detection of Specific Phosphorylation Sites and Protein Expressions
Antibodies used were as follows: acetyl-CoA carboxylase (ACC2) phosphorylation at Ser221: anti-phospho-ACC2 (Ser79; Upstate Biotechnology, #07-303); Akt2 protein expression: anti-Akt2 (D6G4) antibody (Cell Signaling Technology #3063); Akt protein phosphorylation at Thr308: anti-phospho-Akt Thr308 antibody (Cell Signaling Technology #9275); Akt protein phosphorylation at Ser473: anti-phospho-Akt Ser473 antibody (Cell Signaling Technology #9271); AMPKα2 protein expression: anti-AMPKα2-specific antibody raised in sheep as previously described (76), kindly donated by D. G. Hardie, University of Dundee, Scotland, UK; AMPKα subunit phosphorylation at Thr172: anti-phospho-AMPKα antibody Thr172 antibody, Cell Signaling Technology #2531; GLUT4 protein expression: anti-GLUT4 antibody (Pierce Biotechnology #PA1-3031065); hexokinase (HK)II protein expression: anti-hexokinase II (HXK2/HK2) antibody (Alpha Diagnostics #HXK23-A); Rab4 protein expression: anti-Rab4 antibody (Cell Signaling Technology #2167); TBC1 D1/D4 (AS160) protein expression: anti-AS160 protein (Rab-GAP) antibody (Millipore #07-741); TBC1 D1/D4 (AS160) phosphorylation at Thr642: anti-AS160 phospho-Thr642 antibody (Symansis, NZ #3028 P1); and TBC1 D1/D4 (AS160) phosphorylation at Ser711 (71). Secondary HRP-conjugated antibodies used were from Dako (Glostrup, Denmark).
Statistics
To enable comparison of both genotype and metformin treatment effect on the insulin stimulation, three-way repeated-measures ANOVA was performed in SPSS, without post hoc testing. Two-way ANOVA in SigmaStat 3.5 was used for testing genotype and metformin effect on insulin -stimulated increments when interaction was obtained with the three-way repeated-measures ANOVA. Differences between groups were considered statistically significant when P < 0.05. All data are expressed as means ± SE. Specific differences were localized using Student-Newman-Keuls post hoc test.
RESULTS
One Oral Dose of Metformin Treatment Activates AMPK in Mouse Skeletal Muscles
An acute time course experiment was conducted to deduce the time point at which AMPK activation is most pronounced after acute metformin treatment. One dose of metformin (150 mg/kg) or saline (control) was administered by oral gavage to WT mice. The metformin treatment induced a significant AMPK activation 2–10 h (main effect) after the metformin treatment in soleus muscles (Figs. 1, A and B, and 9A) and a borderline (P = 0.069; power = 0.32) significant activation in EDL muscles (Figs. 1, C and D, and 9A) evaluated by AMPK phosphorylation and ACC phosphorylation. The power of the statistics test is not strong enough to select a given time point (with n = 6 in each group) at which AMPK and ACC phosphorylation is significant increased. Consequently, the statistics test does not present a given time point at which p-AMPK and p-ACC were maximally activated after metformin treatment. Nevertheless, based on the seemingly greatest difference between saline and metformin on p-AMPK and p-ACC at 6 h, we chose the 6-h time point for the acute study as described below.
Fig. 1.
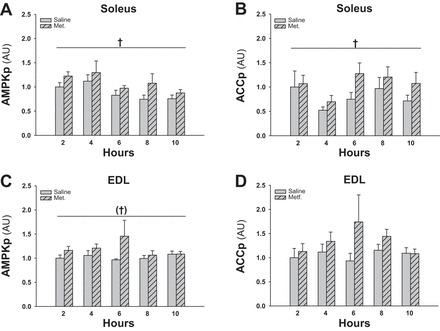
Effect of acute (2–10 h before isolation of muscles) metformin treatment on AMPK activation in soleus and EDL muscles from WT mice. Gray bars without and with hatches show results from saline- and metformin-treated mice, respectively. A: phosphorylated AMPK (AMPKp) in soleus muscles 2–10 h after a single dose metformin/saline. B: phosphorylated acetyl-CoA carboxylase (ACCp) in soleus muscles 2–10 h after a single dose metformin/saline. C: AMPKp in EDL muscles 2–10 h after a single dose metformin/saline. D: ACCp in EDL muscles 2–10 h after single dose metformin/saline. †Significant difference between saline and metformin treatment (main effect, P < 0.05); †P = 0.069; n = 6–8. Values are means ± SE.
Fig. 9.
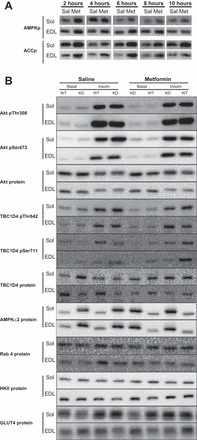
A: representative Western blots of AMPKp and ACCp in soleus and EDL muscles after acute (2–10 h before isolation of muscles) metformin treatment. B: representative Western blots of Aktp Thr308, Ser473, and Akt protein; AS160p Thr642, Ser711, and TBC1D4 protein; AMPKα2 protein, Rab4 protein, HKII protein, and GLUT4 protein in soleus and EDL muscles incubated without or with insulin after chronic saline or metformin treatment.
Acute (6-h) Metformin Treatment Does Not Potentiate Basal or Insulin-Stimulated Glucose Uptake in Soleus and EDL Muscles
To test whether a potential acute effect of in vivo metformin treatment on glucose uptake is dependent on AMPK signaling, WT and AMPK KD littermate mice were treated once orally with metformin or saline. Six hours after treatment, soleus and EDL muscles were isolated for the measurement of ex vivo glucose uptake. Neither basal nor maximal insulin-stimulated glucose uptake in soleus (Fig. 2, A and B) and EDL (Fig. 2C) were affected by metformin treatment. Soleus muscles of the AMPK KD mice had a significantly lower insulin-stimulated increments in glucose uptake compared with WT soleus muscles (Fig. 2B, P < 0.01) independently of the metformin treatment.
Fig. 2.
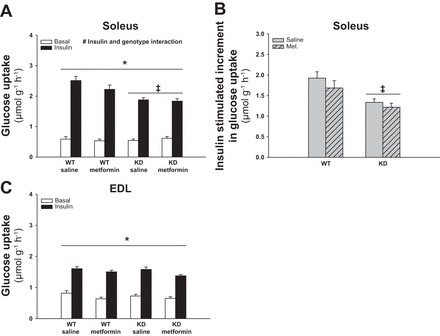
Effect of acute (6-h) metformin treatment on glucose uptake in soleus and EDL muscles from WT and AMPK kinase dead (KD) mice. Filled and open bars show results from muscles incubated with or without insulin (10,000 μU/ml), respectively (A and C). Gray bars without and with hatches show results from saline and metformin treated mice respectively (B). A: basal and insulin-stimulated glucose uptake in soleus muscles from metformin- or saline-treated WT and AMPK KD mice. B: Δ values (insulin-basal) of insulin-stimulated glucose uptake in soleus muscles from metformin- or saline-treated WT and AMPK KD mice. C: basal and insulin-stimulated glucose uptake in EDL muscles from metformin- or saline-treated WT and AMPK KD mice. *Significant difference between basal and insulin-stimulated glucose uptake (main effect, P < 0.01); ‡significant difference between genotypes (main effect, P < 0.01); #significant interaction between insulin and genotype (P < 0.01); n = 12. Values are means ± SE.
Chronic Metformin Treatment Induces AMPK-Dependent Enhancement of Insulin-Stimulated Glucose Uptake in Soleus Muscles
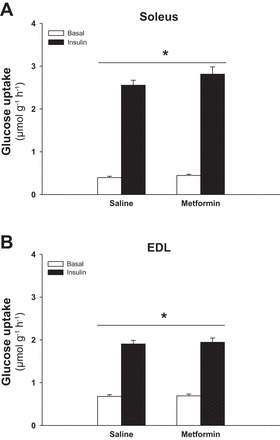
To investigate the role of AMPK in relation to potential chronic effects of metformin treatment on glucose uptake, WT and AMPK KD littermate mice were treated with metformin for 2 wk. The last dose of metformin was administered in the afternoon of the day before soleus and EDL muscles were isolated for measurement of glucose uptake. Maximal insulin-stimulated increments in glucose uptake were enhanced by ∼45% in soleus of metformin-treated mice compared with saline-treated WT mice (Fig. 3, A and B, P < 0.01) and by ∼60% compared with metformin-treated AMPK KD mice (Fig. 3B, P < 0.01). Basal glucose uptake was not affected by metformin treatment in soleus (Fig. 3A). In EDL muscle there was no effect of metformin treatment or genotype on either basal or insulin-stimulated glucose uptake (Fig. 3C). We performed an acute (16-h) control experiment to differentiate outcomes in the chronic metformin experiment from possible effects related to the last dose of metformin administered the afternoon before. As shown in Fig. 4, A and B, the last dose of metformin did not significantly enhance basal or insulin-stimulated glucose uptake in either soleus or EDL, although there was a tendency toward a main effect of metformin in soleus (P = 0.089; power = 0.29).
Fig. 3.
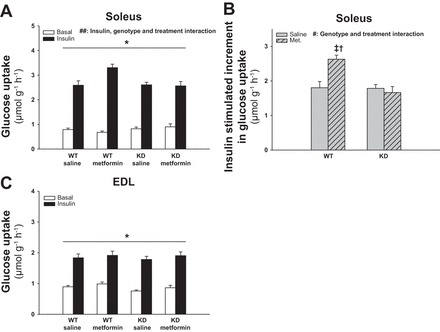
Effect of chronic metformin treatment on glucose uptake in soleus and EDL muscles from WT and AMPK KD mice. Filled and open bars represent results from muscles incubated with or without insulin (10,000 μU/ml), respectively (A and C). Gray bars without and with hatches show results from saline- and metformin-treated mice respectively (B). A: basal and insulin-stimulated glucose uptake in soleus muscles from metformin- or saline-treated WT and AMPK KD mice. B: Δ values (insulin-basal) of insulin-stimulated glucose uptake in soleus muscles from metformin- or saline-treated WT and AMPK KD mice. C: basal and insulin-stimulated glucose uptake in EDL muscles from metformin- or saline-treated WT and AMPK KD mice. *Significant difference between basal and insulin-stimulated glucose uptake (main effect, P < 0.01); †significant difference on insulin-stimulated glucose uptake between interventions in WT mice (P < 0.01); ‡significant difference between genotypes treated with metformin (P < 0.01); #significant interaction between genotype and treatment (P < 0.01); ##significant interaction between insulin, genotype, and treatment (P < 0.01); n = 12–13. Values are means ± SE.
Fig. 4.
Effect of acute metformin treatment (∼16 h before isolation of muscles) on glucose uptake in soleus and EDL muscles from WT mice. Filled and open bars represent results from muscles incubated with or without insulin (10,000 μU/ml) respectively. A: basal and insulin-stimulated glucose uptake in soleus muscles from metformin- or saline-treated WT mice. B: basal and insulin-stimulated glucose uptake in EDL muscles from metformin- or saline-treated WT mice. *Significant difference between basal and insulin-stimulated glucose uptake (main effect, P < 0.01); n = 12. Values are means ± SE.
AMPK and Insulin Signaling Marker Proteins Are Not Affected by Chronic Metformin Treatment
To explore further the metformin-induced AMPK-dependent enhancement of insulin-stimulated glucose uptake, we investigated AMPK activation and proteins related to the insulin signaling cascade and glucose uptake. The chronic treatment did not affect AMPKα2 protein expression (data not shown) and based on measurement of p-AMPK Thr172 and p-ACC Ser212 at the time when muscles where isolated, about 16 h after the last metformin dose, AMPK was no longer activated (data not shown).
We then investigated insulin signaling at the level of Akt Thr308 and Ser473 phosphorylation as indicators of Akt activation. Both sites have increased phosphorylation upon insulin stimulation but were not influenced by genotype or metformin treatment (Figs. 5, A–D, and 9B). Akt protein expression was not regulated by genotype or treatment (data not shown).
Fig. 5.
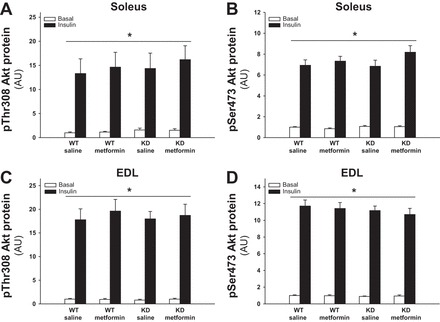
Effect of chronic metformin treatment on Akt Thr308 and Ser473 phosphorylation in soleus and EDL muscles from WT and AMPK KD mice. Filled and open bars represent results from muscles incubated with or without insulin (10,000 μU/ml) respectively. A: Akt Thr308 phosphorylation in basal and insulin-stimulated soleus muscles from metformin- or saline-treated WT and AMPK KD mice. B: Akt Ser473 phosphorylation in basal and insulin-stimulated soleus muscles from metformin- or saline-treated WT and AMPK KD mice. C: Akt Thr308 phosphorylation in basal and insulin-stimulated EDL muscles from metformin- or saline-treated WT and AMPK KD mice. D: Akt Ser473 phosphorylation in basal and insulin-stimulated EDL muscles from metformin- or saline-treated WT and AMPK KD mice. *Significant difference between basal and insulin-stimulated Akt phosphorylation (main effect, P < 0.01); n = 10. Values are means ± SE.
Downstream of Akt, the AS160 (TBC1D4) protein is a potential convergence point for insulin- and AMPK-regulated glucose uptake. TBC1D4 Thr642 and Ser711 phosphorylations were both up regulated by insulin (Figs. 6, A–D, and 9B). Phosphorylation of Ser711 was also dependent on AMPK, where the insulin-stimulated response was significantly decreased in KD compared with WT muscles (Fig. 6, B, D, and E, P < 0.01). Metformin treatment did not influence basal or insulin-stimulated phosphorylation status of TBC1D4. TBC1D4 protein expression in soleus and EDL was not affected by genotype or metformin treatment (data not shown).
Fig. 6.
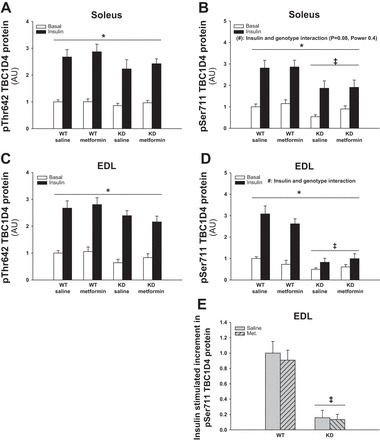
Effect of chronic metformin treatment on TBC1D4 Thr642 and Ser711 phosphorylations in soleus and EDL muscles from WT and AMPK KD mice. Filled and open bars represent results from muscles incubated with or without insulin (10,000 μU/ml), respectively (A–D). Gray bars without and with hatches show results from saline- and metformin-treated mice, respectively (E). A: Thr642 phosphorylation in basal and insulin-stimulated soleus muscles from metformin- or saline-treated WT and AMPK KD mice. B: Ser711 phosphorylation in basal and insulin-stimulated soleus muscles from metformin- or saline-treated WT and AMPK KD mice. C: Thr642 phosphorylation in basal and insulin-stimulated EDL muscles from metformin- or saline-treated WT and AMPK KD mice. D: Ser711 phosphorylation in basal and insulin-stimulated EDL muscles from metformin- or saline-treated WT and AMPK KD mice. E: Δ values (insulin-basal) of insulin-stimulated Ser711 phosphorylation in EDL muscles from metformin- or saline-treated WT mice. *Significant difference between basal and insulin-stimulated TBC1D4 phosphorylation (main effect, P < 0.01); ‡significant difference between genotypes (main effect, P < 0.01); #interaction between insulin and genotype (P < 0.01); (#)tendency for an interaction between insulin and genotype (P = 0.08, power = 0.4); n = 10. Values are means ± SE.
Metformin has recently been associated with AMPK-TBC1D4-dependent GLUT4 translocation via the Rab4 protein (40). We investigated the Rab4 protein expression, but as shown in Figs. 7, A and B, and 9B, the protein is not downregulated in AMPK KD mice, and the chronic metformin treatment did not induce upregulation of the Rab4 protein expression.
Fig. 7.
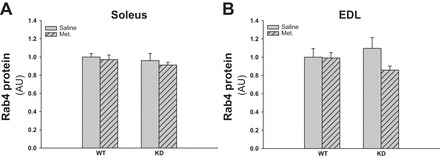
Effect of chronic metformin treatment on Rab4 protein expression in soleus and EDL muscles from WT and AMPK KD mice. Gray bars without and with hatches show results from saline- and metformin-treated mice, respectively. A: Rab4 protein expression in soleus muscles of metformin- or saline-treated WT and AMPK KD mice. B: Rab4 protein expression in EDL muscles of metformin- or saline-treated WT and AMPK KD mice; n = 10. Values are means ± SE.
GLUT4 and HKII Protein Expressions Are Not Regulated by Chronic Metformin Treatment
As shown in Figs. 8, A and C, and 9B, GLUT4 protein expression was not significantly affected in the AMPK KD mice, and metformin did not induce changes in the expression. The HKII protein was downregulated in soleus muscles of the AMPK KD mice, but metformin treatment did not affect the protein expression (Fig. 8, B and D and 9B).
Fig. 8.
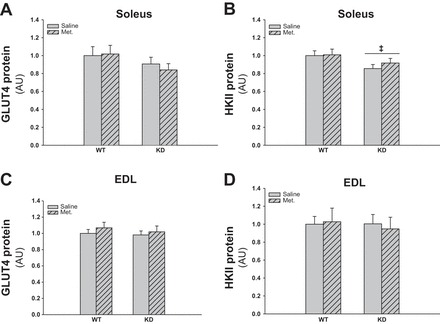
Effect of chronic metformin treatment on GLUT4 and hexokinase (HK)II protein expression in soleus and EDL muscles from WT and AMPK KD mice. Gray bars without and with hatches show results from saline- and metformin-treated mice, respectively. A: GLUT4 protein expression in soleus muscle of metformin- or saline-treated WT and AMPK KD mice. B: HKII protein expression in soleus muscle of metformin- or saline-treated WT and AMPK KD mice. C: GLUT4 protein expression in EDL muscle of metformin- or saline-treated WT and AMPK KD mice. D: HKII protein expression in EDL muscle of metformin- or saline-treated WT and AMPK KD mice. ‡Significant difference between genotypes (main effect, P < 0.05); n = 10. Values are means ± SE.
DISCUSSION
The main finding in the present study was an increased insulin response in soleus muscles after chronic metformin treatment of WT mice that is absent in AMPKα2 KD mice. Although it is impossible to exclude other secondary effects of the AMPK KD construct, our observation strongly indicates that the enhanced insulin-stimulated glucose uptake in skeletal muscles observed after oral metformin treatment for a longer period of time indeed is dependent on AMPK. To our knowledge, it is the first time that such a causal relationship to AMPK has been investigated and reported.
Increased insulin-stimulated glucose uptake in soleus muscle after chronic treatment has been shown earlier, but these studies have primarily been done in insulin-resistant animals (6, 10, 42), and one study failed to detect an effect in soleus (59). Basal glucose uptake was not affected by metformin in the present study. This is in accord with findings in other in vivo rodent studies (6, 10), is supported by clinical observations (52), and is in line with the general view that the effect of metformin is dependent on the presence of insulin (73).
The majority of studies have examined soleus in regard to metformin and glucose uptake, but a single study also used other muscles and observed an enhancing effect on glucose uptake in rat epitrochlearis and extensor carpi muscles (10). Based on this, it is difficult to deduce whether fiber type distribution plays a role for the lack of a metformin effect in EDL in the present study, since EDL and epitrochlearis both contain primarily glycolytic fibers, whereas soleus is dominated by oxidative fibers (1, 3). However, a species-specific difference could also influence the results, as is the case in regard to the effect of chronic AICAR treatment on glucose uptake in rats vs. mice (12, 62), where AICAR enhances insulin-stimulated glucose uptake in EDL muscles of rats, whereas an effect on insulin-stimulated glucose uptake has been shown to be absent in both muscles in mice (62). Clearly, we do not have an explanation for the different results in soleus and EDL muscles in the present study. However, we speculate that it could be related to the different AMPK trimer complex composition in the two muscles (70). It might be that some complexes are more prone to be activated by metformin than other complexes, as shown for another AMPK activating compound (70). Also, metformin uptake and accumulation (74) might differ between the muscles due to different capillarization (17) and possible different membrane metformin transporter content and/or composition (37).
We believe that the metformin effect observed in the soleus muscle after 2 wk of metformin treatment might have physiological relevance. The glycolytic EDL muscle is more representative for the mouse muscles (4) than soleus. Based on this, the physiological significance of our observations might be questioned. However, it is the more oxidative soleus and not the highly glycolytic EDL muscle in mice that in regard to muscle fiber type distribution is most like human skeletal muscle (4, 32). Consequently, the soleus muscle, more than the EDL, may reflect regulation in human skeletal muscle. In that respect, the physiological importance might be acknowledged given that several human studies have demonstrated enhanced insulin-stimulated glucose disposal by use of hyperinsulinemic euglycemic clamp after chronic metformin treatment in T2D patients (45, 52, 57). Although adipose tissue (21) and the intestine (7) may play a role on the effect of metformin on glucose metabolism during hyperinsulinemic euglycemic clamp conditions, skeletal muscle represents the major site of insulin-stimulated glucose uptake (18, 20). Therefore, the effect of metformin on the muscle tissue is very likely to be of greatest importance during the clamp (64, 73). In T2D patients, the impact on glucose disposal has been reported to be between ∼15 and 40% (64), which is comparable with our observations on metformin-induced enhancement of insulin stimulated glucose uptake in the soleus muscle. Although we are aware that our reflections regarding application of our observations in mouse muscle to humans are only speculative, we believe our finding may shed further light into the molecular mechanism by which metformin regulates glucose uptake in human skeletal muscle. In that respect, we think these speculations are supported by the fact that AMPK activation has been shown after chronic metformin treatment in T2D patients (45, 52).
The metformin dose used in the present study and in many other in vivo rodent studies evaluating glucose uptake is about 200–300 mg·kg−1·day−1 (6, 10). The recommended dose used in the clinic to treat T2D patients is 2–3 g/day, corresponding to 30–40 mg·kg−1·day−1 for a 70-kg person (14, 25). However, comparable plasma levels of metformin in humans and mice with the denoted treatment models have been reported (6, 14).
It is a consistent finding that metformin does not influence β-cell secretion of insulin or the insulin receptor binding in skeletal muscle (43, 58), whereas one study reported increased receptor density in soleus after chronic metformin treatment (6). The effect of chronic metformin treatment on the proximal steps of the intracellular insulin signaling is equivocal, since one study showed enhancement of the insulin receptor tyrosine kinase activity (58) whereas other studies reported no effect on PI 3-kinase activity and Akt activity (35, 45) and unaltered IRS-1 and Akt expression (35). In the present study, we observed an insulin effect on Akt phosphorylation but did not detect any changes with metformin treatment. Downstream of Akt, TBC1D4 is thought to be involved in GLUT4 vesicle recruitment to the plasma membrane via regulation of Rab proteins (47, 65). We found an AMPK-dependent enhancement of insulin-stimulated glucose uptake after chronic metformin treatment. Therefore, we measured Thr642 and Ser711 phosphorylation sites as representing Akt and AMPK targets, respectively. Although Ser711 is also shown to be regulated by insulin, it is in an Akt-independent manner (38, 71). We did not detect an effect of the chronic metformin treatment on phosphorylation of these sites. A recent study by Lee et al. (40) reported enhanced AS160 phosphorylation at site Thr642 in an AMPK-dependent manner upon acute metformin treatment of mouse C2C12 cells. However, due to very high metformin concentrations (10 mM) and a long incubation period (6–12 h), it is difficult to compare such in vitro studies with in vivo treatment. Nevertheless, the study by Lee et al. also showed that metformin increases Rab4 expression in these cells via AMPK-TBC1D4 signaling (40). A role for Rab4 in insulin-stimulated GLUT4 vesicle translocation has also been suggested in rat skeletal muscle (2, 61), but, as is evident from our results, we did not see an AMPK dependence or metformin effect on the Rab4 protein content in mouse soleus muscles. The absence of a metformin effect on GLUT4 protein expression in the present study is in accord with the majority of chronic in vivo studies (28, 59, 66, 68), besides studies done with STZ (T1D) and obese fat-fed rats where reduced GLUT4 expression was partially rescued after metformin treatment (13, 42). The glucose phosphorylation step and thus hexokinase can be rate limiting for glucose uptake (24), and metformin treatment has been shown to upregulate hexokinase activity/expression (6, 16, 66); thus, a role for hexokinase in metformin-enhanced glucose uptake is possible. The reason for the discrepancy between the studies by Bailey et al. (6) and Da et al. (16) and our study, showing no metformin effect on hexokinase expression, could be due to the T1D model used in those two studies showing metformin-induced rescue of a depressed expression (6, 16). On the other hand, Suwa et al. (66) examined the consequences of metformin emplying normal rats and actually did not detect a change in soleus and only a moderate increase in white gastrocnemious.
It has been suggested that reduced mitochondrial content as well as protein expressions and activities observed in diabetic muscles may contribute to skeletal muscle insulin resistance (44, 48). However, this is a debatable topic (67), and other studies have not confirmed such a relationship (11, 27). We recently showed that 2-wk metformin treatment did not regulate mitochondrial respiration in tibialis anterior muscles from WT mice but in AMPK KD mice only (39). In that respect, our observations of an AMPK-dependent enhancement of insulin-stimulated glucose uptake after 2-wk metformin treatment could not be explained by regulation of mitochondria function, although caution should be taken, as mitochondria function and glucose transport were measured in different muscles.
Alternatively, we speculate that the lack of changes in protein activation or expression related to insulin signaling at the level of Akt, TBC1D4, Rab4, GLUT4, or HKII in the present study may suggest that AMPK and metformin affect steps further distal related to fusion or docking of GLUT4 (36), which might be affected by membrane fluidity and cholesterol content. The latter is supported by cell studies indicating that both AMPK (26) and metformin (51, 72) play a role in changes in membrane composition that may lead to enhanced insulin action and GLUT4 translocation.
To our knowledge, we are the first to describe the acute effect of oral in vivo metformin treatment on skeletal muscle glucose uptake in rodents. The absence of an effect with acute metformin treatment in the present study is in line with Bailey et al. (5), showing no effect on glucose uptake in soleus measured with an intravenous glucose tolerance test after acute intravenous administration of metformin (250 mg/kg) in rats, and a clinical study by Perriello et al. (56) reporting only an effect on glucose production but not on glucose uptake measured with a hyperinsulemic euglycemic clamp after acute metformin treatment of T2D patients. However, these results are in contrast to recent observations by Sajan et al. (60), showing markedly increased basal glucose uptake in vastus lateralis already 30 min after metformin injection in mice. The dissimilarity could be due to a combination of possible muscle type differences and a higher metformin dose used in the latter study (250 mg/kg), probably resulting in higher peak plasma metformin concentration due to injection instead of oral treatment (25, 55). An unexpected finding in our acute study was decreased insulin-stimulated glucose uptake in soleus muscle from AMPK KD compared with WT mice. We have not previously observed such an AMPK effect in our AMPK KD strain (22), and to our knowledge it has not been demonstrated in similar AMPK mouse strains (41, 49) or in other transgenic AMPK mouse models (8, 53, 63). Furthermore, we did not rediscover this genotype defect in our chronic metformin treated mice, so it is not a consistent characteristic for the AMPK KD model.
We conclude that the potentiating effect of chronic metformin treatment on insulin-stimulated glucose uptake in soleus muscle is dependent on AMPK signaling. At present, we are unable to explain this phenomenon, but it seems to be unrelated to changes in the insulin signaling pathway at the level of Akt and TBC1D4 as well as protein expression of GLUT4 and hexokinase.
GRANTS
The study was supported by grants from the Danish Medical Research Council, The Novo Nordisk Foundation, the Danish Diabetes Association, Nordea Foundation and the Lundbeck Foundation. This work is part of the research program of the UNIK: Food, Fitness & Pharma for Health and Disease (see http://www.foodfitnesspharma.ku.dk/). The UNIK project is supported by the Danish Ministry of Science, Technology and Innovation. J. T. Treebak was supported by a postdoctoral fellowship from The Danish Agency for Science, Technology and Innovation, Denmark.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.M.K., J.T.T., and J.F.W. conception and design of research; J.M.K. and J.T.T. performed experiments; J.M.K., J.T.T., P.S., L.G., and J.F.W. analyzed data; J.M.K., J.T.T., P.S., L.G., and J.F.W. interpreted results of experiments; J.M.K. prepared figures; J.M.K. and J.F.W. drafted manuscript; J.M.K., J.T.T., P.S., L.G., and J.F.W. edited and revised manuscript; J.M.K., J.T.T., P.S., L.G., and J.F.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Prof. D. Grahame Hardie, Dundee University, Scotland, for donation of valuable tools for this study. We also thank Prof. Morris Birnbaum, University of Pennsylvania, Philadelphia, PA, for providing the founder AMPK-KD mice. Betina Bolmgren, Nicoline Resen Andersen and Anja Jokipii are acknowledged for skilled technical assistance.
Current affiliation of J. M. Kristensen: Department of Endocrinology, Odense University Hospital, Institute of Clinical Research, Section of Molecular Diabetes & Metabolism, University of Southern Denmark, Denmark.
Current affiliation of J. T. Treebak: Section of Integrative Physiology, Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, Copenhagen, Denmark.
REFERENCES
- 1.Ai H, Ihlemann J, Hellsten Y, Lauritzen HP, Hardie DG, Galbo H, Ploug T. Effect of fiber type and nutritional state on A. Am J Physiol Endocrinol Metab 282: E1291–E1300, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Aledo JC, Darakhshan F, Hundal HS. Rab4, but not the transferrin receptor, is colocalized with GLUT4 in an insulin-sensitive intracellular compartment in rat skeletal muscle. Biochem Biophys Res Commun 215: 321–328, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Allen DL, Harrison BC, Sartorius C, Byrnes WC, Leinwand LA. Mutation of the IIB myosin heavy chain gene results in muscle fiber loss and compensatory hypertrophy. Am J Physiol Cell Physiol 280: C637–C645, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Allen DL, Harrison BC, Sartorius C, Byrnes WC, Leinwand LA. Mutation of the IIB myosin heavy chain gene results in muscle fiber loss and compensatory hypertrophy. Am J Physiol Cell Physiol 280: C637–C645, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Bailey CJ, Mynett KJ, Page T. Importance of the intestine as a site of metformin-stimulated glucose utilization. Br J Pharmacol 112: 671–675, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey CJ, Puah JA. Effect of metformin on glucose metabolism in mouse soleus muscle. Diabetes Metab 12: 212–218, 1986. [PubMed] [Google Scholar]
- 7.Bailey CJ, Wilcock C, Scarpello JH. Metformin and the intestine. Diabetologia 51: 1552–1553, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem 279: 38441–38447, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Bogachus LD, Turcotte LP. Genetic downregulation of AMPKα isoforms uncovers the mechanism by which metformin decreases FA uptake and oxidation in skeletal muscle cells. Am J Physiol Cell Physiol 299: C1549–C1561, 2010. [DOI] [PubMed] [Google Scholar]
- 10.Borst SE, Snellen HG. Metformin, but not exercise training, increases insulin responsiveness in skeletal muscle of Sprague-Dawley rats. Life Sci 69: 1497–1507, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 50: 790–796, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buhl ES, Jessen N, Schmitz O, Pedersen SB, Pedersen O, Holman GD, Lund S. Chronic treatment with 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside increases insulin-stimulated glucose uptake and GLUT4 translocation in rat skeletal muscles in a fiber type-specific manner. Diabetes 50: 12–17, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Cheng JT, Huang CC, Liu IM, Tzeng TF, Chang CJ. Novel mechanism for plasma glucose-lowering action of metformin in streptozotocin-induced diabetic rats. Diabetes 55: 819–825, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Cusi K, DeFronzo RA. Metformin: a review of its metabolic effects. Diabetes Rev 6: 89–131, 1998. [Google Scholar]
- 15.Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 81: 4059–4067, 1996. [DOI] [PubMed] [Google Scholar]
- 16.Da SD, Zancan P, Coelho WS, Gomez LS, Sola-Penna M. Metformin reverses hexokinase and 6-phosphofructo-1-kinase inhibition in skeletal muscle, liver and adipose tissues from streptozotocin-induced diabetic mouse. Arch Biochem Biophys 496: 53–60, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Davidson YS, Clague JE, Horan MA, Pendleton N. The effect of aging on skeletal muscle capillarization in a murine model. J Gerontol A Biol Sci Med Sci 54: B448–B451, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Dinneen S, Gerich J, Rizza R. Carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N Engl J Med 327: 707–713, 1992. [DOI] [PubMed] [Google Scholar]
- 19.El Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275: 223–228, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Ferrannini E, Smith JD, Cobelli C, Toffolo G, Pilo A, DeFronzo RA. Effect of insulin on the distribution and disposition of glucose in man. J Clin Invest 76: 357–364, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer M, Timper K, Radimerski T, Dembinski K, Frey DM, Zulewski H, Keller U, Muller B, Christ-Crain M, Grisouard J. Metformin induces glucose uptake in human preadipocyte-derived adipocytes from various fat depots. Diabetes Obes Metab 12: 356–359, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Frosig C, Jensen TE, Jeppesen J, Pehmoller C, Treebak JT, Maarbjerg SJ, Kristensen JM, Sylow L, Alsted TJ, Schjerling P, Kiens B, Wojtaszewski JF, Richter EA. AMPK and insulin action—responses to ageing and high fat diet. PLoS One 8: e62338, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 277: 25226–25232, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Fueger PT, Shearer J, Bracy DP, Posey KA, Pencek RR, McGuinness OP, Wasserman DH. Control of muscle glucose uptake: test of the rate-limiting step paradigm in conscious, unrestrained mice. J Physiol 562: 925–935, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE, Timmins P, Williams KM. Clinical pharmacokinetics of metformin. Clin Pharmacokinet 50: 81–98, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Habegger KM, Hoffman NJ, Ridenour CM, Brozinick JT, Elmendorf JS. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 153: 2130–2141, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA 105: 7815–7820, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Handberg A, Kayser L, Hoyer PE, Voldstedlund M, Hansen HP, Vinten J. Metformin ameliorates diabetes but does not normalize the decreased GLUT 4 content in skeletal muscle of obese (fa/fa) Zucker rats. Diabetologia 36: 481–486, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology 131: 973, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Hundal HS, Ramlal T, Reyes R, Leiter LA, Klip A. Cellular mechanism of metformin action involves glucose transporter translocation from an intracellular pool to the plasma membrane in L6 muscle cells. Endocrinology 131: 1165–1173, 1992. [DOI] [PubMed] [Google Scholar]
- 31.Hutber CA, Hardie DG, Winder WW. Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. Am J Physiol Endocrinol Metab 272: E262–E266, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Jensen TE, Leutert R, Rasmussen ST, Mouatt JR, Christiansen ML, Jensen BR, Richter EA. EMG-normalised kinase activation during exercise is higher in human gastrocnemius compared to soleus muscle. PLoS One 7: e31054, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA. Role of AMPKα2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab 292: E331–E339, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 279: 1070–1079, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Kim YB, Ciaraldi TP, Kong A, Kim D, Chu N, Mohideen P, Mudaliar S, Henry RR, Kahn BB. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes 51: 443–448, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Klip A, Schertzer JD, Bilan PJ, Thong F, Antonescu C. Regulation of glucose transporter 4 traffic by energy deprivation from mitochondrial compromise. Acta Physiol (Oxf) 196: 27–35, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24: 1227–1251, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55: 2067–2076, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Kristensen JM, Larsen S, Helge JW, Dela F, Wojtaszewski JF. Two weeks of metformin treatment enhances mitochondrial respiration in skeletal muscle of AMPK kinase dead but not wild type mice. PLoS One 8: e53533, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JO, Lee SK, Jung JH, Kim JH, You GY, Kim SJ, Park SH, Uhm KO, Kim HS. Metformin induces Rab4 through AMPK and modulates GLUT4 translocation in skeletal muscle cells. J Cell Physiol 226: 974–981, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Lee-Young RS, Bonner JS, Mayes WH, Iwueke I, Barrick BA, Hasenour CM, Kang L, Wasserman DH. AMP-activated protein kinase (AMPK)alpha2 plays a role in determining the cellular fate of glucose in insulin-resistant mouse skeletal muscle. Diabetologia 56: 608–617, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Wan Q, Guan Q, Gao L, Zhao J. High-fat diet feeding impairs both the expression and activity of AMPKa in rats' skeletal muscle. Biochem Biophys Res Commun 339: 701–707, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Lord JM, Puah JA, Atkins TW, Bailey CJ. Postreceptor effect of metformin on insulin action in mice. J Pharm Pharmacol 37: 821–823, 1985. [DOI] [PubMed] [Google Scholar]
- 44.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 307: 384–387, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Luna V, Casauban L, Sajan MP, Gomez-Daspet J, Powe JL, Miura A, Rivas J, Standaert ML, Farese RV. Metformin improves atypical protein kinase C activation by insulin and phosphatidylinositol-3,4,5-(PO4)3 in muscle of diabetic subjects. Diabetologia 49: 375–382, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den BG, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 10: 1247–1255, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J 391: 87–93, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moreira PI, Oliveira CR. Mitochondria as potential targets in antidiabetic therapy. Handb Exp Pharmacol (203): 331–356, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Mu J, Barton ER, Birnbaum MJ. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on “lazy mice”. Biochem Soc Trans 31: 236–241, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell 7: 1085–1094, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Muller S, Denet S, Candiloros H, Barrois R, Wiernsperger N, Donner M, Drouin P. Action of metformin on erythrocyte membrane fluidity in vitro and in vivo. Eur J Pharmacol 337: 103–110, 1997. [DOI] [PubMed] [Google Scholar]
- 52.Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51: 2074–2081, 2002. [DOI] [PubMed] [Google Scholar]
- 53.O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, Van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 108: 16092–16097, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348: 607–614, 2000. [PMC free article] [PubMed] [Google Scholar]
- 55.Pentikainen PJ, Neuvonen PJ, Penttila A. Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol 16: 195–202, 1979. [DOI] [PubMed] [Google Scholar]
- 56.Perriello G, Misericordia P, Volpi E, Santucci A, Santucci C, Ferrannini E, Ventura MM, Santeusanio F, Brunetti P, Bolli GB. Acute antihyperglycemic mechanisms of metformin in NIDDM. Evidence for suppression of lipid oxidation and hepatic glucose production. Diabetes 43: 920–928, 1994. [DOI] [PubMed] [Google Scholar]
- 57.Prager R, Schernthaner G, Graf H. Effect of metformin on peripheral insulin sensitivity in non insulin dependent diabetes mellitus. Diabetes Metab 12: 346–350, 1986. [PubMed] [Google Scholar]
- 58.Rossetti L, DeFronzo RA, Gherzi R, Stein P, Andraghetti G, Falzetti G, Shulman GI, Klein-Robbenhaar E, Cordera R. Effect of metformin treatment on insulin action in diabetic rats: in vivo and in vitro correlations. Metabolism 39: 425–435, 1990. [DOI] [PubMed] [Google Scholar]
- 59.Rouru J, Koulu M, Peltonen J, Santti E, Hanninen V, Pesonen U, Huupponen R. Effects of metformin treatment on glucose transporter proteins in subcellular fractions of skeletal muscle in (fa/fa) Zucker rats. Br J Pharmacol 115: 1182–1187, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sajan MP, Bandyopadhyay G, Miura A, Standaert ML, Nimal S, Longnus SL, Van OE, Hainault I, Foufelle F, Kahn R, Braun U, Leitges M, Farese RV. AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. Am J Physiol Endocrinol Metab 298: E179–E192, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sherman LA, Hirshman MF, Cormont M, Le Marchand-Brustel Y, Goodyear LJ. Differential effects of insulin and exercise on Rab4 distribution in rat skeletal muscle. Endocrinology 137: 266–273, 1996. [DOI] [PubMed] [Google Scholar]
- 62.Song XM, Fiedler M, Galuska D, Ryder JW, Fernstrom M, Chibalin AV, Wallberg-Henriksson H, Zierath JR. 5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia 45: 56–65, 2002. [DOI] [PubMed] [Google Scholar]
- 63.Steinberg GR, O'Neill HM, Dzamko NL, Galic S, Naim T, Koopman R, Jorgensen SB, Honeyman J, Hewitt K, Chen ZP, Schertzer JD, Scott JW, Koentgen F, Lynch GS, Watt MJ, Van Denderen BJ, Campbell DJ, Kemp BE. Whole body deletion of AMP-activated protein kinase (beta)2 reduces muscle AMPK activity and exercise capacity. J Biol Chem 285: 37198–37209, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stumvoll M, Haring HU, Matthaei S. Metformin. Endocr Res 32: 39–57, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci USA 107: 19909–19914, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S. Metformin increases the PGC-1α protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol 101: 1685–1692, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 8: 92–103, 2012. [DOI] [PubMed] [Google Scholar]
- 68.Thomas CR, Turner SL, Jefferson WH, Bailey CJ. Prevention of dexamethasone-induced insulin resistance by metformin. Biochem Pharmacol 56: 1145–1150, 1998. [DOI] [PubMed] [Google Scholar]
- 69.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100: 328–341, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPKβ1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol 297: C1041–C1052, 2009. [DOI] [PubMed] [Google Scholar]
- 71.Treebak JT, Taylor EB, Witczak CA, An D, Toyoda T, Koh HJ, Xie J, Feener EP, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am J Physiol Cell Physiol 298: C377–C385, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wiernsperger NF. Membrane physiology as a basis for the cellular effects of metformin in insulin resistance and diabetes. Diabet Metab 25: 110–127, 1999. [PubMed] [Google Scholar]
- 73.Wiernsperger NF, Bailey CJ. The antihyperglycaemic effect of metformin: therapeutic and cellular mechanisms. Drugs 58, Suppl 1: 31–39, 1999. [DOI] [PubMed] [Google Scholar]
- 74.Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 24: 49–57, 1994. [DOI] [PubMed] [Google Scholar]
- 75.Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol 528: 221–226, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Woods A, Salt I, Scott J, Hardie DG, Carling D. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett 397: 347–351, 1996. [DOI] [PubMed] [Google Scholar]
- 77.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]