

Abstract
Kidney injury molecule-1 (KIM-1) is a receptor for the “eat me” signal, phosphatidylserine, on apoptotic cells. The specific upregulation of KIM-1 by injured tubular epithelial cells (TECs) enables them to clear apoptotic cells (also known as efferocytosis), thereby protecting from acute kidney injury. Recently, we uncovered that KIM-1 binds directly to the α-subunit of heterotrimeric G12 protein (Gα12) and inhibits its activation by reactive oxygen species during renal ischemia-reperfusion injury (Ismail OZ, Zhang X, Wei J, Haig A, Denker BM, Suri RS, Sener A, Gunaratnam L. Am J Pathol 185: 1207–1215, 2015). Here, we investigated the role that Gα12 plays in KIM-1-mediated efferocytosis by TECs. We showed that KIM-1 remains bound to Gα12 and suppresses its activity during phagocytosis. When we silenced Gα12 expression using small interefering RNA, KIM-1-mediated engulfment of apoptotic cells was increased significantly; in contrast overexpression of constitutively active Gα12 (QLGα12) resulted in inhibition of efferocytosis. Inhibition of RhoA, a key effector of Gα12, using a chemical inhibitor or expression of dominant-negative RhoA, had the same effect as inhibition of Gα12 on efferocytosis. Consistent with this, silencing Gα12 suppressed active RhoA in KIM-1-expressing cells. Finally, using primary TECs from Kim-1+/+ and Kim-1−/− mice, we confirmed that engulfment of apoptotic cells requires KIM-1 expression and that silencing Gα12 enhanced efferocytosis by primary TECs. Our data reveal a previously unknown role for Gα12 in regulating efferocytosis and that renal TECs require KIM-1 to mediate this process. These results may have therapeutic implications given the known harmful role of Gα12 in acute kidney injury.
Keywords: G protein, phagocytosis, Gα12, kidney injury molecule-1 (KIM-1), kidney
acute kidney injury (AKI) is a serious medical condition that affects up to one in five hospitalized patients globally, and for which there is no effective treatment (12, 13, 97). The most common pathoetiological mechanism causing AKI is ischemia-reperfusion injury (IRI) (7). Renal proximal tubular epithelial cells (TECs) are particularly susceptible to ischemic injury (40, 92). Injury or death of TECs is widely recognized as a major pathogenic step in AKI (7, 63). Recent work suggests that both apoptosis (15, 94) and different forms of necrosis (e.g., necroptosis) coexist in AKI (63). While apoptotic cells (or bodies) are rapidly cleared by efferocytosis without triggering inflammation (66), necrotic cells and uncleared apoptotic cells undergoing secondary necrosis (that lack an intact plasma membrane) (93, 101) passively release immunogenic damage-associated molecular pattern proteins (DAMPs), contributing to inflammation and secondary tissue damage in AKI (64, 105).
During AKI, TECs take on attributes of professional phagocytes and rapidly clear apoptotic and necrotic cells through a process known as efferocytosis (42, 62, 106, 110). Phagocytes recognize apoptotic cells via cell-surface receptors that directly or indirectly bind to “eat me” signals displayed on the surface of dying cells (25, 60). We (42) and others (56, 91) identified kidney injury molecule-1 (KIM-1), a novel phagocytic receptor for phosphatidylserine that is specifically upregulated on apical surface of TECs following injury (37, 43). Recently, we reported that Kim-1-deficient mice succumb to worse tissue injury and mortality after ischemic AKI (48). In addition, Yang et al. (110) have highlighted the importance of phagocytic clearance of apoptotic cells in AKI by showing that mice expressing a mucin-domain deletion mutant of Kim-1 with impaired phagocytic function are more prone to AKI as a result of exaggerated inflammation (110). Thus the upregulation of KIM-1 on TECs seems to play a protective role in AKI via efferocytosis.
KIM-1 is also known as T cell immunoglobulin and mucin domain-containing protein 1 (TIM-1) (68) or hepatitis A virus cellular receptor-1 (HAVCR1) (51), and is encoded by the HAVCR1 gene. Both mouse (termed Kim-1) and human proteins (KIM-1) are small type I transmembrane glycoproteins that belongs to the TIM family of proteins (29). The human TIM family consists of TIM-1 (28, 43, 68, 91), TIM-3 (20, 77), and TIM-4 (56, 91), which are conserved between mice and humans and bind directly to phosphatidylserine (29, 42, 43, 56, 91). KIM-1 is expressed by mast cells, B cells, activated CD4 T cells, and injured TECs (29, 37). The structure of KIM-1 consists of an IgV−like domain, a mucin domain, a transmembrane domain, and an intracellular (C-terminal) tail that has been implicated in cell signaling in differentiated T cells (16, 18, 43, 91) and B cells (112). The crystal structure of the IgV-like domain of KIM-1 has been solved and reveals that it contains a conserved binding pocket termed the metal ion-dependent ligand-binding site (MILIBS) (9, 20, 91) that allows for highly specific recognition of phosphatidylserine (29, 91).
The process of apoptotic cell recognition and engulfment is a highly complex and regulated process (2). Clustering of cell-surface KIM-1 by phosphatidylserine on apoptotic cell leads to formation of phagocytic cups on TECs (29). Ligand recognition triggers cell signaling that ultimately results in reorganization of the cell cytoskeleton to enable the phagocyte to engulf, internalize, and degrade the cell corpses (10, 39). Efferocytosis requires dynamic cytoskeletal remodeling that is mediated by Rho family of small GTPases such as RhoA and Rac1 (60, 65, 76, 84). Like all G proteins, RhoA and Rac1 cycle between GDP-bound inactive and GTP-bound active conformations regulated by GTP exchange factors (GEFs) or GTPase-activating proteins (GAPs), respectively (34, 85, 86). Although both RhoA and Rac1 belong to same family, they differ in their action: Rac1 induces polymerization of actin, leading to membrane ruffles and filopodia formation, while RhoA induces actin assembly into bundles and stress fibers (36). While the activation of Rac1 seems to be an evolutionarily conserved event in efferocytosis (53, 54, 83), some have proposed an inhibitory role for RhoA (27, 79, 83, 114).
Recently, we uncovered that KIM-1 interacts with the α-subunit of heterotrimeric G12 proteins (Gα12) and this interaction suppresses downstream injury pathways triggered by Gα12 activation by reactive oxygen species (ROS) in TECs during renal IRI (48). Gα12 belongs to the G12 class of G proteins (23, 102). It is ubiquitously expressed and has pleiotropic effects on cells, including inducing proliferation, focal adhesion assembly/disassembly, and cytoskeletal reorganization (25, 59, 78). Many of these effects, including actin stress fiber formation, are mediated downstream by Gα12-dependent activation of RhoA via Rho GEFs (98), including LARG (31), PDZ-RhoGEF (58), and p115 RhoGEF (45).
Herein, we investigated the role of Gα12 in KIM-1-mediated efferocytosis using cell culture models and primary TECs isolated from the kidneys of mice. We revealed that KIM-1 expression serves to suppress Gα12 activation to permit phagocytosis of apoptotic cells. In addition, we show that the inhibitory effect of Gα12 on efferocytosis is mediated by downstream activation of RhoA. Finally, we showed that primary TECs rely entirely on KIM-1 for efferocytosis and confirm the inhibitory role of Gα12 in this process.
EXPERIMENTAL PROCEDURES
Cell culture and materials.
Human embryonic kidney 293 (HEK-293) cells and porcine proximal tubule epithelial cells (LLC-PK1) were cultured at 37°C in 5% (vol/vol) CO2 incubator and maintained in DMEM (Invitrogen, Carlsbad, CA) containing 10% FBS (Invitrogen). HEK-293 cells stably expressing KIM-1-green fluorescent protein (GFP; herein referred to as HEK-293-KIM-1-GFP), KIM-1 with no tag (HEK-293-KIM-1), pcDNA-GFP (HEK-293-pcDNA), as well as LLC-PK1 stably expressing KIM-1 (LLC-PK1-KIM-1) and pcDNA (LLC-PK1-pcDNA) were generated by transfecting with plasmid constructs encoding either KIM-1 or a control vector using Lipofectamine 2000 (Life Technologies, Thermo Fisher Scientific, Rockford, IL). Several single-cell clones were selected using 800 μg/ml geneticin (G418) sulfate (Santa Cruz Biotechnology, Santa Cruz, CA). Cells expressing high levels of KIM-1 were used for further experiments and maintained in DMEM containing 10% FBS and 800 μg/ml G418. Primary TECs isolated from 2- to 4-wk-old C57BL/6 mice were cultured for the first 6 days in serum-free DMEM:F-12 mixed media (1:1) supplemented with 5% ITS (Invitrogen), 5% penicillin-streptomycin solution (Invitrogen), 0.5 μg/ml of mouse EGF (Peprotech, Rocky Hill, NJ), and 50 ng/ml hydrocortisone (Thermo Fisher Scientific). Thereafter, TECs were cultured with the same media after further supplementation with 5% FBS as described earlier (95). All chemicals were purchased from Sigma (Sigma-Aldrich, St. Louis, MO) unless stated otherwise. The pH-sensitive dye pHrodo Red succinimidyl ester (pHrodo Red SE), Alexa Fluor 555 goat anti-mouse, and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Life Technologies, and rhodamine phalloidin from Cytoskeleton (Denver, CO). Gα12 (1:500) and actin (1:1,000) antibodies were purchased from Santa Cruz Biotechnology. PE-conjugated anti-human KIM-1 (1:100, clone 1D12) and PE-conjugated mouse IgG1 isotype control (1:100, MOPC-21) were purchased from Biolegend (San Diego, CA). Allophycocyanine (APC)-conjugated anti-mouse Tim-1 rat IgG2B (1:100, clone AF1817A) and APC-conjugated rat IgG2B isotype control (1:100) were obtained from R&D Systems (Minneapolis, MN). RhoA-specific monoclonal antibodies and nonhydrolyzable GTP analog were purchased from Cytoskeleton. Anti-KIM-1 (human) antibody (PA4145) was generated by immunizing rabbits against the cytosolic domain of KIM-1 (Life Technologies) as previously described (3). A KIM-1 antibody binding to the mucin domain of KIM-1 (AKG7) was kindly provided by Dr. J. V. Bonventre (Harvard Medical School, Boston, MA). Complete Mini EDTA-free protease inhibitor cocktail tablets were purchased from Roche (Roche Diagnostics, Basel, Switzerland). A Rho inhibitor (cell-permeable C3 transferase) was purchased from Cytoskeleton, and a Rho kinase (ROCK) inhibitor (Y27632) and Rac1 inhibitor (NSC23766) were purchased from EMD Millipore (Billerica, MA). Plasmid constructs for Gα12, RhoA, and Rac1 were kindly provided by Dr. Bradley Denker (Harvard Medical School, Boston, MA), and plasmid constructs for KIM-1 were from Dr. Bonventre. Either Lipofectamine 2000 and Dharmafect 1 (Thermo Fisher Scientific) were used for transfecting plasmid and small interfering (si) RNA, respectively.
Mice.
Wild-type C57BL/6 (Kim-1+/+) mice were obtained from the Jackson Laboratory. C57BL/6 Kim-1-deficient mice (Kim-1−/−) were obtained from Dr. Andrew N. J. McKenzie (MRC Laboratory of Molecular Biology, Cambridge, UK), and these were generated by targeted disruption of mouse havcr1−/− in mouse embryonic stem cells as described previously (104). All animal procedures were performed in accordance with the Western University Animal Use Subcommittee (protocol 210–230).
Phagocytosis assay and FACS analysis.
To prepare apoptotic cells for phagocytosis assay, thymocytes were harvested from 3- to 6-wk-old C57BL/6 mice, and apoptosis was induced by UV exposure for 5 min followed by incubation overnight at 37°C in a 5% CO2 incubator in DMEM media supplemented by 10% FBS and 1% penicillin-streptomycin solution. Apoptotic and necrotic primary TECs were generated by exposing the cells to UV for 5 min or heat-shocking at 65°C for 15 min, respectively. Apoptotic and necrotic cells were stained with pH-sensitive dye pHrodo Red SE at a final concentration of 150 nM for 30 min at room temperature. Labeled apoptotic and necrotic cells were washed twice with 1× PBS to remove excess dye. The cells were counted, and 3 × 106 were added to each well of six-well plates (15 × 106 for 10-cm plates) and incubated for various time points at 37°C in a 5% CO2 incubator. Cells were then placed on ice for 30 min to reduce nonspecific binding of apoptotic cells. The plates were then washed three times with ice-cold PBS, and adherent cells were collected with 5 mM EDTA-PBS and resuspended with FACS buffer (PBS, 2% calf serum, and 0.1% sodium azide) for flow cytometric analysis. For KIM-1-surface staining, TECs were exposed to blocking solution containing PBS and 10% goat serum for 15 min, followed by incubation for 30 min at room temperature with either PE-conjugated anti-KIM-1 (human) or APC-conjugated anti-Tim-1 (mouse), and their respective isotype control antibodies at a concentration of 1:100. All samples were analyzed using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). The percentage of phagocytic cells that had internalized the apoptotic cell(s) was calculated and graphed as %phagocytosis. Engulfed cells with low fluorescence representing cells that were adherent, but not internalized, were excluded from the analysis (80).
Immunoprecipitation and Western blotting.
HEK-293 cells were lysed with ice-cold lysis buffer [25 mM HEPES, 150 mM NaCl, 15 mM MgCl2, 1% Triton X-100, and Complete Mini EDTA-free protease inhibitor cocktail tablets (Roche Diagnostics)]. For immunoprecipitation experiments, cell extracts containing 1.0–1.5 mg protein/ml were incubated with 10 μg of Gα12, KIM-1 (PA4145), or rabbit IgG (control) antibody and 20 μl of protein-A/G-Sepharose beads (Santa Cruz Biotechnology) at 4°C overnight. The recovered beads were then centrifuged, washed three times with lysis buffer, and suspended in 20 μl of SDS sample buffer and heated at 100°C for 5 min. Lysates (representing 5% of total lysate) and immunoprecipitates were separated under reducing conditions and transferred to polyvinylidene difluoride (PDVF) membranes (EMD Millipore). PDVF membranes were subsequently probed with antibodies specific to surface KIM-1 (AKG7, 1:3 dilution), the cytosolic domain of KIM-1 (PA 4145, 1:1,500 dilution), Gα12 (1:500 dilution), actin (1:1,000 dilution), or RhoA (1:500 dilution). The signal was visualized using the appropriate horseradish peroxidase-conjugated secondary antibodies, Luminata Forte ECL Western blot detection reagent (EMD Millipore), and chemiluminescence using film (Biomax Denville Scientific, South Plainfield, NJ) or the ChemiDoc MP System (Bio-Rad Laboratories, Hercules, CA).
GST-tetratricopeptide repeat pull down.
A construct of GST-fused tetratricopeptide repeat (TPR) domain was kindly provided by Dr. N. Dhanasekaran (Temple University, Philadelphia, PA). GST-TPR protein was purified from Escherichia coli, as described earlier (107). Cells were lysed with ice-cold lysis buffer (20 mM HEPES, pH 8.0), 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.5% Triton X-100, and Complete Mini EDTA-free protease inhibitor cocktail tablets (Roche Diagnostics). One milligram of protein lysate and ≃1 μg of GST-TPR-coupled glutathione-agarose beads were incubated together at 4°C for 5 h (48, 107). Both lysate and pull-down samples were analyzed by SDS-PAGE and Western blotting to represent total and active Gα12 in these samples, respectively. Lysates loaded with the nonhydrolyzable GTP analog (GTPγS) or GDP (Cytoskeleton) were used as a positive or negative control, respectively.
Cell viability and death assays.
TECs were grown to a confluent monolayer and subjected to different treatments before testing for viability using propidium iodide (PI; Biolegend) or annexin V labeling (Biolegend). The samples were then analyzed using a BD LSR II flow cytometer.
Immunofluorescence and confocal microscopy.
HEK-293 cells were cultured at subconfluent density on poly-d-lysine hydrobromide (Sigma-Aldrich)-coated glass coverslips and transfected with various GFP-tagged Gα12 as indicated or small interfering (si) RNA against Gα12. Cells were then fixed with 4% paraformaldehyde followed by counterstaining of the nucleus with DAPI (0.5 μg/ml). For surface staining of KIM-1 using the AKG antibody, the cells were blocked with 1% BSA in 1× PBS for 1 h at room temperature, followed by incubation of the anti-KIM-1 antibody overnight. Cells were then washed and labeled with Alexa 555-conjugated anti-mouse at a concentration of 1:1,000. To stain the actin cytoskeleton, cells were permeabilized with 0.25% Triton X-100 in 1× PBS for 5 min and then stained with rhodamine phalloidin for 30 min according to the manufacturer's recommendation (Cytoskeleton). Coverslips were mounted using Shandon-Mount permanent mounting (Thermo Fisher Scientific) and viewed using a FLUOVIEW X83I confocal microscope (Olympus, Tokyo, Japan). Data were acquired and analyzed using a FLUOVIEW FV10 ASW 4.0 viewer and ImageJ 1.49 software (National Institutes of Health, Bethesda, MD) to determine the Van Steensel score for colocalization of KIM-1 and Gα12. The colocalization score was calculated using five random fields/sample and from three independent experiments. Quantification of the number of bound apoptotic cells was assessed in eight random fields/sample and four independent experiments.
Silencing Gα12 in HEK-293 and TECs using siRNA.
ON Target plus Smart pool siRNA against Gα12-specific and a nontargeting pool (Control siRNA) were purchased from Dharmacon (Thermo Fisher Scientific). Cells were transfected with 50 nM of siRNA using Dharmafect1 for 24 h before further analysis of the knockdown.
RhoA activation.
Rho activation was determined using Rhotekin-RBD (Rho-binding domain) beads according to the manufacturer's instructions (Cytoskeleton) and as described elsewhere (113). Lysate and pull-down samples were analyzed by SDS-PAGE and Western blotting to detect total and active RhoA respectively. Control lysates were incubated with GDP (1 mM, negative control) or GTPγS (200 μM, positive control) for 15 min before addition of Rhotekin-RBD beads. The ratio of active to total RhoA was determined from densitometric values determined by Western blotting. For siRNA experiments where RhoA activation was measured, we used the G-LISA RhoA Activation Assay from Cytoskeleton due to the small amount of the protein available and its higher sensitivity compared with the pull-down assay.
Quantification and statistics.
Western blots were scanned, and the band intensity was quantified using ImageJ 1.49 software after subtraction of the background and determination of the linear range. The relative change in expression of the band intensity was obtained as follows. First, we calculated the ratio of the densitometric reading for the relevant band (e.g., active Gα12) to that of its loading control (e.g., total Gα12). Then, we divided each of the ratios by the ratio obtained for the control treatment to obtain a treatment value relative to the control value (i.e., relative to a control of 1). Statistical analysis was done using GraphPad Prism (Graph Pad Software, La Jolla, CA) or IBM SPSS statistic 22 (IBM, Armonk, NY). Significance was determined by using one-way ANOVA with Tukey's post hoc test (for GST-TPR data) or an unpaired t-test.
RESULTS
KIM-1 interacts with Gα12 during efferocytosis.
KIM-1 is not expressed by healthy TECs but is upregulated more than any other protein in TECs after AKI (1, 55). Exogenous overexpression of KIM-1 in cultured cell lines is often used as a model for KIM-1 upregulation in TECs during AKI and for studying its phagocytic function (32, 42). In choosing an appropriate cell line to study KIM-1-Gα12 interactions in vitro, we first determined the level of endogenous Gα12 expression in a variety of routinely used cell lines for studying phagocytosis (103). We found that HEK-293 cells expressed sufficient Gα12 to enable its detection by Western blotting (Fig. 1A). We failed to detect endogenous Gα12 protein expression in cells that spontaneously express KIM-1, such as the human renal adenocarcinoma cell line, 769-P, which are also routinely used to study KIM-1-mediated efferocytosis (32, 42, 56). To study the interaction of KIM-1 and Gα12 during KIM-1-mediated efferocytosis, we generated HEK-293 cells that stably expressed human KIM-1 bearing a C-terminal GFP-tag (HEK-293-KIM-1-GFP) and determined the efficiency and kinetics of apoptotic engulfment of these cells (43, 48). To distinguish between bound and internalized apoptotic cells by flow cytometry, apoptotic targets were labeled with a pH-dependent fluorescent dye (pHrodo) that exhibits higher fluorescence upon entry of apoptotic cells into acidic compartments such as phagolysosomes (32, 72, 81). HEK-293-KIM-1-GFP cells were significantly more efficient at engulfing apoptotic cells (40 vs. 8%, P < 0.05) relative to cells stably transfected with control vector (pcDNA-GFP) (Fig. 1B). We also visualized the HEK-293-KIM-1-GFP cells during efferocytosis using confocal microscopy (Fig. 1C). Phagocytic cups formed at the cell surface within 15 min of adding labeled apoptotic cells. The inhibition of apoptotic cell engulfment by KIM-1-expressing HEK-293 with cytochalasin D further confirmed that the uptake of apoptotic cells required actin polymerization that is typical of efferocytosis (Fig. 1D) (50).
Fig. 1.
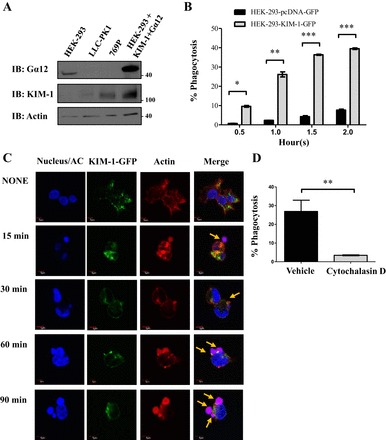
Kidney injury molecule-1 (KIM-1) mediates the uptake of apoptotic cells (AC). A: various cell lines were screened for level of KIM-1 and Gα12 expression via Western blotting. These cells included human embryonic kidney-293 (HEK-293), porcine proximal tubular epithelial cells (LLC-PK1), and human renal adenocarcinoma cell lines (769P). HEK-293 transfected with human KIM-1 and Gα12 served as a positive control. Lysates were obtained and run on Western blots and immunoblotted (IB) for KIM-1, Gα12, and actin. B: HEK-293 stably expressing either a control vector (pcDNA-GFP) or KIM-1-green fluorescent protein (GFP) were fed pHrodo-labeled (Red) AC for various time points (in hours) as indicated. The percentage of phagocytosis was determined by flow cytometry measurement of GFP- and high pHrodo Red double-positive cells (n = 3). *P < 0.05, **P < 0.005, ***P < 0.01 by unpaired t-test. C: HEK-293 cells stably expressing KIM-1-GFP were fed pHrodo-labeled AC (red) for various time points. Cells were fixed and stained with rhodamine-phalloidin to visualize F-actin (red) and 4,6-diamidino-2-phenylindole (DAPI) to visualize the nucleus and apoptotic cells (blue; ×600; bars = 5 μm). AC labeled with both pHrodo Red and DAPI (blue) appear as purple/pink in the merged image. Arrows indicates the AC in the vicinity of KIM-1. D: HEK-293 stably expressing KIM-1-GFP were treated with vehicle (DMSO) or the actin polymerization inhibitor cytochalasin D (1 μM) for 30 min and were then fed fluorescently labeled AC for 90 min. The percentage of phagocytosis represents the percentage of KIM-1-GFP-positive cells with high fluorescence of pHrodo Red of engulfed AC as measured by flow cytometry (n = 3). **P < 0.005 by unpaired t-test.
We previously demonstrated that the cytosolic domain of KIM-1 interacts with Gα12 and inhibits its activation by various Gα12 agonists (48). To investigate whether KIM-1 interacts with Gα12 during efferocytosis, we performed co-immunoprecipitation of Gα12 with KIM-1 in HEK-293-KIM-1-GFP cells before and after exposure to apoptotic cells. We found that the interaction between Gα12 with KIM-1 in cells did not depend on apoptotic cell stimulation of HEK-293-KIM-1-GFP cells (Fig. 2A). Using densitometric analysis, we found that the interaction of Gα12 with KIM-1 after stimulation with apoptotic cells was strongest at 30 min, which correlated with formation of phagocytic cups (Fig. 1C). These data were further confirmed using GST-Gα12 pull down of KIM-1 in lysates prepared from HEK-293 cell stably expressing KIM-1 before and after exposure to apoptotic cells (Fig. 2B).
Fig. 2.
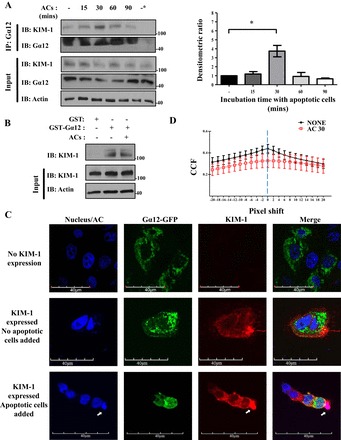
KIM-1 interacts with Gα12 during efferocytosis. A: HEK-293 cells stably expressing human KIM-1-GFP with a high level of endogenous Gα12 were stimulated with AC for indicated time points and used for coimmunoprecipitation (IP) using antibody against Gα12. Densitometric ratio of the coimmunoprecipitated KIM-1 during AC stimulation compared with unstimulated sample was quantified (n = 3). *P < 0.05 1-way ANOVA. B: glutathione-S-transferase (GST) and GST-Gα12 pull down was performed on HEK-293-KIM-1-GFP cells that were left unstimulated or stimulated with AC for 90 min. All samples were analyzed by SDS-PAGE followed by IB with antibodies against KIM-1, Gα12, and actin. The input lane represents 5% of the lysate. The data represent 3 independent experiments. C: HEK-293 cells were transfected with KIM-1-wild-type (WT) and a GFP-tagged Gα12 construct and fed pHrodo Red fluorescently labeled AC (blue/red) for 30 min before fixation, and detection of surface KIM-1 using anti-KIM-1 antibody against mucin domain (AKG) and Alexa 555-conjugated secondary antibody (×600; bars = 40 μm). Arrows indicate where phagocytic cup formed. D: images of colocalization of both KIM-1 and Gα12 were analyzed using the Van Steensel approach, where the cross correlation function (CCFs) was calculated with a pixel shift ± 20. The results shown are representative of 4 images of 3 independent experiments (1-way ANOVA).
To visualize the interaction of Gα12 with KIM-1 in cells, we cotransfected HEK-293 cells with KIM-1 and GFP- tagged Gα12, and examined the kinetics of Gα12-KIM-1 colocalization before and after addition of apoptotic cells (Fig. 2C). In keeping with our previous work (48), we observed that the colocalization of KIM-1 with Gα12 in these cells neither required nor was it perturbed by exposure to apoptotic cells. Moreover, we calculated the quantification of the change in colocalization at the vicinity of apoptotic cells using the Van Steensel approach (6). We failed to reveal any significant difference in colocalization between apoptotic cell-unstimulated and apoptotic cell-stimulated conditions (Fig. 2D). Together, the above results suggested that KIM-1 constitutively interacts with Gα12 in cells and remains so during efferocytosis.
KIM-1 downregulates Gα12 activity during KIM-1-mediated efferocytosis.
Efferocytosis involves a series of events that begins with recognition of eat me signals on apoptotic cells by the phagocytic receptor(s) on the phagocyte, which then leads to phagocytic cup formation, which then sets off a cascade of intracellular signaling events including activation of various GTPases that mediate corpse engulfment (24, 54, 83). Since Gα12 is a GTPase, which cycles between active (GTP-bound) and inactive (GDP-bound) status, we sought to investigate whether and how Gα12 activity was altered during efferocytosis in our model system. We therefore measured the fraction of active-to-total Gα12 in cell lysates of HEK-293-KIM-1-GFP cells after feeding them apoptotic cells. We used a well-established active-Gα12 pull-down assay which utilizes the TPR domain of the downstream effector of Gα12, Ser/Thr phosphatase type 5 (PP5), fused to GST (107). Western blotting was used to detect both active and total Gα12 (107). In KIM-1-expressing cells, we observed a steady decrease in the level of active Gα12 during efferocytosis (Fig. 3A). This decrease in the level of active Gα12 was more pronounced during later stages of efferocytosis (60–90 min after adding apoptotic cells). On the other hand, we observed no significant change in Gα12 activity in control HEK-293 cells exposed to apoptotic cells (Fig. 3B). Consistent with these results, we observed similar kinetics of Gα12 activation when porcine renal proximal TECs (LLC-PK1) stably expressing a C-terminal heme agglutinin (HA)-tagged KIM-1 or control vector (pcDNA) were cotransfected with a Gα12 expression construct (data not shown). Together, these data suggested that endogenous Gα12 activity may be suppressed by KIM-1 expression in HEK-293 cells during efferocytosis. Since we had already demonstrated in Fig. 1B that KIM-1 expression is required for efferocytosis, these data begged the question of whether Gα12 inhibition by KIM-1 was required for efficient efferocytosis.
Fig. 3.
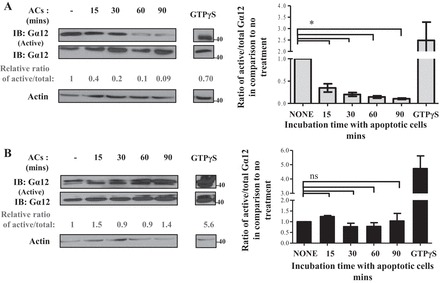
KIM-1 downregulates Gα12 activity during KIM-1-mediated efferocytosis. HEK-293 stably expressing KIM-1-GFP (A) or pcDNA (control vector; B) were either left untreated (none) or fed AC for various time points (15, 30, 60, and 90 min). Cell lysate were subjected to a GST-tetratricopeptide repeat (TPR) pull-down assay to measure active Gα12 as described in experimental procedures. Samples were analyzed by SDS-PAGE followed by IB with antibodies against Gα12 and actin. The total Gα12 lanes represent 5% of the lysate were used for the GST-TPR pull down. Densitometric analysis of the relative ratio of the active Gα12 to total Gα12 compared with nontreated samples is shown as numbers below blots. The average of the ratio of the active to total Gα12 compared with no treatment was blotted as a representation of 3 independent experiments (n = 3). *P < 0.05 by unpaired t-test.
Gα12 is a negative regulator of KIM-1-mediated efferocytosis.
Although data presented thus far demonstrate that Gα12 activity is inhibited during phagocytosis, they did not necessarily imply a functional role for Gα12 in KIM-1-mediated efferocytosis. To test this, HEK-293 cells stably expressing KIM-1 (HEK-293-KIM-1) were transfected with a control vector (pcDNA-GFP), vector encoding WT GFP-tagged Gα12 (WTGα12-GFP), or a vector encoding the GFP-tagged constitutively active (GTPase-deficient) mutant of Gα12 (QLGα12-GFP) (70), and efferocytosis efficiency was studied using flow cytometry. Overexpression of QLGα12-GFP significantly inhibited engulfment of apoptotic cells by the HEK-293-KIM-1 cells, whereas WTGα12-GFP expression had little effect on phagocytosis (Fig. 4A). We obtained similar results when we transfected LLC-PK1 cells with QLGα12-GFP (Fig. 4B). We once again confirmed the effect of QLGα12-GFP transfection on the phagocytic capacity of HEK-293-KIM-1 cells using confocal imaging by counting the number of apoptotic cells present inside GFP-positive cells (pcDNA-GFP or pcDNA-QLGα12-GFP) (Fig. 4C). To test how suppression of Gα12 expression would affect efferocytosis, we silenced endogenous Gα12 in HEK-293-KIM-1-GFP cells using siRNA (Fig. 4D) and performed phagocytosis assays as described above. We found a significant increase in the uptake of apoptotic cells (Fig. 4E) in conjunction with effective knockdown of Gα12 in these cells (Fig. 4D). Evaluation of phagocytosis by confocal microscopy helped confirm the above results (Fig. 4F). Together, these data suggested that active Gα12 has a negative effect on KIM-1-mediated engulfment of apoptotic cells.
Fig. 4.
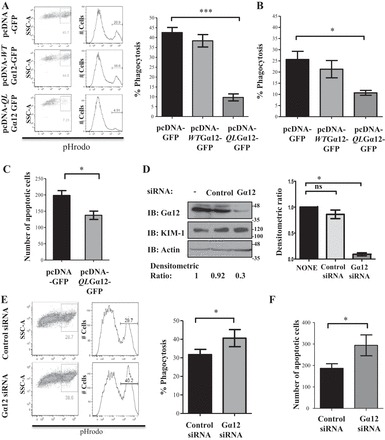
Gα12 negatively regulates KIM-1-mediated efferocytosis. A: HEK-293 cells stably expressing wild-type KIM-1 were transfected with either a control vector encoding GFP (pcDNA-GFP), wild-type GFP-tagged Gα12 (pcDNA-WTGα12-GFP), or constitutively active GFP-tagged Gα12 (pcDNA-QLGα12-GFP). B: LLC-PK1 cells stably expressing KIM-1 were transfected with the same constructs as in A. Cells in A and B were fed AC that are fluorescently labeled with pHrodo Red for 90 min. The percentage of uptake of AC was determined by flow cytometry as described in experimental procedures. The ratio of phagocytosis compared with the control vector was determined by flow cytometry after gating on GFP-positive cells (n = 3). *P < 0.05, ***P < 0.001 by unpaired t-test. C: HEK-293 cells stably expressing wild-type KIM-1 were grown on coverslips and transfected with either pcDNA-GFP or pcDNA-QLGα12-GFP and fed fluorescently labeled (pHrodo Red) AC for 90 min. Confocal microscopy images were taken to quantify the number of AC bound to the phagocytic cells. The results were plotted as a representation of 8 sections from 4 independent experiments (n = 4; ×400; bars = 5 μm). *P < 0.05. D: HEK-293 cells stably expressing KIM-1 were transfected with 50 pmol of control small interfering (si) RNA or siRNA against Gα12 for 24 h, and level of Gα12, KIM-1, and actin was determined by IB with indicated antibodies. A graph of the densitometric ratio of the Gα12 in siRNA-treated samples to nontransfected samples represents 4 independent experiments (n = 4). *P < 0.05 by unpaired t-test. E: HEK-293-KIM-1-GFP-expressing cells transfected with either control or Gα12 siRNA were fed fluorescently labeled (pHrodo Red) AC to measure phagocytosis as in A and B. F: cells in E were visualized with confocal microscopy to count the number of AC. The number of AC bound to the cells that were seen in each field was plotted based on analysis of 8 representative sections from each of 4 independent experiments with ∼120 GFP-positive (KIM-1-GFP) cells counted for each section (n = 4). *P < 0.05 by unpaired t-test.
RhoA acts downstream of Gα12 to inhibit KIM-1-mediated efferocytosis.
Gα12 has been shown to transduce signals from GPCR to activate RhoA via Rho guanine nucleotide exchange factors (98). RhoA belongs to the Rho family of GTPases (11, 35, 100), which regulate the actin cytoskeleton and efferocytosis (74, 76, 83). To determine the role of RhoA in KIM-1-dependent engulfment of apoptotic cells, we inhibited RhoA signaling using a cell-permeable Rho inhibitor (C3 transferase) (30) or its downstream effector ROCK using Y27632 (46, 47). The inhibition of either RhoA or ROCK activity significantly increased the uptake of apoptotic cells in our system compared with the respective controls (Fig. 5A). The comparable effects of both inhibitors on apoptotic cell uptake suggested that ROCK might be mediating the inhibitory effect of RhoA on efferocytosis in KIM-1-expressing HEK-293 cells. HEK-293-KIM-1 cells transfected with a vector encoding either WT RhoA or a constitutively activated RhoA mutant (Q62L) significantly inhibited the phagocytic efficiency of these cells compared with the HEK-293-KIM-1 cells transfected with a control vector (Fig. 5B). Importantly, we were able to reproduce the effects of chemical inhibition of RhoA on efferocytosis when HEK-293-KIM-1 cells were transfected with dominant negative RhoA (T19N), which resulted in a significant increase in phagocytosis (Fig. 5B).
Fig. 5.
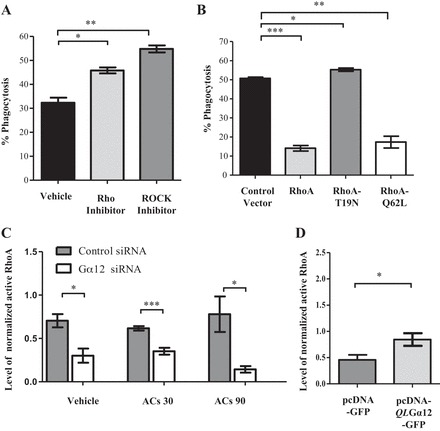
RhoA is a downstream mediator of Gα12 in KIM-1-mediated phagocytosis. A: HEK-293 cells stably expressing KIM-1 were treated for 4 h with vehicle (DMSO), Rho inhibitor (cell-permeable C3 transferase; 0.5 μg/ml), or Rho kinase (ROCK) inhibitor Y27632 (10 μM). B: cells were transfected with cyan fluorescent protein (CFP)-tagged control vector, CFP-tagged wild-type RhoA, CFP-tagged dominant negative RhoA (T19N), or CFP-tagged constitutively active RhoA (Q62L). These cells (A and B) were incubated with pHrodo Red-labeled AC for 90 min, and the percentage of phagocytosis by CFP-positive cells was measured by flow cytometry (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 by unpaired t-test. C: HEK-293 cells stably expressing KIM-1 were transfected with either control siRNA or siRNA against Gα12 for 24 h and then stimulated with AC for 30 or 90 min. D: HEK-293-KIM-1 cells were transfected with a control vector or constitutively active Gα12 (QLGα12). Levels of active RhoA (in C and D) were measured using a G-LISA RhoA activation assay kit. Total RhoA levels in lysates were measured using an RhoA ELISA. The levels of normalized active RhoA were calculated and graphed (n = 3). *P < 0.05, ***P < 0.001 by unpaired t-test.
The parallel and negative effects of both activated Gα12 and RhoA on KIM-1-mediated efferocytosis suggested that RhoA likely mediated the Gα12-inhibitory effect on phagocytosis. To formally test whether the endogenous RhoA activity was due to Gα12 in our cells, we silenced endogenous Gα12 expression in HEK-293-KIM-1-GFP cells and measured the RhoA activation using a commercially available pull-down assay that employs the binding domain of Rho effector protein (Rhotekin) fused to GST (46). Endogenous RhoA activity was significantly inhibited in HEK-293-KIM-1-GFP cells treated with Gα12 siRNA but not control siRNA at baseline, at 30 min (early phagocytosis), and at 90 min (late phagocytosis) after of exposure to apoptotic cells (Fig. 5C). Additionally, HEK-293-KIM-1-GFP cells transfected with a constitutively active mutant of Gα12 (QLGα12) exhibited significantly increased RhoA activity (Fig. 5D).
We also examined dynamic RhoA activity during phagocytosis. Following stimulation with apoptotic cells, we observed a gradual increase in the level of active RhoA that peaked at ∼90 min after the addition of apoptotic cells (Fig. 6A). On the other hand, there was no discernible pattern of RhoA activation observed in cells not expressing KIM-1 that were also fed apoptotic cells (Fig. 6B). Taken together, the above results suggested that Gα12-RhoA signaling negatively regulates KIM-1-dependent efferocytosis. Our data are consistent with reports of RhoA and ROCK being negative regulators of efferocytosis mediated by other phagocytic receptors (76, 100).
Fig. 6.
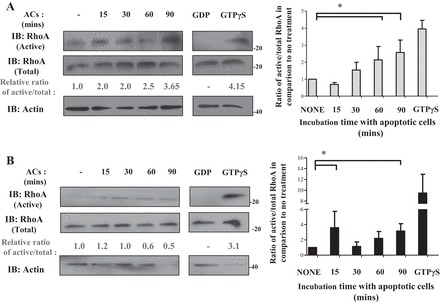
RhoA activity is increased during the course of KIM-1-mediated efferocytosis. HEK-293 stably expressing KIM-1-GFP (A) or pcDNA (control vector; B) were fed AC for various time points (15, 30, 60, and 90 min). Cell lysate were loaded with nonhydrolyzable GTP (GTPyS) as a positive control or GDP as a negative control. The level of active RhoA was measured using an active RhoA (RhoA-GTP) pull-down assay as described in experimental procedures. Samples were analyzed by SDS-PAGE followed by IB with antibodies against RhoA and actin. Densitometric analysis of the relative ratio of active RhoA to the respective total fractions compared with the untreated sample is shown as numbers below blots. The average ratio of active to total RhoA compared with no treatment was graphed as a representation of 3 independent experiments (n = 3). *P < 0.05 by unpaired t-test.
Unlike RhoA, Rac1 has been shown to have a conserved and positive role in efferocytosis in both professional and semiprofessional phagocytes that employ a variety of phagocytic receptors to engulf apoptotic cells (82, 83). Thus we sought to determine the role of Rac1 in KIM-1-mediated efferocytosis. Consistent with previous reports, transfection of KIM-1-expressing cells with a vector encoding dominant negative Rac1 (T17N) or treatment of KIM-1-expressing cells with a Rac1 inhibitor significantly inhibited phagocytic uptake of apoptotic cells compared with the respective controls (Fig. 7, A and B). These data suggested that Rac1 is required for KIM-1-dependent efferocytosis and plays an opposite role to RhoA in corpse engulfment in our cell culture model.
Fig. 7.
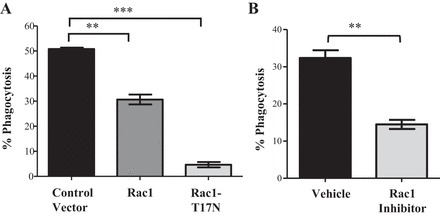
Rac1 is involved in KIM-1-mediated efferocytosis. A: HEK-293 cells were cotransfected with a construct encoding wild-type KIM-1 and either a CFP-tagged control vector (control) or CFP-tagged dominant negative Rac1 (Rac1-T17N). B: HEK-293-KIM-1 cells were treated for 4 h with vehicle (DMSO) or a Rac1 inhibitor (NSC23766; 50 μM). Cells in A and B were fed to pHrodo-labeled AC for 90 min, and the percentage of uptake was measured by flow cytometry. In A, the percentage of pHrodo Red high fluorescent cells was determined for CFP-positive cells (n = 3). **P < 0.01, ***P < 0.001 by unpaired t-test.
KIM-1 is required for efferocytosis by primary proximal TECs and is inhibited by Gα12.
Taken together with our previous report (48) showing that KIM-1-expression on TECs suppresses Gα12 activation by blocking GTP-loading, our present data suggest that KIM-1-mediated inhibition of endogenous Gα12 activation in KIM-1-expressing cells enables efficient efferocytosis. To confirm the inhibitory role of Gα12 in a physiologically relevant cell type, we studied primary TECs isolated from mouse kidneys. First, we wished to determine whether KIM-1 is the major efferocytosis receptor in renal TECs, because our findings regarding Gα12 would be generalizable to any efferocytosis by TECs. To formally determine whether KIM-1 is absolutely required for efferocytosis by primary TECs or whether there is redundancy (in apoptotic cell receptors) in efferocytosis receptors in TECs, we compared the phagocytic efficiency between primary TECs isolated from WT (Kim-1+/+) C57BL/6 and previously generated Kim-1-deficient mice (Kim-1−/−) mice (104). As previously reported, ex vivo culturing of TECs from WT mice readily resulted in spontaneous Kim-1 upregulation, presumably because cell culturing is a form cell stress or injury (1, 41, 42). Compared with Kim-1+/+ TECs, there was an absence of both total Kim-1 protein level and Kim-1 cell-surface expression in TECs isolated from Kim-1−/−, as determined by Western blotting and flow cytometry, respectively (Fig. 8, A and B). Next, apoptotic cells were fed to the TECs and efferocytosis was measured at different time periods over 24 h. We observed a significant difference in phagocytosis between Kim-1−/− TECs and Kim-1+/+ TECs (Fig. 8C). Surprisingly, Kim-1-deficient TECs were virtually incapable of uptaking apoptotic cells. To verify these findings using an alternate method, TECs were fed fluorescently labeled apoptotic cells for 3 h and cells were stained for surface Kim-1 and imaged by confocal microscopy (Fig. 8D). Similar to what was observed using flow cytometry, Kim-1+/+ TECs readily bound apoptotic cells and formed phagocytic cups around them in contrast to Kim-1-deficient TECs (arrows, Fig. 8D).
Fig. 8.
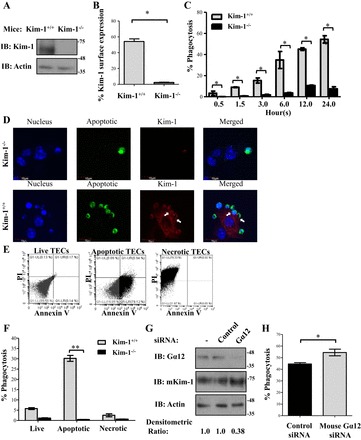
KIM-1 is a major phagocytic receptor in proximal tubular epithelial cells (TECs), and Gα12 is a negative regulator. A: proximal TECs isolated from C57BL/6 wild-type (Kim-1+/+) and Kim-1 knockout (Kim-1−/−) mice were cultured, and the level of mouse Kim-1 expressed was measured by Western blotting. B: surface expression of mouse Kim-1 on TECs was determined by flow cytometry using PE-conjugated anti-mouse Tim-1/Kim-1 antibody (R&D Systems; n = 3). *P < 0.05 by unpaired t-test. C: TECs were fed pHrodo red-labeled AC for various time points (in hour), and the percentage of phagocytosis of fluorescently labeled AC was measured by flow cytometry (n = 3). *P < 0.05 by 1-way ANOVA. D: TECs were cultured on glass coverslips and fed green-labeled AC for 3 h. Cells were fixed and surface stained for mouse Kim-1 using an anti-mouse Kim-1 antibody and Cy5-conjugated secondary antibody (×600; bars = 10 μm). E: TECs isolated from wild-type C57BL/6 mice were left untreated (live), subjected to UV for 3 min, followed by incubation overnight (to stimulate apoptosis), or subjected to heat shock at 65°C for 15 min (to stimulate necrosis). The different type of cell death was confirmed using PI and annexin V staining followed by flow cytometry. Data represent 3 independent experiments. F: live, apoptotic, or necrotic cells labeled with pHrodo Red were fed to either Kim-1+/+ or Kim-1−/− TECs, and the percentage of phagocytosis was measured using flow cytometry (n = 3) **P < 0.005 by unpaired t-test. G: TECs isolated from wild-type C57BL/6 mice were transfected with 50 pmol of control siRNA or siRNA against mouse Gα12 for 24 h, and the level of Gα12, mouse Kim-1, and actin was determined by Western blotting. H: Gα12 siRNA-treated cells were subjected to phagocytosis assay, and the percentage of phagocytosis of pHrodo-labeled AC was measured by flow cytometry (n = 4). *P < 0.5 by unpaired t-test.
A major limitation of our data thus far was that we used apoptotic thymocytes to study efferocytosis, when in AKI the source of apoptotic cells would have been neighboring TECs subjected to lethal ischemia and/or reperfusion injury (15). To formally test whether primary TECs can efficiently engulf apoptotic or necrotic TECs, we generated apoptotic or necrotic primary TECs from Kim-1+/+ mice and confirmed cell death using annexin V and PI staining (Fig. 8E). We then fed equal numbers of apoptotic or necrotic TECs to live primary TECs isolated from either Kim-1+/+ or Kim-1−/− mice, respectively. In keeping with our data thus far, we observed that TECs from Kim-1+/+ mice efficiently engulfed apoptotic TECs. Much to our surprise, we observed no significant phagocytosis of necrotic TECs by live TECs (Fig. 8F). Moreover, TECs from Kim-1−/− were unable to take up either apoptotic or necrotic cells.
Finally, to test whether Gα12 negatively regulates KIM-1-dependent efferocytosis in primary TECs, we treated TECs isolated from Kim-1+/+ mice with Gα12 siRNA or control siRNA and compared their relative phagocytic efficiencies (104). In keeping with what was observed in KIM-1-expressing HEK-293 cells, silencing Gα12 expression significantly increased efferocytosis by primary TECs (Fig. 8, G and H).
DISCUSSION
KIM-1 is a phosphatidylserine receptor (29) that is specifically upregulated by TECs during ischemic or toxic AKI (42–44), but the relevant intracellular signaling pathway(s) had not been appreciated until now. Here, we reveal for the first time that Gα12 negatively regulates KIM-1-dependent efferocytosis. Furthermore, we showed that Gα12 mediates this inhibition via downstream activation of RhoA. Given that KIM-1-mediated efferocytosis has been shown to protect against AKI (48, 110) and our data demonstrate that KIM-1 is indispensable for efferocytosis (Fig. 8C) by renal TECs, we conclude that Gα12-RhoA signaling likely plays an important role in regulating efferocytosis by TECs during AKI.
Our findings here are supported by our previous work demonstrating that KIM-1, when upregulated following IRI, binds directly to Gα12 and inhibits its activation in TECs by blocking GTP loading onto it (48). We also showed that Kim-1-deficient mice exhibit higher levels of active Gα12 (due to ROS) in the kidneys and worse kidney function following renal ischemic injury. Since ROS-mediated activation of Gα12 had been shown to activate downstream injury pathways (e.g., destabilizes tight junctions via Src kinase) downstream (113), we deduced that KIM-1-mediated inhibition of Gα12 was at least partly important for renal protection from IRI.
Our findings here suggest that the ability of KIM-1 to suppress Gα12 activation is likely also crucial for TECs to carry out efferocytosis during AKI. This finding is particularly relevant because Yang et al. (110) recently showed that KIM-1-mediated efferocytosis by TECs reduced acute injury to the kidneys during IRI. Thus we propose that the upregulation of KIM-1 on TECs during AKI and subsequent inhibition of Gα12 protect from renal damage during renal IRI via at least two potential mechanisms. The first mechanism would be by blocking ROS-mediated activation of injury pathways via activated Gα12 that lead to worse tissue damage and impaired repair (109, 113). The second mechanism would be by enhancing the phagocytic clearance of apoptotic cells by surviving TECs (32, 42). It is likely that by inhibiting Gα12 in TECs KIM-1 would indirectly help limit inflammation and secondary tissue damage caused by spillage of intracellular contents from uncleared apoptotic cells undergoing secondary necrosis (24, 84, 96). Interestingly, Kim-1-deficient mice (48) exhibited much more tissue damage and organ dysfunction from renal IRI compared with mice expressing a mutant form of Kim-1 (mucin domain deletion mutant) with an intact cytosolic domain (110) capable of interacting with endogenous Gα12 in TECs. Overall, we propose that KIM-1 (via its action on Gα12) profoundly influences TEC biology and thereby the renal response to AKI (7, 44).
Actin cytoskeletal rearrangement is crucial for engulfment of apoptotic cells by phagocytes (10, 11). Rac1 and RhoA belong to the RHO family of GTPases that regulate actin cytoskeletal reorganization (85) and have been implicated in efferocytosis (83). The reciprocal role for Rac1 and RhoA in KIM-1-dependent phagocytosis by TECs proposed by our work is novel and is consistent with what has been reported by several others studying other phosphatidylserine receptors (27, 76, 100). In these studies, the inhibitory effect of RhoA on apoptotic cell clearance was mediated by ROCK, as was the case in our experiments (100). Activated RhoA increases the kinase activity of ROCK, which in turn phosphorylates myosin light chain and promotes contractility via actin stress fiber formation (87). Therefore, one potential mechanism by which RhoA inhibited efferocytosis in our studies might be by increasing cell contractility via stress fiber formation (19, 83). In contrast to RhoA, activated Rac1 is known to have an evolutionarily conserved and positive role in efferocytosis (38, 53, 75). Rac1 is recruited to help form phagocytic cups composed of actin patches and generate membrane ruffles necessary to facilitate corpse engulfment (57, 75). Rac1 is then downregulated when the phagocytic cup closes in conjunction with abrupt disruption of the actin patches. Our data are consistent with the requirement for Rac1 in efferocytosis. Whether one or more of the conserved phagocytic signaling pathways that regulate Rac1 activation in other phagocytes are involved downstream of KIM-1 in TECs remains to be tested (53, 99). Moreover, the opposing roles of RhoA and Rac1 in KIM-1-mediated efferocytosis might suggest a spatiotemporal pattern of activation that may be better visualized through use of biosensors (and fluorescence resonance energy transfer) that respond to RhoA and Rac1 activation.
To our knowledge, this is the first report describing phagocytic signaling pathways that govern KIM-1-mediated engulfment of apoptotic cells despite the fact that members of the TIM family of phosphatidylserine receptors that include KIM-1 [also known at T-cell immunoglobulin and mucin domain protein-1 (TIM-1)], TIM-3, and TIM-4 have been the subject of much research (4, 5, 14, 29, 67, 112). Studies on TIM-1 signaling thus far have focused primarily on its nonphagocytic functions, for instance, on costimulation of T cell responses (4, 7, 16, 61, 67). Balasubramanian et al. (4) reported that TIM-1 mediates the degradation of NUR77, a nuclear receptor implicated in apoptosis and cell survival, but did not investigate efferocytic signaling in their studies. Additionally, Yang et al. (110) demonstrated that KIM-1 interacts with p85 in TECs and downmodulates NF-κB signaling. TIM-3 is expressed by T cells and dendritic cells where it mediates phagocytosis of apoptotic cells, but very little is known regarding how it mediates phagocytic signaling (73, 77, 90). Tim-4 on the other hand has been shown to not directly mediate phagocytic signaling (81). Although various GTPases make up fundamental components of the phagocytic signaling machinery (39, 83), to our knowledge our study is the first one to implicate Gα12 in efferocytosis. Given the various fundamental processes that Gα12 regulates, such as motility (52), modulation of cell-cell junctions (69), cellular transformation (22), and contractility (8, 33), our study might suggest a relationship between these processes and efferocytosis in TECs, particularly during AKI. For example, pathological activation of Gα12 during IRI results in activation of injury pathways via destabilization tight junctions (89) through its interaction with zonula occludens-1 (ZO-1) and Src kinase (69, 70, 88). The disruption of tight junctions has been shown to be a hallmark of epithelial cell damage in IRI and is believed to be a contributor to kidney dysfunction that often ensues (21, 71). The relationship between tight junction assembly and disassembly during efferocytosis has not been studied to date.
All in all, deciphering KIM-1 signaling pathway(s) may be of clinical significance to those treating AKI. Our work presented here and previously suggests that targeting Gα12 might be a potential strategy to treat AKI by enhancing clearance of apoptotic cells and simultaneously inhibiting downstream injury pathways. Unfortunately, there is no specific Gα12 inhibitor available to currently test this in vivo. Nonetheless, our detailed work uncovering the importance of KIM-1 in TEC efferocytosis and how it is regulated offers important insights into TEC biology that might be relevant to AKI.
GRANTS
This work was supported by operating grants from the Canadian Institutes of Health Research (HDK 232429 and 244945) and from the Department of Medicine (Western University).
O. Ismail is the recipient of a Studentship from the Lawson Health Research Institute. L. Gunaratnam is the recipient of a New Investigator award from the Kidney Research Scientist Core Education and Training (KRESCENT) program and is also supported by the Academic Medical Organization of Southern Ontario.
DISCLOSURES
J. V. Bonventre is a co-inventor on KIM-1 patents that are assigned to Partners Healthcare and licensed by Partners to Johnson and Johnson, Sekisui, Biogen Idec, and a number of research reagent companies. J. V. Bonventre is a consultant for Sekisui.
AUTHOR CONTRIBUTIONS
Author contributions: O.I., X.Z., and L.G. performed experiments; O.I. and L.G. analyzed data; O.I. and L.G. interpreted results of experiments; O.I. and L.G. prepared figures; O.I. and L.G. drafted manuscript; O.I., J.V.B., and L.G. edited and revised manuscript; O.I. and L.G. approved final version of manuscript; L.G. provided conception and design of research.
REFERENCES
- 1.Ajay AK, Kim TM, Ramirez-Gonzalez V, Park PJ, Frank DA, Vaidya VS. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J Am Soc Nephrol 25: 105–118, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16: 907–917, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem 277: 39739–39748, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Balasubramanian S, Kota SK, Kuchroo VK, Humphreys BD, Strom TB. TIM family proteins promote the lysosomal degradation of the nuclear receptor NUR77. Sci Signal 5: ra90, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binne LL, Scott ML, Rennert PD. Human TIM-1 associates with the TCR complex and up-regulates T cell activation signals. J Immunol 178: 4342–4350, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL. G alpha 12 and G alpha 13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J Biol Chem 270: 24631–24634, 1995. [DOI] [PubMed] [Google Scholar]
- 9.Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, Zencheck WD, Lary JW, Cole JL, Deng H, Xiao H, Dilorenzo TP, Allison JP, Nathenson SG, Almo SC. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity 26: 311–321, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Castellano F, Chavrier P, Caron E. Actin dynamics during phagocytosis. Semin Immunol 13: 347–355, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Chimini G, Chavrier P. Function of Rho family proteins in actin dynamics during phagocytosis and engulfment. Nat Cell Biol 2: E191–E196, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53: 961–973, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curtiss ML, Hostager BS, Stepniak E, Singh M, Manhica N, Knisz J, Traver G, Rennert PD, Colgan JD, Rothman PB. Fyn binds to and phosphorylates T cell immunoglobulin and mucin domain-1 (Tim-1). Mol Immunol 48: 1424–1431, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daemen MA, van 't Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P, Buurman WA. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 104: 541–549, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Souza AJ, Oak JS, Jordanhazy R, DeKruyff RH, Fruman DA, Kane LP. T cell Ig and mucin domain-1-mediated T cell activation requires recruitment and activation of phosphoinositide 3-kinase. J Immunol 180: 6518–6526, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Souza AJ, Oriss TB, O'Malley KJ, Ray A, Kane LP. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci USA 102: 17113–17118, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deery WJ, Heath JP. Phagocytosis induced by thyrotropin in cultured thyroid cells is associated with myosin light chain dephosphorylation and stress fiber disruption. J Cell Biol 122: 21–37, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, Karisola P, Pichavant M, Kaplan GG, Umetsu DT, Freeman GJ, Casasnovas JM. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol 184: 1918–1930, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. Am J Physiol Renal Physiol 274: F1–F9, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Dermott JM, Reddy MR, Onesime D, Reddy EP, Dhanasekaran N. Oncogenic mutant of Galpha12 stimulates cell proliferation through cyclooxygenase-2 signaling pathway. Oncogene 18: 7185–7189, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Dhanasekaran N, Dermott JM. Signaling by the G12 class of G proteins. Cell Signal 8: 235–245, 1996. [DOI] [PubMed] [Google Scholar]
- 24.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol 189: 1059–1070, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ 15: 243–250, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Erwig LP, McPhilips KA, Wynes MW, Ivetic A, Ridley AJ, Henson PM. Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin-radixin-moesin (ERM) proteins. Proc Natl Acad Sci USA 103: 12825–12830, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feigelstock D, Thompson P, Mattoo P, Zhang Y, Kaplan GG. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J Virol 72: 6621–6628, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 235: 172–189, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fritz G, Just I, Wollenberg P, Aktories K. Differentiation-induced increase in Clostridium botulinum C3 exoenzyme-catalyzed ADP-ribosylation of the small GTP-binding protein Rho. Eur J Biochem 223: 909–916, 1994. [DOI] [PubMed] [Google Scholar]
- 31.Fukuhara S, Chikumi H, Gutkind JS. Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G(12) family to Rho. FEBS Lett 485: 183–188, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Gandhi R, Yi J, Ha J, Shi H, Ismail O, Nathoo S, Bonventre JV, Zhang X, Gunaratnam L. Accelerated receptor shedding inhibits kidney injury molecule-1 (KIM-1)-mediated efferocytosis. Am J Physiol Renal Physiol 307: F205–F221, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gavard J, Gutkind JS. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J Biol Chem 283: 29888–29896, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geyer M, Wittinghofer A. GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr Opin Struct Biol 7: 786–792, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Hall A. Rho GTPases and the actin cytoskeleton. Science 279: 509–514, 1998. [DOI] [PubMed] [Google Scholar]
- 36.Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos Trans R Soc Lond B Biol Sci 355: 965–970, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62: 237–244, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Henson PM. Engulfment: ingestion and migration with Rac, Rho and TRIO. Curr Biol 15: R29–R30, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol 5: a008748, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hughes J, Cailhier JF, Watson S, Savill JS. Apoptosis in glomerulonephritis. Rheum Dis Clin North Am 30: 655–676, xi-xii, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Humphreys BD, Xu F, Sabbisetti V, Grgic I, Naini SM, Wang N, Chen G, Xiao S, Patel D, Henderson JM, Ichimura T, Mou S, Soeung S, McMahon AP, Kuchroo VK, Bonventre JV. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 123: 4023–4035, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273: 4135–4142, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Ichimura T, Brooks CR, Bonventre JV. Kim-1/Tim-1 and immune cells: shifting sands. Kidney Int 81: 809–811, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iguchi T, Sakata K, Yoshizaki K, Tago K, Mizuno N, Itoh H. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J Biol Chem 283: 14469–14478, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Ishizaki T, Naito M, Fujisawa K, Maekawa M, Watanabe N, Saito Y, Narumiya S. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett 404: 118–124, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol 57: 976–983, 2000. [PubMed] [Google Scholar]
- 48.Ismail OZ, Zhang X, Wei J, Haig A, Denker BM, Suri RS, Sener A, Gunaratnam L. Kidney injury molecule-1 protects against Galpha12 activation and tissue damage in renal ischemia-reperfusion injury. Am J Pathol 185: 1207–1215, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang S, Ohtani K, Fukuoh A, Yoshizaki T, Fukuda M, Motomura W, Mori K, Fukuzawa J, Kitamoto N, Yoshida I, Suzuki Y, Wakamiya N. Scavenger receptor collectin placenta 1 (CL-P1) predominantly mediates zymosan phagocytosis by human vascular endothelial cells. J Biol Chem 284: 3956–3965, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone SM. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J 15: 4282–4296, 1996. [PMC free article] [PubMed] [Google Scholar]
- 52.Kelly P, Stemmle LN, Madden JF, Fields TA, Daaka Y, Casey PJ. A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J Biol Chem 281: 26483–26490, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H, Schnabel R, Hengartner MO. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature 434: 93–99, 2005. [DOI] [PubMed] [Google Scholar]
- 54.Kinchen JM, Ravichandran KS. Journey to the grave: signaling events regulating removal of apoptotic cells. J Cell Sci 120: 2143–2149, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Ko GJ, Grigoryev DN, Linfert D, Jang HR, Watkins T, Cheadle C, Racusen L, Rabb H. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am J Physiol Renal Physiol 298: F1472–F1483, 2010. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27: 927–940, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science 290: 333–337, 2000. [DOI] [PubMed] [Google Scholar]
- 58.Kuner R, Swiercz JM, Zywietz A, Tappe A, Offermanns S. Characterization of the expression of PDZ-RhoGEF, LARG and G(alpha)12/G(alpha)13 proteins in the murine nervous system. Eur J Neurosci 16: 2333–2341, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Kurose H. Galpha12 and Galpha13 as key regulatory mediator in signal transduction. Life Sci 74: 155–161, 2003. [DOI] [PubMed] [Google Scholar]
- 60.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell 14: 277–287, 2004. [DOI] [PubMed] [Google Scholar]
- 61.Lee HH, Meyer EH, Goya S, Pichavant M, Kim HY, Bu X, Umetsu SE, Jones JC, Savage PB, Iwakura Y, Casasnovas JM, Kaplan G, Freeman GJ, DeKruyff RH, Umetsu DT. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. J Immunol 185: 5225–5235, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Gong Q, Zhong S, Wang L, Guo H, Xiang Y, Ichim TE, Wang CY, Chen S, Gong F, Chen G. Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against renal ischaemia-reperfusion injury. Nephrol Dial Transplant 26: 469–478, 2011. [DOI] [PubMed] [Google Scholar]
- 63.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol 25: 2689–2701, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant 13: 2797–2804, 2013. [DOI] [PubMed] [Google Scholar]
- 65.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature 461: 99–103, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manfredi AA, Iannacone M, D'Auria F, Rovere-Querini P. The disposal of dying cells in living tissues. Apoptosis 7: 153–161, 2002. [DOI] [PubMed] [Google Scholar]
- 67.Mariat C, Degauque N, Balasubramanian S, Kenny J, DeKruyff RH, Umetsu DT, Kuchroo V, Zheng XX, Strom TB. Tim-1 signaling substitutes for conventional signal 1 and requires costimulation to induce T cell proliferation. J Immunol 182: 1379–1385, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, Freeman GJ, Umetsu DT, DeKruyff RH. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol 2: 1109–1116, 2001. [DOI] [PubMed] [Google Scholar]
- 69.Meyer TN, Hunt J, Schwesinger C, Denker BM. Gα12 regulates epithelial cell junctions through Src tyrosine kinases. Am J Physiol Cell Physiol 285: C1281–C1293, 2003. [DOI] [PubMed] [Google Scholar]
- 70.Meyer TN, Schwesinger C, Denker BM. Zonula occludens-1 is a scaffolding protein for signaling molecules. Galpha(12) directly binds to the Src homology 3 domain and regulates paracellular permeability in epithelial cells. J Biol Chem 277: 24855–24858, 2002. [DOI] [PubMed] [Google Scholar]
- 71.Meyer TN, Schwesinger C, Ye J, Denker BM, Nigam SK. Reassembly of the tight junction after oxidative stress depends on tyrosine kinase activity. J Biol Chem 276: 22048–22055, 2001. [DOI] [PubMed] [Google Scholar]
- 72.Miksa M, Komura H, Wu R, Shah KG, Wang P. A novel method to determine the engulfment of apoptotic cells by macrophages using pHrodo succinimidyl ester. J Immunol Methods 342: 71–77, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature 450: 435–439, 2007. [DOI] [PubMed] [Google Scholar]
- 74.Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, Xiao YQ, Kench JA, Henson PM, Vandivier RW. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol 176: 7657–7665, 2006. [DOI] [PubMed] [Google Scholar]
- 75.Nakaya M, Kitano M, Matsuda M, Nagata S. Spatiotemporal activation of Rac1 for engulfment of apoptotic cells. Proc Natl Acad Sci USA 105: 9198–9203, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem 281: 8836–8842, 2006. [DOI] [PubMed] [Google Scholar]
- 77.Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113: 3821–3830, 2009. [DOI] [PubMed] [Google Scholar]
- 78.Needham LK, Rozengurt E. Galpha12 and Galpha13 stimulate Rho-dependent tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130 Crk-associated substrate. J Biol Chem 273: 14626–14632, 1998. [DOI] [PubMed] [Google Scholar]
- 79.Olazabal IM, Caron E, May RC, Schilling K, Knecht DA, Machesky LM. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr Biol 12: 1413–1418, 2002. [DOI] [PubMed] [Google Scholar]
- 80.Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, Ravichandran KS. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477: 220–224, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol 19: 346–351, 2009. [DOI] [PubMed] [Google Scholar]
- 82.Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 35: 445–455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol 7: 964–974, 2007. [DOI] [PubMed] [Google Scholar]
- 84.Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ 5: 563–568, 1998. [DOI] [PubMed] [Google Scholar]
- 85.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol 11: 471–477, 2001. [DOI] [PubMed] [Google Scholar]
- 86.Ridley AJ. Stress fibres take shape. Nat Cell Biol 1: E64–E66, 1999. [DOI] [PubMed] [Google Scholar]
- 87.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Sabath E, Negoro H, Beaudry S, Paniagua M, Angelow S, Shah J, Grammatikakis N, Yu AS, Denker BM. Galpha12 regulates protein interactions within the MDCK cell tight junction and inhibits tight-junction assembly. J Cell Sci 121: 814–824, 2008. [DOI] [PubMed] [Google Scholar]
- 89.Samarin S, Nusrat A. Regulation of epithelial apical junctional complex by Rho family GTPases. Front Biosci 14: 1129–1142, 2009. [DOI] [PubMed] [Google Scholar]
- 90.Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK, Gutierrez-Ramos JC, Coyle AJ, Strom TB. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol 4: 1093–1101, 2003. [DOI] [PubMed] [Google Scholar]
- 91.Santiago C, Ballesteros A, Martinez-Munoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity 27: 941–951, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanz AB, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol 19: 1634–1642, 2008. [DOI] [PubMed] [Google Scholar]
- 93.Saraste A. Morphologic criteria and detection of apoptosis. Herz 24: 189–195, 1999. [DOI] [PubMed] [Google Scholar]
- 94.Schumer M, Colombel MC, Sawczuk IS, Gobe G, Connor J, O'Toole KM, Olsson CA, Wise GJ, Buttyan R. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol 140: 831–838, 1992. [PMC free article] [PubMed] [Google Scholar]
- 95.Sharpe CC, Dockrell ME. Primary culture of human renal proximal tubule epithelial cells and interstitial fibroblasts. Methods Mol Biol 806: 175–185, 2012. [DOI] [PubMed] [Google Scholar]
- 96.Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett 584: 4491–4499, 2010. [DOI] [PubMed] [Google Scholar]
- 97.Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL, and Acute Kidney Injury Advisory Group of the American Society of Nephrology. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol 8: 1482–1493, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tanabe S, Kreutz B, Suzuki N, Kozasa T. Regulation of RGS-RhoGEFs by Galpha12 and Galpha13 proteins. Methods Enzymol 390: 285–294, 2004. [DOI] [PubMed] [Google Scholar]
- 99.Tosello-Trampont AC, Kinchen JM, Brugnera E, Haney LB, Hengartner MO, Ravichandran KS. Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells. Cell Death Differ 14: 963–972, 2007. [DOI] [PubMed] [Google Scholar]
- 100.Tosello-Trampont AC, Nakada-Tsukui K, Ravichandran KS. Engulfment of apoptotic cells is negatively regulated by Rho-mediated signaling. J Biol Chem 278: 49911–49919, 2003. [DOI] [PubMed] [Google Scholar]
- 101.Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 17: 922–930, 2010. [DOI] [PubMed] [Google Scholar]
- 102.Vara Prasad MV, Shore SK, Dhanasekaran N. Activated mutant of G alpha 13 induces Egr-1, c-fos, and transformation in NIH 3T3 cells. Oncogene 9: 2425–2429, 1994. [PubMed] [Google Scholar]
- 103.Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, Thorp EB. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 113: 1004–1012, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wong SH, Barlow JL, Nabarro S, Fallon PG, McKenzie AN. Tim-1 is induced on germinal centre B cells through B-cell receptor signalling but is not essential for the germinal centre response. Immunology 131: 77–88, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, Chadban SJ. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol 21: 1878–1890, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu X, Molinaro C, Johnson N, Casiano CA. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthritis Rheum 44: 2642–2652, 2001. [DOI] [PubMed] [Google Scholar]
- 107.Yamaguchi Y, Katoh H, Mori K, Negishi M. Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol 12: 1353–1358, 2002. [DOI] [PubMed] [Google Scholar]
- 109.Yanamadala V, Negoro H, Gunaratnam L, Kong T, Denker BM. Galpha12 stimulates apoptosis in epithelial cells through JNK1-mediated Bcl-2 degradation and up-regulation of IkappaBalpha. J Biol Chem 282: 24352–24363, 2007. [DOI] [PubMed] [Google Scholar]
- 110.Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, Ichimura T, Kuchroo V, Bonventre JV. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest 125: 1620–1636, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yeung MY, Ding Q, Brooks CR, Xiao S, Workman CJ, Vignali DA, Ueno T, Padera RF, Kuchroo VK, Najafian N, Rothstein DM. TIM-1 signaling is required for maintenance and induction of regulatory B cells. Am J Transplant 15: 942–953, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu W, Beaudry S, Negoro H, Boucher I, Tran M, Kong T, Denker BM. H2O2 activates G protein, alpha 12 to disrupt the junctional complex and enhance ischemia reperfusion injury. Proc Natl Acad Sci USA 109: 6680–6685, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou Z, Caron E, Hartwieg E, Hall A, Horvitz HR. The C. elegans PH domain protein CED-12 regulates cytoskeletal reorganization via a Rho/Rac GTPase signaling pathway. Dev Cell 1: 477–489, 2001. [DOI] [PubMed] [Google Scholar]