

Abstract
Morphological and functional alterations of hepatic mitochondria have been documented in patients with alcoholic liver disease (ALD). Our recent study demonstrated that zinc level was decreased in whole liver and mitochondria by chronic alcohol feeding. The present study was undertaken to determine whether zinc deficiency mediates alcohol-induced mitochondrial electron transport chain (ETC) defect and whether defective ETC function may lead to generation of reactive oxygen species (ROS). Male Wistar rats were pair fed with the Lieber-DeCarli control or ethanol diet for 5 mo. Chronic alcohol exposure increased hepatic triglyceride, free fatty acid, and 4-hydroxynonenal (4HNE) levels; meanwhile hepatic mitochondrial 4HNE level was also increased. Moreover, hepatic mitochondrial respiratory complexes I, III, IV, and V and hepatic ATP production were decreased by chronic alcohol exposure. Chronic alcohol feeding decreased peroxisome proliferator-activated receptor gamma coactivator-1-alpha (PGC1α), nuclear respiratory factor 1 (NRF1), mitochondrial transcription factor A (TFAM), and mitochondrial DNA. HepG2 cells were treated with N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) for 6 h. Zinc deficiency significantly decreased mitochondrial respiratory complexes I, III, and IV. In addition, PGC1α, NRF1, and TFAM levels as well as mitochondrial DNA were significantly decreased by TPEN treatment. Knockdown of mitochondrial respiratory complexes I, III, or IV by shRNA caused a decrease in mitochondrial membrane potential and an increase in ROS production. These results suggest that alcohol-induced hepatic zinc deficiency could inactivate mitochondrial biogenesis pathway and decrease mitochondrial DNA replication, which, in turn, decreases mitochondrial complex protein expression. The defect of mitochondrial respiratory complexes may worsen alcohol-induced ROS production.
Keywords: alcoholic liver disease, zinc, mitochondrial complex, ROS
excessive alcohol consumption can exert serious adverse health effects, including the development of alcoholic liver disease (ALD). The spectrum of ALD includes alcoholic steatosis, alcoholic hepatitis, and alcoholic cirrhosis, which may ultimately progress to carcinoma (17). The simple steatosis manifests as lipid accumulation and enlarged liver, and alcoholic hepatitis is characterized by inflammatory cell infiltration in the liver (28, 54). Cirrhosis is the most severe stage of ALD, and at this stage normal liver tissue is replaced by scar tissue and proper function of liver is lost (32). It has been proposed that alcohol-induced generation of reactive oxygen species (ROS) contributes to the transition of simple steatosis to advanced stage of ALD (60). Studies demonstrated that alcohol-induced massive production of ROS results in inflammasome formation, cell injury, and apoptotic signal activation (57, 58). Meanwhile, deletion of genes that promote hydrogen peroxide generation ameliorated alcohol-induced steatosis and cell injury (25).
The mitochondrion is the organelle well known for its role in energy generation from nutrients and alcohol oxidation. However, the generation of ATP accompanies with ROS production, which is the inevitable byproducts of oxidative phosphorylation (OXPHOS) (40). Mechanistic studies demonstrated that defect in mitochondrial OXPHOS process results in further generation of ROS (3, 11). In addition, mitochondria are dynamic organelles involved in biogenesis. The regulation of mitochondrial biogenesis is achieved by PGC1α, which promotes nuclear respiratory factor (NRF)-mediated mitochondrial transcription factor A (TFAM) expression (4). The translocation of TFAM to mitochondria stimulates mitochondrial biogenesis and mtDNA replication (4). Alterations in morphology and function of mitochondria have been reported in the liver of patients with ALD (13).
Zinc deficiency is well documented in patients with advanced ALD (35). Zinc is required to maintain normal structure of proteins and for activities of more than 300 enzymes (15). In addition, zinc participates in antioxidant defense (35). Thus zinc deficiency links with severe health consequences. It has been documented that chronic alcohol feeding significantly decreases zinc levels in the plasma and liver of mice and rats (23, 42), whereas zinc supplementation to mice chronically fed alcohol diet ameliorates ROS generation in liver (23). Our recent study demonstrated that zinc level is most abundant in the mitochondria compared with other subcellular compartments but is significantly reduced after alcohol exposure (43). Therefore, this study was undertaken to investigate whether there is a mechanistic link between alcohol-induced hepatic zinc deficiency and mitochondrial electron transport chain (ETC) defect and whether ETC defect contributes to ROS production.
MATERIALS AND METHODS
Animal and alcohol feeding experiments.
Male Wistar rats were obtained from Harlan (Indianapolis, IN). The animal protocol was approved by the Institutional Animal Care and Use Committee of the North Carolina Research Campus. Eight-week-old male rats were pair fed a modified Lieber-DeCarli alcohol (Dyets, Bethlehem, PA, no. 7100027) or isocaloric maltose dextrin control liquid diet (Dyets, no. 710260) for 5 mo (n = 6 for each group) with a stepwise feeding procedure, as described previously (43). In brief, the ethanol content (%, wt/vol) in the diet was started with 1.6 and increased by 1 every 2 days to reach 3.6 at the end of prefeeding. On the day of feeding, the ethanol content in the diet was 5.0 (36% of total calories) and gradually increased to 6.3 (44% of total calories). At the end of 5 mo of feeding, rats were anesthetized with inhalational isoflurane. Left lobe of liver was collected for organelle isolation process, and the rest of the liver were fixed for pathology or stored at −80°C.
Assessment of hepatic lipid accumulation.
Liver tissues were frozen in Tissue-Tek OCT (Optimum Cutting Temperature) Compound (VWR, Batavia, IL). Cryostat liver tissue sections were cut at 7 μm, fixed, and processed with Oil Red O solution to stain neutral lipid. Hepatic triglycerides and free fatty acid levels were quantitatively measured by biochemical assay with BioVision assay kits.
Cell culture and treatment.
Human HepG2 hepatoma cells obtained from the American Type Culture Collection (Manassas, VA) were grown in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin sulfate (100 μg/ml) (Invitrogen, Carlsbad, CA). HepG2 cells were seeded overnight at 5×105 cells per well for six-well plates, or 1×105 cells per well for eight-well chamber slides, or 2×104 cells per well for 96-well plates. Then cells were treated with 3 μM N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) (Sigma-Aldrich) for 6 h in absence of FBS.
Determination of ROS.
Superoxide levels in the liver of rats or HepG2 cells were detected with dihydroethidium (DHE) fluorescence microscopy. DHE exhibits blue fluorescence in the cytosol until oxidized by superoxide anions to ethidium, which intercalates into DNA and gives nucleus a bright red fluorescence (8). The frozen liver tissue sections or HepG2 cell chamber slides were incubated with 5 μM DHE (Life Technologies, Carlsbad, CA, no. D-1168) for 30 min and washed twice with PBS.
Hydrogen peroxide levels in the cell culture medium were quantitatively measured by spectrometry with Amplex Red Hydrogen Peroxide/Peroxidase Assay kit (Invitrogen, Waltham, MA, no. A22188). Briefly, cells were seeded in a 96-well plate overnight. In the next morning, fresh media were changed for each well. Six hours later, 50 μl of medium was transferred to a clean 96-well plate subject to hydrogen peroxide assay according to the instruction provided by the manufacturer. Then the level of hydrogen peroxide was measured with plate reader at OD 560 nm.
Immunohistochemical staining.
Hepatic myeloperoxidase (MPO), 4-hydroxynonenal (4HNE), OXPHOS, MTCO1, AMPK, PGC1α, NRF1, and TFAM levels were detected by immunohistochemical peroxidase staining. Briefly, liver tissue paraffin sections were incubated with 3% hydrogen peroxide for 10 min to inactivate endogenous peroxidases. Tissue sections were then incubated with a polyclonal rabbit anti-MPO (LSBio, Seattle, WA, no. LS-B6699, 1:100), a monoclonal mouse anti-4HNE antibody (Northwest Life Science Specialties, Vancouver, WA, no. NWA-HNE020, 1:100), a monoclonal mouse anti-OXPHOS (Abcam, Cambridge, MA, no. ab110413, 1:100), a monoclonal mouse anti-MTCO1 (Abcam, no. ab14705, 1:100), a monoclonal rabbit anti-p-AMPK (Cell Signaling Technology, Danvers, MA, no. 2537, 1:100), a monoclonal mouse anti-PGC1α (Calbiochem, no. ST1202, 1:100), a polyclonal rabbit anti-NRF1 (Boster, Pleasanton, CA, no. PA1948, 1:200), or a monoclonal mouse anti-TFAM (Novus Biological, Littleton, CO, no. NBP1-71648, 1:100). Tissue sections were then incubated with horseradish peroxidase (HRP)-labeled corresponding secondary antibodies, followed by visualization with 3,3′-diaminobenzidine (DAB). Hepatic CD68-positive cells were detected by immunofluorescent staining with a monoclonal mouse anti-CD68 (AbD Serotec no. MCA341GA, 1:50). The CD68-positive cells were visualized by incubation with Alexa Fluor 594-conjugated donkey anti-mouse IgG (Invitrogen). Negative controls were conducted by replacing the primary antibody with PBS, and no positive staining was found. Quantification of positive staining was conducted by Image J (NIH).
Immunoblot analysis.
Liver tissue or purified hepatic mitochondrial proteins were extracted by T-PER tissue extraction reagent (Thermo Scientific) containing protease inhibitors (Sigma-Aldrich, St. Louis, MO). Aliquots containing 80 μg proteins were loaded onto an 8–15% sodium dodecyl sulfate-polyacrylamide gel. After electrophoresis, proteins were transferred to a polyvinylidene fluoride membrane. The membrane was probed with antibodies, including anti-4HNE (1:500), anti-superoxide dismutase 2 (SOD2, Millipore, Billerica, MA, no. 06984, 1:1,000), anti-heat shock protein 60 (hsp60, BD, Franklin Lakes, NJ, no. 611562, 1:1,000), anti-total OXPHOS cocktail (1:2,000), anti-PGC1α (1:1,000), anti-NRF1 (1:1,000), and anti-TFAM (1:1,000), respectively. The membrane was then incubated with HRP-conjugated goat anti-rabbit IgG or goat anti-mouse IgG antibody. The protein bands were visualized by an Enhanced Chemiluminescence detection system (GE Healthcare, Piscataway, NJ, no. 34076) and quantified by densitometry analysis with Image J (NIH).
Measurement of hepatic ATP.
Hepatic ATP level was measured by commercial ATP Assay Kit (Abcam, no. ab83355). Briefly, liver tissue (∼20–30 mg) was freeze clamped by use of an aluminum block precooled in liquid nitrogen and immediately immersed in liquid nitrogen. Liver samples were stored at −80°C. On the day of assay, liver sample was pulverized under liquid nitrogen and homogenized with 6% perchloric acid. Homogenates were centrifuged at 13,000 g for 5 min at 4°C, and the supernatants were neutralized to pH 7.8 with 2 M KOH, placed on ice for 1 h, and centrifuged at 13,000 g for 5 min at 4°C. The supernatants were subjected to ATP assay according to the manufacturer's instruction. The amount of tissue ATP was determined at OD 570 nm.
qPCR.
The DNA was extracted by QIAamp DNA Mini Kit (Qiagen, Venlo, Limburg, no. 51306). The forward and reverse primers were purchased from Integrated DNA Technologies (Coralville, IA). Primer sequences of rat and human NADH dehydrogenase for the quantitative PCRs (qPCRs) are described elsewhere (53). qPCR analysis with SYBR Green PCR Master Mix (Qiagen, Valencia, CA, no. 203445) was performed on the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems).
Silence of mitochondrial respiratory complex in HepG2 cells.
HepG2 cells were transfected with human CI-NDUFB8, CIII-UQCRC2 (Santa Cruz Biotechnologies, Santa Cruz, CA), or CIV-MTCO1 shRNA (Applied Biological Materials, Richmond, BC). Stable clones were generated by delivering shRNA lentivirus into cells. The selection of stable silenced clones was started 48 h later with 2 μM puromycin.
Measurement of mitochondrial membrane potential.
The mitochondrial membrane potential of live cells was assessed by TMRE mitochondrial kit (Abcam, no. ab113852), as described previously (43).
Statistics.
Results are expressed as means ± standard deviation. Differences between two groups were analyzed by two-tailed Student's t-test. Differences among multiple groups were analyzed by analysis of variance followed by Tukey's test. The significance between groups was defined as P < 0.05.
RESULTS
Chronic alcohol feeding induces hepatic lipid accumulation and inflammation.
The serum alanine aminotransferase (ALT), aspartate aminotransferase, and hepatic and subcellular zinc levels have been reported previously (43). CD68 and MPO staining illustrated that chronic alcohol feeding significantly increased the number of Kupffer cells and neutrophil infiltration in the liver of alcohol-fed rats (Fig. 1, A and B). Oil Red O staining demonstrated that 5 mo of alcohol feeding increased the number and size of lipid droplets (arrows) in the liver (Fig. 1A). Quantitative measurement of hepatic lipid contents showed that 5-mo alcohol feeding significantly increased hepatic triglyceride (P < 0.05) and free fatty acid (P < 0.05) concentrations (Fig. 1, C and D).
Fig. 1.
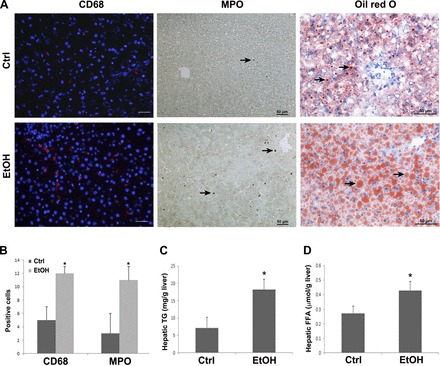
Inflammatory cell infiltration and lipid accumulation in rats chronically fed alcohol for 5 mo. A: immunofluorescent staining of CD68 in the liver, which was counterstained with DAPI; immunohistochemical staining of MPO in the liver; Oil Red O staining of the neutral lipids in the liver. CD68-positive cells are showed as red staining in the cytosol. The brownish staining shows MPO-positive cells, as indicated by arrows. Lipid droplets are stained with red color, as indicated by arrows. B: quantitative measurements of positive cells of CD68 and MPO. C and D: quantitative measurements of hepatic concentration of triglycerides (TG; C) and free fatty acids (FFA; D). Data are expressed as means ± SD from 6 rats. Statistical difference (P < 0.05) was analyzed by Student's t-test. Ctrl, control; EtOH, ethanol.
Chronic alcohol feeding induces oxidative stress in the whole liver and purified hepatic mitochondria.
To determine whether liver and mitochondrial oxidative stress is significantly increased by chronic alcohol feeding, ROS in the liver and purified hepatic mitochondria were analyzed. DHE fluorescence microscopy showed that alcohol feeding caused ROS accumulation in the liver, as indicated by strong red fluorescence formation in the nuclei (Fig. 2A). A lipid peroxidation product, 4HNE, was detected by immunohistochemistry. As shown in Fig. 2A, the intensity of 4HNE staining was increased in the liver of rats after alcohol feeding. Moreover, Western blot analysis showed that alcohol feeding significantly increased 4HNE, SOD2, and hsp60 in the purified hepatic mitochondria compared with the controls (Fig. 2B).
Fig. 2.
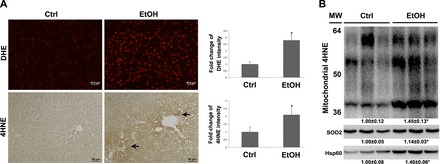
Oxidative stress in the whole liver and purified mitochondria in rats chronically fed alcohol for 5 mo. A: reactive oxygen species (ROS) accumulation in the liver was detected by dihydroethidium (DHE), as indicated by red fluorescence in the nuclei (top). Scale bar: 20 μm. The formation of 4-hydroxynonenal (4HNE) adduct (arrows) was detected by immunohistochemistry (bottom). Scale bar: 50 μm. B: immunoblot bands of 4HNE, superoxide dismutase 2 (SOD2), and heat shock protein 60 (hsp60) in purified mitochondria. Quantification of DHE and 4HNE staining intensity was conducted by Image J. Data are expressed as means ± SD from 6 rats. Statistical difference (*P < 0.05) was analyzed by Student's t-test. MW, molecular weight.
Chronic alcohol feeding impairs mitochondrial OXPHOS and biogenesis.
To determine whether alcohol feeding affects mitochondrial OXPHOS, ETC was assessed at protein level. The protein levels of mitochondrial complex I, complex III, complex IV, and complex V were significantly decreased by chronic alcohol feeding (Fig. 3A). Hepatic ATP level was measured to assess the productivity of ETC. Alcohol feeding reduced hepatic ATP level to 87% of that of the controls (Fig. 3B). In addition, immunohistochemical staining of total OXPHOS proteins and complex IV (MTCO1) showed a weaker staining intensity in the liver of alcohol-fed rats compared with the controls (Fig. 3C). Quantification of positive staining further showed significantly decreased expression of total OXPHOS proteins and complex IV protein in the liver of rats after 5-mo alcohol exposure (Fig. 3D).
Fig. 3.
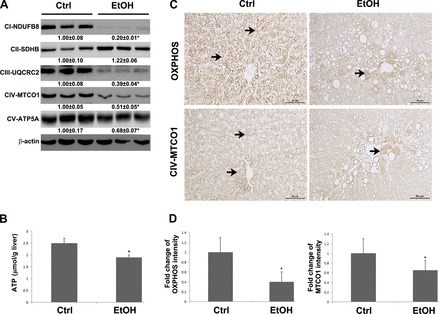
Protein level of mitochondrial respiratory complexes and ATP production in the liver of rats chronically fed alcohol for 5 mo. A: immunoblot bands of oxidative phosphorylation (OXPHOS) proteins, subunit of complex I (CI-NDUFB8), subunit of complex II (CII-SDHB), subunit of complex III (UQCRC2), subunit of complex IV (MTCO1), and subunit of complex V (ATP5A). B: ATP level in the liver of rat was detected by commercial ATP assay kit. C: immunohistochemical staining of total mitochondrial respiratory complex proteins (top) and complex IV MTCO-1 proteins (bottom). Arrows indicate positive staining. D: fold change of immunohistochemical staining intensity. The quantification was conducted by Image J. Data are expressed as means ± SD from 6 rats. Statistical difference (*P < 0.05) was analyzed by Student's t-test.
To further explore the possible mechanisms by which chronic alcohol feeding decreases OXPHOS, major mitochondrial biogenesis regulators were evaluated. As shown in Fig. 4A, chronic alcohol feeding significantly decreased hepatic p-AMPK, PGC1α, NRF1, and TFAM at protein levels. Meanwhile, immunohistochemical staining demonstrated that the intensities of p-AMPK, nuclear PGC1α and NRF1, and mitochondrial TFAM were reduced in the alcohol-fed rats compared with that of the controls (Fig. 4, B and C). In accordance, hepatic mitochondrial DNA level was significantly decreased in the alcohol fed rats compared with that of the controls (Fig. 4D).
Fig. 4.
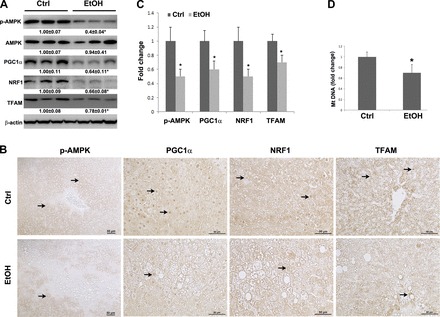
Protein level of mitochondrial biogenesis regulators and mtDNA level in the liver of rats chronically fed alcohol for 5 mo. A: immunoblot bands of p-AMPK, AMPK, PGC1α, NRF1, and TFAM. B: immunohistochemical staining of p-AMPK, PGC1α, NRF1, and TFAM proteins. Arrows indicate positive staining. C: fold change of immunohistochemical staining intensity. The quantification was conducted by Image J. D: mtDNA level was measured with NADH dehydrogenase subunit 6 by quantitative PCR (qPCR). Data are expressed as means ± SD from 6 rats. Statistical difference (*P < 0.05) was analyzed by Student's t-test.
Zinc deprivation perturbs mitochondrial biogenesis and oxidative phosphorylation in HepG2 cells.
We have previously reported that mitochondrial zinc level was significantly decreased in the liver of rats chronically fed alcohol; therefore the possible link between zinc deficiency and mitochondrial ETC reduction was evaluated in this study. Zinc deprivation in HepG2 cells was achieved by TPEN treatment. Western blot analysis showed that zinc deprivation did not affect hsp60 but significantly upregulated SOD2 expression (Fig. 5A). Then the expression of mitochondrial respiratory complexes was assessed. TPEN treatment significantly decreased the expression of complex I, complex III, and complex IV but increased the expression of complex V at protein level (Fig. 5B). To understand the mechanism by which zinc deprivation perturbs mitochondrial respiratory complexes, mitochondrial biogenesis regulators were evaluated. As shown in Fig. 5C, p-AMPK, PGC1α, NRF1, and TFAM were significantly decreased by TPEN treatment. Meanwhile, mitochondrial DNA level in TPEN-treated cells was dramatically decreased to 47% of that in controls (Fig. 5D).
Fig. 5.
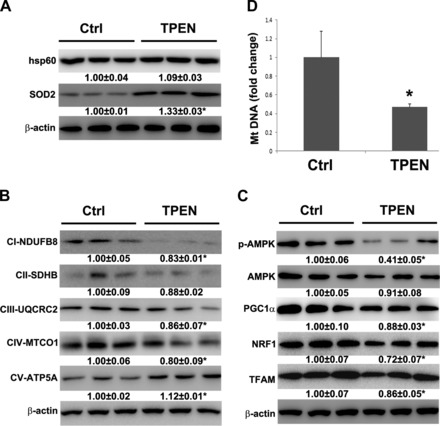
Effect of TPEN on hsp60, SOD2, and OXPHOS proteins, mitochondrial biogenesis regulators, and mtDNA level in human HepG2 cells. Human HepG2 cells were treated with 3 μM TPEN without FBS for 6 h. A: immunoblot bands of hsp60 and SOD2. B: immunoblot bands of OXPHOS proteins, subunit of complex I (CI-NDUFB8), subunit of complex II (CII-SDHB), subunit of complex III (CIII-UQCRC2), subunit of complex IV (CIV-MTCO1), and subunit of complex V (CV-ATP5A). C: immunoblot bands of p-AMPK, AMPK, PGC1α, NRF1, and TFAM. D: mtDNA level was measured with NADH dehydrogenase subunit 6 by qPCR. Data are expressed as means ± SD (n = 3). Statistical difference (*P < 0.05) was analyzed by Student's t-test.
Zinc deficiency-induced defect of mitochondrial ETC leads to a decreased MMP and increased ROS production.
We have previously shown that zinc deprivation induced generation of ROS and loss of mitochondrial membrane potential (MMP) and thereby caused hepatocyte apoptosis. In the present study, we tested whether ETC protein defect due to zinc deprivation is a cause of increased ROS and decreased MMP in hepatocytes. HepG2 cells were genetically manipulated with scrambled shRNAs, complex I-NDUFB8 shRNA, complex III-UQCRC2 shRNA, or complex IV-MTCO1 shRNA, respectively. As shown in Fig. 6, transfection with all the three mitochondrial complex shRNAs effectively silenced their target genes, respectively.
Fig. 6.
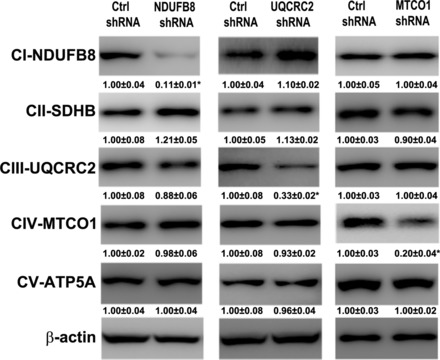
The efficiency of transfection with shRNAs. The human HepG2 cells were transfected with scrambled, CI-NDUFB8, CIII-UQCRC2, CIV-MTCO1 shRNA lentivirus for 48 h and the stable clone was selected with 2 μM puromycin. Immunoblot bands of OXPHOS proteins in stable transfected cells. Data are expressed as means ± SD (n = 3). Statistical difference (*P < 0.05) was analyzed by Student's t-test.
MMP and ROS were assessed by fluorescence microscopy, and the red fluorescence represents active mitochondria and ROS accumulation, respectively. As shown in Fig. 7A, HepG2 cells transfected with NDUFB8 shRNA, UQCRC2 shRNA, or MTCO1 shRNA showed a decreased MMP but an increased ROS production compared with the HepG2 cells transfected with the scrambled shRNA. The hydrogen peroxide concentration in the cell culture medium was measured as well. As shown in Fig. 7B, HepG2 cells transfected with NDUFB8 shRNA, UQCRC2 shRNA, or MTCO1 shRNA had a higher hydrogen peroxide concentration in the culture media compared with the HepG2 cells transfected with the scrambled shRNA.
Fig. 7.
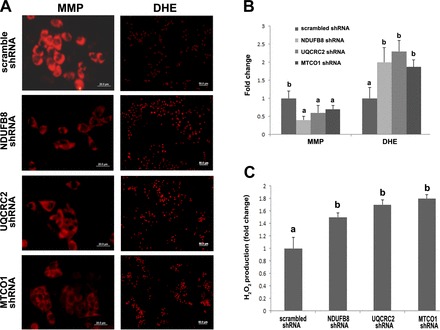
Effect of defective mitochondrial respiratory complex on mitochondrial membrane potential (MMP) and ROS generation. HepG2 cells transfected with scrambled shRNA, NDUFB8 shRNA, UQCRC2 shRNA, or MTCO shRNA were stained with TMRE or DHE for assessing MMP and ROS, respectively. A: red fluorescence represents TMRE-labeled active mitochondria (left, scale bar 20 μm) or cells with ROS production (right, scale bar 50 μm). B: fold change of fluorescence intensity. Quantification was conducted by Image J. C: hydrogen peroxide secreted into cell cultural medium was detected by hydrogen peroxide/peroxidase assay kit. Data are expressed as means ± SD (n = 3). Results for bars that do not share a letter differ significantly (P < 0.05). Significant differences among groups were determined by ANOVA followed by Tukey's test.
DISCUSSION
Increased generation of ROS is a major cause of alcohol consumption-induced liver damage (31). Reduced ROS production by genetic manipulation or dietary interventions effectively protected liver injury induced by alcohol consumption. A previous study showed that p47 knockout mice had a decreased ROS, plasma ALT, hepatic lipid accumulation, and inflammation compared with wild-type mice after alcohol feeding (26). In addition, our group previously demonstrated that zinc supplementation effectively ameliorated ROS generation and partially reversed plasma ALT, hepatic lipid accumulation, and apoptosis after alcohol consumption (23). In the present study, we found not only an increased ROS level in the liver but also accumulated 4HNE in the purified mitochondria. Meanwhile, the increased production of SOD2 was detected. SOD2 is a mitochondrial-specific antioxidant that converts superoxide to hydrogen peroxide and the upregulated expression indicates an increased ROS accumulation in the mitochondria. Therefore, the present study demonstrated that ROS not only accumulated in the liver but also in the mitochondria.
The mitochondrion has been referred as a power plant while passing electrons through ETC to produce ATP. Mitochondrial ETC is composed of four multisubunit complexes. The subunits of each mitochondrial respiratory complex are encoded by both nuclei DNA (nuDNA) and mtDNA, except for complex II (38). Subunits of complex II are completely encoded by nuDNA. Previous studies showed that chronic alcohol exposure repressed OXPHOS by decreased synthesis of mitochondrial respiratory complexes, including complexes I, III, IV, and V in rats (9, 10). Accordingly, mitochondrial respiration (state III) and respiratory control ratio (state III/state IV) were decreased (9), which indicated a decreased ability to convert ADP to ATP. In the present study, we found that chronic alcohol feeding decreased the expression of complexes I, III, IV, and V, but not complex II; meanwhile the production of ATP was decreased. Interestingly, complex IV was constantly reduced, but complex II was always unchanged in all of the chronically alcohol feeding studies on rats (10, 45, 46). It seems that complex IV is one of the most sensitive components to oxidative damage in response to alcohol; however, complex II is the most resistant component to alcohol-induced toxicity in rats. Moreover, previous studies suggest that impairment of mtDNA and mitochondrial ribosomes accounts for the decreased synthesis of the subunits encoded by mtDNA (6, 7, 36). It is still unclear how alcohol represses the synthesis of respiratory subunits encoded by nuDNA. It is possible that the decreased synthesis of respiratory subunits encoded by mtDNA links with the reduced importation and/or the increased degradation of those subunits encoded by nuDNA in mitochondria. Therefore, the unchanged complex II may be due to the lack of respiratory subunits encode by mtDNA. In addition, we found that chronic alcohol consumption increased hepatic hsp60. It is known that hsp60 is a mitochondrial-specific chaperone protein, and the increased hsp60 level indicates a folding crisis of mitochondrial proteins (51, 56). The sustained folding crisis may lead to increased protein degradation (48). Accordingly, the results suggest that ATP reduction after chronic alcohol exposure may result from the decreased OXPHOS function, as demonstrated by reduced mitochondrial respiratory complexes I, III, IV, and V proteins.
Mitochondrial biogenesis is a process by which cells replenish the damaged and degraded mitochondria to maintain their mitochondrial number and function. The regulation of mitochondrial biogenesis involves more than 1,000 genes encoded by both mtDNA and nuDNA. The master regulator of mitochondrial biogenesis is PGC1α (47), which can be regulated by p-AMPK at both posttranslational and transcriptional levels (22, 33, 44). AMPK is well known to be a sensor of cellular energy status and could be activated by a decreased cellular ATP level or an increased AMP-to-ATP ratio (20). Activation of AMPK functions to restore energy homeostasis by shifting ATP-consuming pathway to ATP-producing pathway. Although chronic alcohol consumption decreased hepatic ATP levels (52), hepatic p-AMPK levels were reduced in alcohol-fed mice and rats (1, 24). A mechanistic study found that hepatocytes from chronic ethanol-fed rats showed significantly lower sensitivity to AMPK activator, AICAR (18). AMPK in the liver of ethanol-fed rats also showed a decreased specific activity and a lower sensitivity to changes in the AMP-to-ATP ratio (18). In addition, decreased phosphorylation of AMPK may attribute to activation of protein phosphatase 2A, which inactivates LKB1, the upstream kinase of AMPK (29, 30, 50). The downstream factors of PGC1α include NRF1, peroxisome proliferator receptors and estrogen related receptors; they all can activate TFAM to promote mitochondrial gene expression (4). It has been reported that PGC1α and TFAM positively correlate with mitochondrial respiratory complexes (19, 55). In consistent with previous findings, we demonstrated that alcohol consumption decreases both PGC1α and mitochondrial respiratory complexes in the liver. In addition to PGC1α and TFAM, the present study demonstrates, for the first time, that chronic alcohol feeding also reduces hepatic NRF1, a key nuclear transcription factor for mitochondrial respiration. It has been reported that NRF1 binding sites are found in a numerous genes involved in the respiratory chain (12). Most of these genes were involved in mitochondrial respiratory complexes, but others related to expression and assembly of the respiratory apparatus. NRF1 plays an important role in nucleomitochondrial communication by which the proteins involved in mitochondrial respiration can be produced accordingly in both nuclear and mitochondria. Moreover, it has been reported that homozygous NRF1-null blastocysts have impaired mitochondrial membrane potential and severely reduced mtDNA level (21). Thus the decreased NRF1 level may play an important role in alcohol-induced decrease in mitochondrial respiration and function in addition to regulation of mitochondrial biogenesis. Thus the present study suggests that inactivation of PGC1α, NRF1, and TFAM represents the key mechanism underlying chronic alcohol consumption-induced reduction of mitochondrial biogenesis and respiration.
Zinc deficiency has been reported constantly in human and animals with ALD, and zinc supplement to alcohol fed animals showed beneficial effect (23, 35). A previous study demonstrated that zinc supplementation could partially rescue alcohol consumption-induced morphological change of mitochondria (59). In addition, it has been shown that zinc is required to maintain mitochondrial complex IV content and ATP production in mammary gland (41). In the present study, we found that zinc deficiency reduced the expression of mitochondrial respiratory complexes I, III, IV, and V. Moreover, zinc deficiency also decreased the expression of PGC1α, NRF1, and TFAM and reduced the level of mtDNA. It has been reported that zinc plays an important role in cell proliferation via zinc proteins (39, 49); thus it is not surprising that zinc regulates mitochondrial biogenesis. However, the exact molecular mechanisms need to be further explored. It is noteworthy to mention that experimental zinc deprivation induced complex V expression instead of a decreased expression as found in the in vivo study. This controversial result indicates that other factors in combination with zinc deficiency may contribute to the reduction of mitochondrial respiratory complexes in rats chronically fed alcohol. In addition, we did not find an altered hsp60 in the in vitro study, which further suggests that other factors in addition to zinc must play a role in mitochondrial dysfunction after alcohol exposure. Further investigations are required to define the mechanisms by which alcohol reduces the mitochondrial complex V. Furthermore, oxidative stress, in addition to PGC1α inactivation, may also contribute to the reduction of mtDNA level by experimental zinc deficiency based on the observation from our group that experimental zinc deficiency by TPEN treatment increased MDA level in HepG2 cells (23). As a whole, we demonstrated that chronic alcohol consumption downregulates hepatic PGC1α, NRF1, and TFAM and mitochondrial respiratory complexes I, III, and IV, and zinc deficiency is likely a mediator of the alcohol effects.
The mitochondrion is the major site of ROS production. Normally, ∼1–2% of the oxygen consumed in mitochondria is converted into ROS (14, 27). It has been shown that mitochondrial respiratory complex I and III are the sites leaking electrons, which, in turn, react with oxygen to generate ROS in the mitochondrial matrix (38). However, under disease conditions, other mitochondrial respiratory complexes in addition to complex I and III may also contribute to mitochondrial origin of ROS generation. A previous study showed that defective complex II was mainly responsible for the overproduction of mitochondria-oriented ROS in mice with Rett syndrome (11). Furthermore, although it is unlikely that ROS can be generated during enzyme turnover of complex IV, one study demonstrated that cytochrome oxidase contributed to the production of ROS after myocardial ischemia/perfusion (37). In addition, Galati et al. (16) further showed that knockdown of cytochrome c oxidase subunit Vb increased mitochondrial ROS level. The exact site where defective cytochrome oxidase generates ROS is still unknown. We reported coexistence of ROS overproduction and MMP impairment in the ALD model, which seems contradictory to the existing theory that increased MMP is necessary for ROS production via enhancing proton backpressure and inducing electron leakage. Interestingly, previous studies also reported the same finding as ours in their disease models (5, 11). It is likely that, under disease conditions, the more ROS is generated through defective ETC, the more mitochondria are damaged. As a result, the total number of mitochondria is decreased and leads to decreased MMP. Meanwhile, the remaining mitochondria with impaired ETC generates more than usual ROS.
It is noteworthy to mention that NDUFB8 and UQCRC2 are accessory subunits of the complex I and complex III, respectively (34), and they are required to anchor the corresponding complexes. Previous studies have shown that selective inhibition of electron transfer from iron-sulfur center of complex I or cytochrome bH of complex III to ubiquinone with rotenone- or antimycin-induced ROS overproduction (2). In the present study, we selectively knocked down accessory subunit of complex I and III to understand whether noncatalytic subunit deficiency also contributes to dysfunction of complexes and subsequent ROS overproduction. In addition, MTCO1 is one of the core subunits of complex IV and plays catalytic role for the function of complex IV. It has been shown that increased ROS cause reduction of complex IV (3); meanwhile a few studies also showed that dysfunction of complex IV, under disease conditions, may contribute to ROS production (37). Therefore, the role of MTCO1 was assessed. In the present study, selective knockdown of either NDUFB8 (subunit of complex I), UQCRC2 (subunit of complex III), or MTCO1 (subunit of complex IV) increased ROS production and decreased MMP. The results demonstrated, for the first time, that defective complex IV, like defective complexes I and III, could cause overproduction of ROS in vitro. In accordance, the data suggests that alcohol-induced defect of ETC could be an important factor in the generation of oxidative stress after chronic alcohol feeding.
In summary, the present study demonstrated that chronic alcohol consumption impairs mitochondrial biogenesis and perturbs ETC, leading to generation of ROS. As illustrated in Fig. 8, experimental zinc deprivation reduced the expression of mitochondrial biogenesis regulators PGC1α, NRF1, and TFAM as well as mtDNA, which accounts for the reduction of mitochondrial respiratory complex proteins. Another important finding in the present study is that the reduction of mitochondrial respiratory complex IV may also contributes to ROS generation. However, it is still unclear whether increased production of ROS is directly produced through defective complex IV or other mechanisms are also involved. These results provide a novel insight into the role of zinc in mitochondrial biogenesis and function and a new evidence of zinc therapy for treating ALD.
Fig. 8.
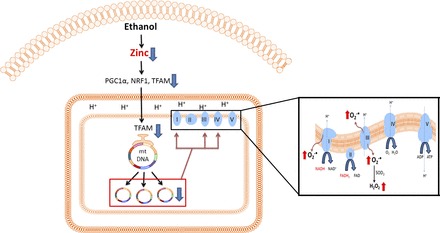
Schematic hypothesis on zinc deprivation-induced accumulation of ROS based on the results of the present study. Alcohol consumption induces a decrease in zinc level in the liver. Zinc deficiency downregulates expression of PGC1α, NRF1, and TFAM, which results in reduced mitochondrial biogenesis and mtDNA replication. Impairment of mtDNA leads to defect of mitochondrial respiratory complexes, namely complexes I, III, and IV. As a result, defective complexes I, III, and IV causes ROS overproduction in alcoholic liver disease.
GRANTS
This research was supported by the National Institutes of Health (R01AA020212 and 2R01AA018844).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Q.S. and Z.Z. conception and design of research; Q.S. performed experiments; Q.S. analyzed data; Q.S. and Z.Z. interpreted results of experiments; Q.S. prepared figures; Q.S. drafted manuscript; Q.S., W. Zhong, W. Zhang, and Z.Z. edited and revised manuscript; Q.S., W. Zhong, W. Zhang, and Z.Z. approved final version of manuscript.
REFERENCES
- 1.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 295: G833–G842, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med 32: 11–16, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bansal S, Srinivasan S, Anandasadagopan S, Chowdhury AR, Selvaraj V, Kalyanaraman B, Joseph J, Avadhani NG. Additive effects of mitochondrion-targeted cytochrome CYP2E1 and alcohol toxicity on cytochrome c oxidase function and stability of respirosome complexes. J Biol Chem 287: 15284–15297, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 61: 599–610, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobba A, Amadoro G, Valenti D, Corsetti V, Lassandro R, Atlante A. Mitochondrial respiratory chain Complexes I and IV are impaired by beta-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 13: 298–311, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Cahill A, Baio DL, Ivester P, Cunningham CC. Differential effects of chronic ethanol consumption on hepatic mitochondrial and cytoplasmic ribosomes. Alcohol Clin Exp Res 20: 1362–1367, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Cahill A, Wang X, Hoek JB. Increased oxidative damage to mitochondrial DNA following chronic ethanol consumption. Biochem Biophys Res Commun 235: 286–290, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Cai H, Dikalov S, Griendling KK, Harrison DG. Detection of reactive oxygen species and nitric oxide in vascular cells and tissues: comparison of sensitivity and specificity. Methods Mol Med 139: 293–311, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Coleman WB, Cunningham CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim Biophys Acta 1058: 178–186, 1991. [DOI] [PubMed] [Google Scholar]
- 10.Coleman WB, Cunningham CC. Effects of chronic ethanol consumption on the synthesis of polypeptides encoded by the hepatic mitochondrial genome. Biochim Biophys Acta 1019: 142–150, 1990. [DOI] [PubMed] [Google Scholar]
- 11.De Filippis B, Valenti D, de Bari L, De Rasmo D, Musto M, Fabbri A, Ricceri L, Fiorentini C, Laviola G, Vacca RA. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: protective effect of CNF1. Free Radic Biol Med 83: 167–177, 2015. [DOI] [PubMed] [Google Scholar]
- 12.Evans MJ, Scarpulla RC. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev 4: 1023–1034, 1990. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann G, Maurice M, Husson JM, Fiessinger JN, Camilleri JP, Benhamou JP, Housset E. Hepatocyte giant mitochondria: an almost constant lesion in systemic scleroderma. Virchows Arch A Pathol Anat Histol 374: 215–227, 1977. [DOI] [PubMed] [Google Scholar]
- 14.Forman HJ, Kennedy JA. Role of superoxide radical in mitochondrial dehydrogenase reactions. Biochem Biophys Res Commun 60: 1044–1050, 1974. [DOI] [PubMed] [Google Scholar]
- 15.Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J Biol Inorg Chem 16: 1123–1134, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galati D, Srinivasan S, Raza H, Prabu SK, Hardy M, Chandran K, Lopez M, Kalyanaraman B, Avadhani NG. Role of nuclear-encoded subunit Vb in the assembly and stability of cytochrome c oxidase complex: implications in mitochondrial dysfunction and ROS production. Biochem J 420: 439–449, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141: 1572–1585, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Villafranca J, Guillen A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 90: 460–466, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Han D, Ybanez MD, Johnson HS, McDonald JN, Mesropyan L, Sancheti H, Martin G, Martin A, Lim AM, Dara L, Cadenas E, Tsukamoto H, Kaplowitz N. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem 287: 42165–42179, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol 33: 1–7, 2015. [DOI] [PubMed] [Google Scholar]
- 21.Huo L, Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol 21: 644–654, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104: 12017–12022, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, Zhou Z. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology 50: 1241–1250, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ki SH, Choi JH, Kim CW, Kim SG. Combined metadoxine and garlic oil treatment efficaciously abrogates alcoholic steatosis and CYP2E1 induction in rat liver with restoration of AMPK activity. Chem Biol Interact 169: 80–90, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Koch OR, Fusco S, Ranieri SC, Maulucci G, Palozza P, Larocca LM, Cravero AA, Farre SM, De Spirito M, Galeotti T, Pani G. Role of the life span determinant P66(shcA) in ethanol-induced liver damage. Lab Invest 88: 750–760, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest 106: 867–872, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem 279: 4127–4135, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Levene AP, Goldin RD. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 61: 141–152, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Liangpunsakul S, Rahmini Y, Ross RA, Zhao Z, Xu Y, Crabb DW. Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. Am J Physiol Gastrointest Liver Physiol 302: G515–G523, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liangpunsakul S, Sozio MS, Shin E, Zhao Z, Xu Y, Ross RA, Zeng Y, Crabb DW. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol 298: G1004–G1012, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med 44: 723–738, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mann RE, Smart RG, Govoni R. The epidemiology of alcoholic liver disease. Alcohol Res Health 27: 209–219, 2003. [PMC free article] [PubMed] [Google Scholar]
- 33.McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 57: 860–867, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta 1817: 851–862, 2012. [DOI] [PubMed] [Google Scholar]
- 35.Mohammad MK, Zhou Z, Cave M, Barve A, McClain CJ. Zinc and liver disease. Nutr Clin Pract 27: 8–20, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel VB, Cunningham CC. Altered hepatic mitochondrial ribosome structure following chronic ethanol consumption. Arch Biochem Biophys 398: 41–50, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem 281: 2061–2070, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raimundo N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol Med 20: 282–292, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Sandstead HH, Lanier VC Jr, Shephard GH, Gillespie DD. Zinc and wound healing. Effects of zinc deficiency and zinc supplementation. Am J Clin Nutr 23: 514–519, 1970. [DOI] [PubMed] [Google Scholar]
- 40.Schiavi A, Ventura N. The interplay between mitochondria and autophagy and its role in the aging process. Exp Gerontol 56: 147–153, 2014. [DOI] [PubMed] [Google Scholar]
- 41.Seo YA, Lopez V, Kelleher SL. A histidine-rich motif mediates mitochondrial localization of ZnT2 to modulate mitochondrial function. Am J Physiol Cell Physiol 300: C1479–C1489, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun Q, Li Q, Zhong W, Zhang J, Sun X, Tan X, Yin X, Sun X, Zhang X, Zhou Z. Dysregulation of hepatic zinc transporters in a mouse model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 307: G313–G322, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Q, Zhong W, Zhang W, Li Q, Sun X, Tan X, Sun X, Dong D, Zhou Z. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am J Physiol Gastrointest Liver Physiol 308: G757–G766, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan X, Sun X, Li Q, Zhao Y, Zhong W, Sun X, Jia W, McClain CJ, Zhou Z. Leptin deficiency contributes to the pathogenesis of alcoholic fatty liver disease in mice. Am J Pathol 181: 1279–1286, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thayer WS, Rubin E. Molecular alterations in the respiratory chain of rat liver after chronic ethanol consumption. J Biol Chem 256: 6090–6097, 1981. [PubMed] [Google Scholar]
- 46.Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, Bailey SM. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J Biol Chem 279: 22092–22101, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79: 208–217, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Voos W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta 1833: 388–399, 2013. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe T, Arakawa T, Fukuda T, Higuchi K, Kobayashi K. Zinc deficiency delays gastric ulcer healing in rats. Dig Dis Sci 40: 1340–1344, 1995. [DOI] [PubMed] [Google Scholar]
- 50.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem 282: 9777–9788, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 117: 4055–4066, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Young TA, Bailey SM, Van Horn CG, Cunningham CC. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol 41: 254–260, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, Zhong W, Sun X, Sun Q, Tan X, Li Q, Sun X, Zhou Z. Visceral white adipose tissue is susceptible to alcohol-induced lipodystrophy in rats: role of acetaldehyde. Alcohol Clin Exp Res 39: 416–423, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao L, Zou X, Feng Z, Luo C, Liu J, Li H, Chang L, Wang H, Li Y, Long J, Gao F, Liu J. Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp Gerontol 56: 3–12, 2014. [DOI] [PubMed] [Google Scholar]
- 56.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao Y, Zhong W, Sun X, Song Z, Clemens DL, Kang YJ, McClain CJ, Zhou Z. Zinc deprivation mediates alcohol-induced hepatocyte IL-8 analog expression in rodents via an epigenetic mechanism. Am J Pathol 179: 693–702, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, Tan X, Sun X, Jia W, Zhou Z. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J Hepatol 62: 1375–1381, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol 166: 1681–1690, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med (Maywood) 237: 474–480, 2012. [DOI] [PubMed] [Google Scholar]