

Abstract
The inflammatory diseases ulcerative colitis and Crohn's disease constitute the two main forms of inflammatory bowel disease (IBD). They are characterized by chronic, relapsing inflammation of the gastrointestinal tract, significantly impacting on patient quality of life and often requiring prolonged treatment. Existing therapies for IBD are not effective for all patients, and an unmet need exists for additional therapies to induce and maintain remission. Here we describe the mechanism of action of the Janus kinase (JAK) inhibitor, tofacitinib, for the treatment of IBD and the effect of JAK inhibition on the chronic cycle of inflammation that is characteristic of the disease. The pathogenesis of IBD involves a dysfunctional response from the innate and adaptive immune system, resulting in overexpression of multiple inflammatory cytokines, many of which signal through JAKs. Thus JAK inhibition allows multiple cytokine signaling pathways to be targeted and is expected to modulate the innate and adaptive immune response in IBD, thereby interrupting the cycle of inflammation. Tofacitinib is an oral, small molecule JAK inhibitor that is being investigated as a targeted immunomodulator for IBD. Clinical development of tofacitinib and other JAK inhibitors is ongoing, with the aspiration of providing new treatment options for IBD that have the potential to deliver prolonged efficacy and clinically meaningful patient benefits.
Keywords: Crohn's disease, inflammatory bowel disease, ulcerative colitis, inflammation
inflammatory bowel disease (IBD) is a chronic, idiopathic condition with two main forms: ulcerative colitis (UC) and Crohn's disease (CD). A recent review of the global epidemiology of IBD showed increasing incidence and prevalence of both forms of the disease worldwide; for UC, the highest reported prevalence values were 505, 168, and 249 cases per 100,000 people for Europe, Asia and the Middle East, and North America, respectively; and for CD, 322, 68, and 319 cases per 100,000 people, respectively (36). In addition to a small associated increase in mortality (7), the impact of IBD on patient quality of life is high (24), with alternating periods of relapse and remission a feature of both UC and CD (3, 47).
UC is associated with diffuse mucosal inflammation affecting the colon (47), while, in CD, inflammation may be transmural and can affect any region of the gastrointestinal tract (3). The exaggerated immune response observed in affected tissues, characteristic of both forms of IBD, has a multifactorial pathogenesis driven by the imbalanced production of proinflammatory cytokines (53). The purpose of this article is to discuss the cytokine pathways central to IBD pathogenesis and, subsequently, to review the evidence and hypotheses for the mechanism of action of Janus kinase (JAK) inhibitors for the treatment of IBD.
CURRENT TREATMENTS AND GUIDELINES FOR IBD
Appropriate choice of therapy for IBD depends on multiple factors, including disease severity, response to previous treatment, and comorbidities, with the goals of therapeutic intervention being to prevent intestinal damage and to induce and maintain steroid-free remission of symptoms (63, 64). More recently, the promotion of mucosal healing has emerged as a new goal of treatment and predicts sustained clinical remission without surgical intervention (45, 57).
Current Therapies and Unmet Needs for the Treatment of IBD
Existing conventional therapies for the treatment of IBD include aminosalicylates, corticosteroids, and immunosuppressive agents, such as azathioprine, mercaptopurine, and methotrexate (29, 35). Advances made during the 1990s in understanding the inflammatory cascade and the role of cytokines and cell adhesion molecules in IBD pathogenesis led to the introduction of the first disease-modifying biological drugs for the treatment of the disease. Tumor necrosis factor inhibitors (TNFi) infliximab, adalimumab, and golimumab are available for the treatment of UC (29), whereas infliximab, adalimumab, and certolizumab are indicated for the treatment of CD (73). TNFi therapy inhibits signaling elicited by TNF-α, one of the key mediating cytokines of intestinal tract inflammation in IBD, and is effective in the management of acute disease for many patients. Therapies that target integrin molecules (responsible for mediating cell-cell interactions between endothelial cells and leukocytes) represent a novel approach to the treatment of IBD. Vedolizumab, a humanized monoclonal antibody against the α4β7-integrin, was efficacious in UC and CD (14, 65), and further agents within this class of drug are in clinical development (11).
The efficacy of combination therapy for the treatment of IBD has been demonstrated in two recent clinical trials. In the UC SUCCESS (49) and SONIC trials (10), combination therapy with infliximab and azathioprine was superior to azathioprine monotherapy in inducing steroid-free remission in patients with moderate to severe UC and CD, respectively.
Despite the range of treatment options available, induction and maintenance of remission remains problematic with both conventional and biological therapies (56). In randomized controlled trials of infliximab, aminosalicylates, and thiopurines, maintenance of remission was observed in 33, 53, and 60%, respectively, of patients with UC (23, 62, 72). Similarly, 43, 45, 70, and 71% of patients with CD maintained remission in randomized controlled trials of adalimumab, infliximab, methotrexate, and azathioprine, respectively (52, 55, 59). Furthermore, ∼20% of patients with CD or UC still require surgical treatment, despite biological therapy (15, 56, 70). Hence, there remains a need for new therapies, resulting in better tolerability and greater long-term efficacy.
Currently approved TNFi therapies target inflammation through inhibition of a single cytokine, with long-lasting effect. Given the involvement of multiple cytokine-driven inflammatory pathways in the pathogenesis of IBD, a therapeutic approach blocking multiple cytokines could be effective (16, 37). Such an approach could be achieved with a JAK inhibitor.
The discovery of the JAK family of intracellular tyrosine kinases (TYKs), reviewed by Ghoreschi et al. (22), and elucidation of their role in cytokine signaling pathways has led to their identification as potential therapeutic targets in the treatment of IBD. Successful development of JAK inhibitors will rely on further understanding of their mode of action based on a sound understanding of the pathogenesis of IBD.
INFLAMMATORY CYTOKINES INVOLVED IN THE PATHOGENESIS OF IBD
The pathogenesis of IBD is multifactorial and comprises genetic, environmental, microbial, and immune response components (79). In health, the gut microbiota and immune cells resident in the lining of the gastrointestinal tract exist in equilibrium. The intestinal mucosa forms a barrier layer mediating this equilibrium by preventing microorganisms within the gut lumen from entering host tissue, with intact barrier function necessary for the prevention of uncontrolled inflammation. The pathogenesis of IBD is related to altered barrier function in the form of structural changes to intestinal epithelial cells (44). Subsequently, substantial changes in the composition of gut microbiota are observed, termed “dysbiosis,” eliciting an inflammatory response from the immune system. The initial innate immune response in IBD involves activation of immune cells, including macrophages, dendritic cells, neutrophils, and natural killer cells, in reaction to the presence of microbial antigens. Subsequent antigen presentation to T cells stimulates the adaptive immune response, which varies between UC and CD, according to the prevailing T helper (Th) cell profile in each disease (69, 79). Th cells mediate the continued inflammatory response in both cases, with Th17 cells having been identified as key effectors in the immunopathogenesis of IBD. They are involved in the production of proinflammatory cytokine interleukin (IL)-17 and also display functional plasticity, exhibiting a regulatory Th17/Th1 phenotype in which IL-17 production is suppressed and interferon-γ (IFN-γ) is secreted (18, 34). Thus there is a balance between the proinflammatory Th17 response and anti-inflammatory regulatory response, which may be important when considering treatment with IL-17-suppressing agents.
Dysfunction of both innate and adaptive immune response pathways contributes to the inflammatory response (20), and cytokines are central to the mediation of the cell-cell interactions at the heart of these responses (20, 71). A continuing immune response to commensal microbes results in the cytokine-mediated chronic cycle of inflammation characteristic of IBD, challenging mucosal homeostasis (38, 53). In the late stage of the disease process, continued accumulation of myofibroblasts at locations where barrier function has been compromised may lead to intestinal fibrosis and the formation of fistulae (38). The optimal timing of intervention with a JAK inhibitor with respect to disease stage remains to be studied in the clinic, but may be an important concept, given the potential for variation in IBD immunophenotypes between early and late-stage disease (31). The immunopathogenesis of IBD is detailed in Fig. 1, and cytokines involved in the innate and adaptive immune responses in the disease are summarized in Fig. 2. Key cytokines in the immune response include TNF-α, IFN-γ, IL-1β, IL-6, IL-12, IL-13, IL-17, and IL-2 (1, 25, 69), as well as IL-15 (48). Thus therapeutic approaches targeting multiple cytokines in combination may be a viable new approach for the treatment of IBD (5).
Fig. 1.
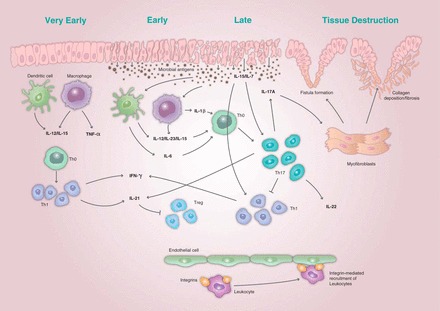
The immunopathogenesis of inflammatory bowel disease (IBD) showing the initial immune response and amplification of the immune response leading to the phenotypic expression of the disease and tissue destruction. Recruitment of leukocytes is mediated by interactions between adhesion molecules known as integrins and the intestinal endothelium. Structural changes to the epithelial barrier result in microbial antigens from the gut lumen entering the host tissue. The initial immune response to the presence of these antigens involves production of interleukin (IL)-7, IL-12, and IL-15, and activation of immune cells, including dendritic cells and macrophages. This results in a T helper (Th) 1 cell response and production of tumor necrosis factor (TNF)-α and interferon (IFN)-γ. The subsequent adaptive immune response results in an amplification of the initial inflammatory response characterized by high levels of IL-6 and IL-23 and the Th17-mediated production of IL-17A, IL-21, and IL-22. The accumulation of myofibroblasts in response to the proliferation of multiple inflammatory cytokines leads to an imbalance in the normal epithelial healing process, resulting in fibrosis and the formation of fistulae. Treg, T regulatory cell.
Fig. 2.
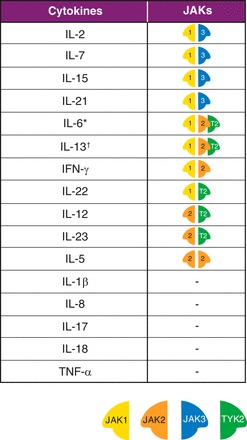
Key cytokines in IBD and Janus kinase (JAK) combinations for cytokines that depend on JAK pathways for signaling. The pairing of JAKs is significant to their role in cytokine signaling, with each member of the JAK family exhibiting preferential binding to the intracellular domains of individual cytokine receptor chains. JAK1 pairs with JAK3 to control signaling of the γ-common cytokines, including IL-2, IL-7, IL-15, and IL-21. Type II cytokine receptors, such as those for the gp130 subunit-sharing receptors, include IL-6 signal mainly through JAK1, but are also associated with JAK2 and tyrosine kinase (TYK) 2. IL-13 signals are through receptors associated with JAK1 and either JAK2 or TYK2. JAK2 is unique in pairing with itself and controls signaling for IL-5. Other key cytokines in IBD that signal using JAKs include IFN-γ (JAK1/JAK2), IL-22 (JAK1/TYK2), and IL-12 and IL-23 (JAK2/TYK2). IL-1β, IL-8, IL-17, IL-18, and TNF-α are other important cytokines in IBD, but do not signal directly using JAKs. An important indirect effect of JAK2/TYK2 blockade for IBD is the downstream inhibition of IL-17 production achieved through direct inhibition of IL-23 signaling. *Type II cytokine receptors, such as those for the gp130 subunit-sharing receptors, including IL-6, signal mainly through JAK1, but are also associated with JAK2 and TYK2. †IL-13 signals are through receptors associated with JAK1 and either JAK2 or TYK2.
JAK INHIBITION OF MULTIPLE INFLAMMATORY PATHWAYS
A large number of cytokines operate by activating JAK-signal transducers and activators of transcription (JAK-STAT) pathways (46). The JAK family comprises four intracellular protein TYKs: JAK1, JAK2, JAK3, and TYK2 (22). Cytokine binding to its receptor at the surface of a cell leads to intracellular activation of JAKs, followed by phosphorylation and activation of STATs. Phosphorylated dimerized STATs then translocate to the cell nucleus, where they modulate gene transcription that ultimately alters aspects of cellular function, including growth, maturation, differentiation, and survival, in addition to inflammatory and immune responses (39). Cytokines signal through activation of JAKs in pairs, with each JAK associated with different intracellular domains of individual cytokine receptor chains. As a consequence, different combinations of JAKs are associated with different cytokine receptors. Figure 2 shows cytokine signaling pathways in IBD and the associated JAK combinations.
Cellular functions associated with the immune response in IBD are regulated by multiple different cytokines, which, in turn, signal through different combinations of JAKs. IL-12 and IL-23 signal through the combination of JAK2 and TYK2 and regulate innate immune responses, differentiation of Th1 and Th17, and production of proinflammatory cytokines (32). IFN-γ is an important cytokine in the intestinal response against bacterial pathogens and activates the JAK1/JAK2 combination. The γ-common cytokines, IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, all signal through the activation of JAK1/JAK3 and are associated with modulation of the adaptive immune function, including B-cell maturation and Th1, Th2, and Th17 differentiation (27). IL-13 signals through JAK1 and either JAK2 or TYK2 and is an important effector cytokine associated with impaired barrier function in IBD (25). In addition to direct inhibition of cytokine signaling, JAK blockade may also achieve downstream inhibition of cytokine synthesis by reducing the production of activator cytokines; an important example in the IBD disease process is the IL-23-stimulated production of IL-17 (28). As a result, JAK pathways are central to both innate and adaptive immune responses in IBD, playing critical roles in T-cell differentiation, B-cell development, and production of mucus and antibodies required to maintain anti-viral and anti-bacterial defenses at the mucosal surface (26). Accordingly, a blockade of JAKs results in modulation of the adaptive and immune response found in IBD and may, therefore, be effective in interrupting the chronic cycle of gastrointestinal inflammation (8, 16, 21, 66).
Owing to the role that JAKs play in functions such as cell growth, maturation, differentiation, and hematopoiesis, it is necessary to balance the inhibition of JAK pathways involved in disease pathogenesis with the potential negative effects of JAK inhibition. Each of the protein kinases is responsible for various aspects of the innate and adaptive immune responses. As such, JAK inhibition may lead to increased risk of infection adverse events. Inhibition of JAK2 may potentially lead to neutropenia, anemia, and thrombocytopenia due to the involvement of JAK2 in erythropoiesis (41). JAK3 associates only with a single cytokine receptor chain (the common γ-chain) and appears to have the most discrete function, having no role outside of hematopoietic cells. As a consequence, targeting JAK3 may be advantageous in suppressing the inflammatory response associated with the γ-common cytokines, with less scope for side effects resulting from the inhibition of cellular processes associated with other JAKs (54).
Existing biological therapies inhibit cytokine signaling by binding and neutralizing either the cytokine itself or its receptor (61). None of the currently approved biologic therapies targets the intracellular components of cytokine signaling pathways. Thus targeting JAK-STAT pathways offers an alternative approach to combination therapy, with the potential to block multiple cytokine signals by inhibition of a common signal transduction pathway. In addition to targeting multiple cytokines, a downstream approach to cytokine inhibition is characterized by its ability to achieve partial and reversible inhibition through appropriate dosing. In contrast to biological therapies, which effect near-complete inhibition of cytokine signaling during a dosing period, intracellular inhibition with a small molecule affords the ability to modulate JAK-dependent signaling downstream, allowing the potential for greater control of the inflammatory and immunomodulatory response throughout the dosing period (9). This partial and reversible inhibition of multiple cytokine pathways with a small molecule, compared with inhibition of a single cytokine pathway with a biological therapy, could offer a differentiated therapeutic profile.
JAK Inhibition with Tofacitinib
Tofacitinib is an oral, small-molecule, ATP-competitive reversible inhibitor of JAKs that is being investigated as a targeted immunomodulator for IBD. It has a short pharmacokinetic half-life of ∼3 h and is rapidly absorbed (time to peak concentration is ∼0.5 h) and eliminated (13). In a cellular setting, tofacitinib demonstrates preferential inhibition of signaling pathways associated with JAK1 and/or JAK3 (43). With tofacitinib 5 mg twice daily (bid), average inhibition of preferentially inhibited cytokines is 50–60%, declining to 10–30% at trough concentration during each dosing period (12). Reversibility of pharmacodynamic effects is generally seen within 14 days of tofacitinib discontinuation (19). Unlike biological therapies, tofacitinib does not elicit neutralizing antibodies through immunogenicity, and data for tofacitinib in rheumatoid arthritis (RA) are available, showing sustained responses for up to 7 yr (77). It is likely, therefore, that tofacitinib may have utility in patients experiencing inadequate response to biological therapies, as well as those not yet exposed to biologicals. It is currently approved in many territories for the treatment of adults with moderate to severe RA and is being investigated for a number of other inflammatory diseases, including psoriasis, psoriatic arthritis, UC, and CD.
As with IBD, the pathogenesis of RA is mediated by a number of inflammatory cytokines (40). Inhibition of JAK1 and JAK3 with tofacitinib blocks signaling for IL-2, IL-4, IL-6, IL-7, IL-9, IL-15, IL-21, and IFN-γ cytokines, some of which are integral to the activation of the immune response in RA pathophysiology (22, 42). The efficacy of tofacitinib, either as monotherapy or in combination with nonbiological disease-modifying antirheumatic drugs, for the treatment of RA has been demonstrated across phase 3 and long-term extension studies (6, 17, 30, 33, 75, 76, 78).
In psoriasis, JAK inhibition is expected to attenuate T-cell function and differentiation and the production of IL-6, IL-21, IL-23, and IFN-γ cytokines, which play a key role in the activation of Th1 and Th17 cells, characteristic of the inflammatory psoriatic phenotype (60). One phase 2 study (50) and four phase 3 studies (2, 4, 51) have demonstrated the efficacy of oral tofacitinib compared with placebo for the treatment of plaque psoriasis, highlighting the importance of JAKs as therapeutic targets in this immune-mediated inflammatory disease.
Based on the modulation of the cytokine-mediated immune response observed in other inflammatory diseases, targeting the JAK pathway in IBD with tofacitinib represents a promising therapeutic approach, and initial clinical trials investigating tofacitinib treatment for UC are encouraging and, taken together with the potential signals of efficacy observed with tofacitinib in CD, provide a rationale for the importance of the ongoing clinical development program in IBD (UC: NCT01465763, NCT01458951, NCT01458574, NCT01470612; CD: NCT01470599). In an 8-wk placebo-controlled phase 2 trial of 194 patients with moderate to severely active UC (66), patients received tofacitinib at a dose between 0.5 and 15 mg bid. The primary endpoint of clinical response was defined as an absolute decrease of at least three points from baseline total Mayo score, or relative decrease of at least 30%, with an accompanying decrease in rectal bleeding subscore of at least 1 point or absolute subscore of 0 or 1. Significant improvement vs. placebo in the primary endpoint and secondary endpoints (clinical remission, endoscopic response, and endoscopic remission) were observed with tofacitinib 15 mg bid. Tofacitinib 10 mg bid demonstrated significantly greater efficacy in the majority of secondary endpoints. Reductions in fecal calprotectin and C-reactive protein, and improvements in the IBD Questionnaire score were also observed with tofacitinib treatment. Frequently reported adverse events were nasopharyngitis and influenza, and there were dose-dependent increases in serum lipids. In a shorter 4-wk placebo-controlled study of patients with CD (67), tofacitinib was administered at a dose between 1 and 15 mg bid. The primary efficacy endpoint was clinical response defined as a decrease from baseline ≥70 points in Crohn's Disease Activity Index at week 4. Tofacitinib did not demonstrate a clinical response compared with placebo; however, response and remission rates with placebo were higher than expected, and observed reductions in C-reactive protein and fecal calprotectin (68) associated with tofacitinib treatment were suggestive of its biological activity.
Given its novel immunomodulatory mode of action, evaluation of the long-term safety of tofacitinib has been a focus of its clinical development program. In patients with moderate to severe RA, consistent safety up to 72 mo has been demonstrated in open-label extension studies of tofacitinib dosed at 5 mg bid and tofacitinib 10 mg bid (77, 78). Incidence rates for safety events of special interest, including serious infections, opportunistic infections, tuberculosis, cardiovascular events, malignancy, and mortality, were stable over the duration of phase 3 and open-label extension studies and were consistent with rates observed with other biological disease-modifying antirheumatic drugs (6, 17, 30, 33, 75, 76, 78). Changes in clinical laboratory parameters, including neutrophil, lymphocyte and platelet counts, hemoglobin, creatine kinase, serum creatinine, and serum lipids (low-density lipoprotein and high-density lipoprotein cholesterol), were generally stable during long-term treatment and reversible with appropriate medical management (78). In phase 2 and phase 3 studies of up to 52 wk of tofacitinib for patients with moderate to severe chronic plaque psoriasis (2, 4, 50, 51, 58), tofacitinib 5 mg bid and tofacitinib 10 mg bid have been well tolerated, with no new safety findings observed with respect to the RA program. In phase 2 studies of UC and CD (66, 67), dose-dependent increases in low-density lipoprotein and high-density lipoprotein cholesterol levels were observed with tofacitinib, consistent with observations in tofacitinib RA and psoriasis studies; however, the UC and CD studies had relatively short durations and sample sizes.
Other JAK Inhibitors in Development
The current stage of development and associated clinical programs of other JAK inhibitors are summarized in Fig. 3. As the clinical development programs of tofacitinib and other JAK inhibitors progress, they will provide further mechanistic information regarding the role of JAK pathways in cytokine-mediated diseases. Comparisons of their relative efficacy, tolerability, and safety will be important in determining optimal dosing and the specificity of their effect.
Fig. 3.
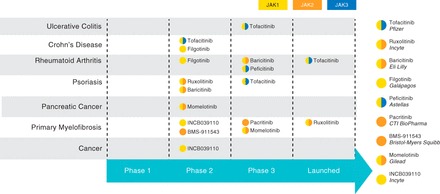
JAK inhibitors, showing all JAK inhibitor profiles, their current stage of development, and their reported JAK selectivity. Currently, tofacitinib (a JAK1/JAK3 inhibitor) and filgotinib (a JAK1 inhibitor) are the only agents undergoing clinical testing for IBD indications. Other JAK inhibitors in advanced stages of clinical development for the treatment of cytokine-mediated diseases include JAK1/JAK2 inhibitors baricitinib, momelotinib, and ruxolitinib (the latter of which is approved for the treatment of myelofibrosis), JAK1/JAK3 inhibitor peficitinib, and JAK2 inhibitor pacritinib [Source: Thomson Reuters Cortellis (74)]. Reported selectivity is indicated for each JAK inhibitor.
CONCLUSIONS
IBD is a chronic, inflammatory disease regulated by multiple cytokine signaling pathways central to the innate and adaptive response of the immune system. Rates of induction and maintenance of remission in IBD indicate that there is still a need for new therapies with alternative modes of action that are able to demonstrate long-term efficacy and allow steroid-free remission. JAK-STAT pathways play an important role in the inflammatory response characteristic of IBD and represent a promising therapeutic target for treatment of the disease. Importantly, JAK inhibition targets multiple cytokine pathways. Potential benefits of JAK inhibition relating to the intracellular mechanism of action include the ability to achieve inhibition with a small molecule, offering the potential for oral dosing, and the ability to modulate the extent of cytokine inhibition during a dosing period. Potential side effects of JAK inhibitors arising from the pleiotropic nature of JAK-STAT signaling pathways include increased rate of infections and malignancies and changes in laboratory parameters, including neutrophil and lymphocyte cell counts, platelets, and liver enzymes. Initial results from clinical development programs investigating JAK inhibitors have demonstrated signals of efficacy in both UC and CD, with safety profiles generally similar to existing therapies, and no new safety signals with respect to their use in other indications. With continued clinical development, it is hoped that new therapies will move IBD treatment forward and provide additional clinically meaningful benefits to patients with IBD. Key areas to consider in the future development of JAK inhibitors for IBD will be the potentially disparate effect of JAK inhibitors between UC and CD, a greater understanding of the safety and tolerability of JAK inhibitors specific to their use in IBD indications, and the positioning of JAK inhibitors within treatment guidelines.
GRANTS
M. Grisham is grateful for support provided by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-091269 and Department of Defense Grant PR100317.
DISCLOSURES
J.-B. Telliez and J. Hodge are employees of Pfizer Inc. M. Grisham has worked on contract projects for Fibrogen Inc. Medical writing support was provided under the guidance of the authors by Daniel Binks, PhD, of Complete Medical Communications and was funded by Pfizer Inc.
AUTHOR CONTRIBUTIONS
Author contributions: S.D., M.B.G., J.H., and J.-B.T. conception and design of research; S.D., M.B.G., J.H., and J.-B.T. prepared figures; S.D., M.B.G., J.H., and J.-B.T. drafted manuscript; S.D., M.B.G., J.H., and J.-B.T. edited and revised manuscript; S.D., M.B.G., J.H., and J.-B.T. approved final version of manuscript.
REFERENCES
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med 361: 2066–2078, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachelez H, van de Kerkhof PCM, Strohal R, Kubanov A, Valenzuela F, Lee JH, Yakusevich V, Chimenti S, Papacharalambous J, Proulx J, Gupta P, Tan H, Tawadrous M, Valdez H, Wolk R. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet 386: 552–561, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart D, Sandborn WJ. Crohn's disease. Lancet 380: 1590–1605, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Bissonnette R, Iversen L, Sofen H, Griffiths CEM, Foley P, Romiti R, Bachinsky M, Rottinghaus ST, Tan H, Proulx J, Valdez H, Gupta P, Mallbris L, Wolk R. Tofacitinib withdrawal and retreatment in moderate-to-severe chronic plaque psoriasis: a randomized controlled trial. Br J Dermatol 172: 1395–1406, 2015. [DOI] [PubMed] [Google Scholar]
- 5.Boland BS, Sandborn WJ, Chang JT. Update on Janus kinase antagonists in inflammatory bowel disease. Gastroenterol Clin North Am 43: 603–617, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, Gruben D, Wallenstein G, Krishnaswami S, Zwillich SH, Koncz T, Soma K, Bradley J, Mebus C; ORAL Step Investigators . Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 381: 451–460, 2013. [DOI] [PubMed] [Google Scholar]
- 7.Card T, Hubbard R, Logan RF. Mortality in inflammatory bowel disease: a population-based cohort study. Gastroenterology 125: 1583–1590, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Changelian PS, Moshinsky D, Kuhn CF, Flanagan ME, Munchhof MJ, Harris TM, Whipple DA, Doty JL, Sun J, Kent CR, Magnuson KS, Perregaux DG, Sawyer PS, Kudlacz EM. The specificity of JAK3 kinase inhibitors. Blood 111: 2155–2157, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem 57: 5023–5038, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, Lichtiger S, D'Haens G, Diamond RH, Broussard DL, Tang KL, van der Woude CJ, Rutgeerts P. Infliximab, azathioprine, or combination therapy for Crohn's disease. N Engl J Med 362: 1383–1395, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Danese S, Panes J. Development of Drugs to Target Interactions Between Leukocytes and Endothelial Cells and Treatment Algorithms for Inflammatory Bowel Diseases. Gastroenterology 147: 981–989, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Dowty ME, Jesson MI, Ghosh S, Lee J, Meyer DM, Krishnaswami S, Kishore N. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J Pharmacol Exp Ther 348: 165–173, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Dowty ME, Lin J, Ryder TF, Wang W, Walker GS, Vaz A, Chan GL, Krishnaswami S, Prakash C. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a Janus kinase inhibitor, in humans. Drug Metab Dispos 42: 759–773, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, Van AG, Axler J, Kim HJ, Danese S, Fox I, Milch C, Sankoh S, Wyant T, Xu J, Parikh A. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 369: 699–710, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Filippi J, Allen PB, Hebuterne X, Peyrin-Biroulet L. Does anti-TNF therapy reduce the requirement for surgery in ulcerative colitis? A systematic review. Curr Drug Targets 12: 1440–1447, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Flanagan ME, Blumenkopf TA, Brissette WH, Brown MF, Casavant JM, Shang-Poa C, Doty JL, Elliott EA, Fisher MB, Hines M, Kent C, Kudlacz EM, Lillie BM, Magnuson KS, McCurdy SP, Munchhof MJ, Perry BD, Sawyer PS, Strelevitz TJ, Subramanyam C, Sun J, Whipple DA, Changelian PS. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem 53: 8468–8484, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, Gruben D, Wallenstein GV, Zwillich SH, Kanik KS; ORAL Solo Investigators . Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 367: 495–507, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Galvez J. Role of Th17 cells in the pathogenesis of human IBD. ISRN Inflamm 2014: 928461, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese MC, Kawabata T, Soma K, Menon S, Clark JD, Hodge J, Takiya L, Riese R, Krishnaswami S. Reversibility of pharmacodynamic effects after short- and long-term treatment with tofacitinib in patients with rheumatoid arthritis (Abstract). Arthritis Rheum 65: S193, 2013. [Google Scholar]
- 20.Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev 13: 3–10, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, Warner JD, Tanaka M, Steward-Tharp SM, Gadina M, Thomas CJ, Minnerly JC, Storer CE, LaBranche TP, Radi ZA, Dowty ME, Head RD, Meyer DM, Kishore N, O'Shea JJ. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol 186: 4234–4243, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev 228: 273–287, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gisbert JP, Linares PM, McNicholl AG, Mate J, Gomollon F. Meta-analysis: the efficacy of azathioprine and mercaptopurine in ulcerative colitis. Aliment Pharmacol Ther 30: 126–137, 2009. [DOI] [PubMed] [Google Scholar]
- 24.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis 12, Suppl 1: S3–S9, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, Zeitz M, Fuss I, Strober W, Schulzke JD. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129: 550–564, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Heneghan AF, Pierre JF, Kudsk KA. JAK-STAT and intestinal mucosal immunology. JAKSTAT 2: e25530, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofmann SR, Ettinger R, Zhou YJ, Gadina M, Lipsky P, Siegel R, Candotti F, O'Shea JJ. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr Opin Allergy Clin Immunol 2: 495–506, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity 21: 467–476, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Kornbluth A, Sachar DB. Ulcerative colitis practice guidelines in adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol 105: 501–523, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin-Mola E, Isaacs JD, Gruben D, Wallenstein G, Krishnaswami S, Zwillich SH, Koncz T, Riese R, Bradley J. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 159: 253–261, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Kugathasan S, Saubermann LJ, Smith L, Kou D, Itoh J, Binion DG, Levine AD, Blumberg RS, Fiocchi C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut 56: 1696–1705, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langrish CL, McKenzie BS, Wilson NJ, de Waal MR, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev 202: 96–105, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley J, Gruben D, Koncz T, Krishnaswami S, Wallenstein G, Zang C, Zwillich S, van Vollenhoven R, on behalf of the ORAL Start Investigators . Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 370: 2377–2386, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity 30: 92–107, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lichtenstein GR, Hanauer SB, Sandborn WJ. Management of Crohn's disease in adults. Am J Gastroenterol 104: 465–483, 2009. [DOI] [PubMed] [Google Scholar]
- 36.M'Koma AE. Inflammatory bowel disease: an expanding global health problem. Clin Med Insights Gastroenterol 6: 33–47, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacDonald TT, Biancheri P, Sarra M, Monteleone G. What's the next best cytokine target in IBD? Inflamm Bowel Dis 18: 2180–2189, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474: 298–306, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Mavers M, Ruderman EM, Perlman H. Intracellular signal pathways: potential for therapies. Curr Rheumatol Rep 11: 378–385, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 7: 429–442, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Menet CJ, Rompaey LV, Geney R. Advances in the discovery of selective JAK inhibitors. Prog Med Chem 52: 153–223, 2013. [DOI] [PubMed] [Google Scholar]
- 42.Meyer D, Head R, Thompson J, Ghosh S, LaBranche T, Storer C, Minnerly J, Jesson M, Kishore N. Mechanism of action of the JAK inhibitor, CP-690550, in rheumatoid arthritis. In: Proceedings of the 8th Cytokines and Inflammation Conference, San Diego, California, 28 January, 2010. [Google Scholar]
- 43.Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, Gross CJ, Dowty ME, Ramaiah SK, Hirsch JL, Saabye MJ, Barks JL, Kishore N, Morris DL. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm (Lond) 7: 41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol 7: 6–19, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut 61: 1619–1635, 2012. [DOI] [PubMed] [Google Scholar]
- 46.O'Shea JJ. Targeting the Jak/STAT pathway for immunosuppression. Ann Rheum Dis 63, Suppl 2: ii67–ii71, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ordàs I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet 380: 1606–1619, 2012. [DOI] [PubMed] [Google Scholar]
- 48.Pagliari D, Cianci R, Frosali S, Landolfi R, Cammarota G, Newton EE, Pandolfi F. The role of IL-15 in gastrointestinal diseases: a bridge between innate and adaptive immune response. Cytokine Growth Factor Rev 24: 455–466, 2013. [DOI] [PubMed] [Google Scholar]
- 49.Panaccione R, Ghosh S, Middleton S, Marquez JR, Scott BB, Flint L, van Hoogstraten HJ, Chen AC, Zheng H, Danese S, Rutgeerts P. Combination therapy with infliximab and azathioprine is superior to monotherapy with either agent in ulcerative colitis. Gastroenterology 146: 392–400, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Papp KA, Menter A, Strober B, Langley RG, Buonanno M, Wolk R, Gupta P, Krishnaswami S, Tan H, Harness JA. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol 167: 668–677, 2012. [DOI] [PubMed] [Google Scholar]
- 51.Papp KA, Menter MA, Abe M, Elewski B, Feldman SR, Gottlieb AB, Langley R, Luger T, Thaci D, Buonanno M, Gupta P, Proulx J, Lan S, Wolk R. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two, randomised, placebo-controlled, Phase 3 trials. Br J Dermatol 173: 949–961, 2015. [DOI] [PubMed] [Google Scholar]
- 52.Patel V, Macdonald JK, McDonald JW, Chande N. Methotrexate for maintenance of remission in Crohn's disease. Cochrane Database Syst Rev 8: CD006884, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pedersen J, Coskun M, Soendergaard C, Salem M, Nielsen OH. Inflammatory pathways of importance for management of inflammatory bowel disease. World J Gastroenterol 20: 64–77, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev 223: 132–142, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn's disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 6: 644–653, 2008. [DOI] [PubMed] [Google Scholar]
- 56.Peyrin-Biroulet L, Lemann M. Review article: remission rates achievable by current therapies for inflammatory bowel disease. Aliment Pharmacol Ther 33: 870–879, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Pineton de Chambrun G, Peyrin-Biroulet L, Lemann M, Colombel JF. Clinical implications of mucosal healing for the management of IBD. Nat Rev Gastroenterol Hepatol 7: 15–29, 2010. [DOI] [PubMed] [Google Scholar]
- 58.Ports WC, Khan S, Lan S, Lamba M, Bolduc C, Bissonnette R, Papp K. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol 169: 137–145, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prefontaine E, Sutherland LR, Macdonald JK, Cepoiu M. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn's disease. Cochrane Database Syst Rev 1: CD000067, 2009. [DOI] [PubMed] [Google Scholar]
- 60.Racz E, Prens EP. Molecular pathophysiology of psoriasis and molecular targets of antipsoriatic therapy. Expert Rev Mol Med 11: e38, 2009. [DOI] [PubMed] [Google Scholar]
- 61.Rosman Z, Shoenfeld Y, Zandman-Goddard G. Biologic therapy for autoimmune diseases: an update. BMC Med 11: 88, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJ, Present D, Sands BE, Colombel JF. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 353: 2462–2476, 2005. [DOI] [PubMed] [Google Scholar]
- 63.Sandborn WJ. Step-up versus top-down therapy in the treatment of ulcerative colitis. Gastroenterol Hepatol (N Y) 3: 16–17, 2007. [PMC free article] [PubMed] [Google Scholar]
- 64.Sandborn WJ, Feagan BG, Lichtenstein GR. Medical management of mild to moderate Crohn's disease: evidence-based treatment algorithms for induction and maintenance of remission. Aliment Pharmacol Ther 26: 987–1003, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, Lukas M, Fedorak RN, Lee S, Bressler B, Fox I, Rosario M, Sankoh S, Xu J, Stephens K, Milch C, Parikh A. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med 369: 711–721, 2013. [DOI] [PubMed] [Google Scholar]
- 66.Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, Niezychowski W; Study A3921063 Investigators. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med 367: 616–624, 2012. [DOI] [PubMed] [Google Scholar]
- 67.Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn's disease. Clin Gastroenterol Hepatol 12: 1485–1493, 2014. [DOI] [PubMed] [Google Scholar]
- 68.Sandborn WJ, Panes J, Zhang H, Yu D, Niezychowski W, Su C. Correlation between concentrations of fecal calprotectin and outcomes of patients with ulcerative colitis in a phase 2 trial. Gastroenterology. In press. [DOI] [PubMed] [Google Scholar]
- 69.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol 3: 390–407, 2006. [DOI] [PubMed] [Google Scholar]
- 70.Schnitzler F, Fidder H, Ferrante M, Noman M, Arijs I, Van Assche G, Hoffman I, Van Steen K, Vermeire S, Rutgeerts P. Long-term outcome of treatment with infliximab in 614 patients with Crohn's disease: results from a single-centre cohort. Gut 58: 492–500, 2009. [DOI] [PubMed] [Google Scholar]
- 71.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140: 1756–1767, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sutherland L, Macdonald JK. Oral 5-aminosalicylic acid for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev 2: CD000544, 2006. [DOI] [PubMed] [Google Scholar]
- 73.Terdiman JP, Gruss CB, Heidelbaugh JJ, Sultan S, Falck-Ytter YT. American Gastroenterological Association Institute guideline on the use of thiopurines, methotrexate, and anti-TNF-alpha biologic drugs for the induction and maintenance of remission in inflammatory Crohn's disease. Gastroenterology 145: 1459–1463, 2013. [DOI] [PubMed] [Google Scholar]
- 74.Thomson Reuters Cortellis. Thomson Reuters Cortellis (Online). https://cortellis.thomsonreuterslifesciences.com [30 March 2015].
- 75.van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzová D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA; ORAL Scan Investigators . Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum 65: 559–570, 2013. [DOI] [PubMed] [Google Scholar]
- 76.van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, Forejtova S, Zwillich SH, Gruben D, Koncz T, Wallenstein GV, Krishnaswami S, Bradley JD, Wilkinson B; ORAL Standard Investigators . Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 367: 508–519, 2012. [DOI] [PubMed] [Google Scholar]
- 77.Wollenhaupt J, Silverfield J, Lee EB, Terry K, Kwok K, lazariciu I, Nduaka C, Connell C, DeMasi R, Wang L. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: safety and clinical and radiographic efficacy in open-label, long-term extension studies over 7 years. Arthritis Rheumatol 67, Suppl 10, 2015. [Google Scholar]
- 78.Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K, Nduaka CI, Benda B, Gruben D, Nakamura H, Komuro Y, Zwillich SH, Wang L, Riese RJ. Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, long-term extension studies. J Rheumatol 41: 837–852, 2014. [DOI] [PubMed] [Google Scholar]
- 79.Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol 20: 91–99, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]