

Abstract
Background: phosphorylation of AS160 and TBC1D1 plays an important role for GLUT4 mobilization to the cell surface. The phosphorylation of AS160 and TBC1D1 in humans in response to acute exercise is not fully characterized. Objective: to study AS160 and TBC1D1 phosphorylation in human skeletal muscle after aerobic exercise followed by a hyperinsulinemic euglycemic clamp. Design: eight healthy men were studied on two occasions: 1) in the resting state and 2) in the hours after a 1-h bout of ergometer cycling. A hyperinsulinemic euglycemic clamp was initiated 240 min after exercise and in a time-matched nonexercised control condition. We obtained muscle biopsies 30 min after exercise and in a time-matched nonexercised control condition (t = 30) and after 30 min of insulin stimulation (t = 270) and investigated site-specific phosphorylation of AS160 and TBC1D1. Results: phosphorylation on AS160 and TBC1D1 was increased 30 min after the exercise bout, whereas phosphorylation of the putative upstream kinases, Akt and AMPK, was unchanged compared with resting control condition. Exercise augmented insulin-stimulated phosphorylation on AS160 at Ser341 and Ser704 270 min after exercise. No additional exercise effects were observed on insulin-stimulated phosphorylation of Thr642 and Ser588 on AS160 or Ser237 and Thr596 on TBC1D1. Conclusions: AS160 and TBC1D1 phosphorylations were evident 30 min after exercise without simultaneously increased Akt and AMPK phosphorylation. Unlike TBC1D1, insulin-stimulated site-specific AS160 phosphorylation is modified by prior exercise, but these sites do not include Thr642 and Ser588. Together, these data provide new insights into phosphorylation of key regulators of glucose transport in human skeletal muscle.
Keywords: AS160, TBC1D4, TBC1D1, endurance exercise, insulin sensitivity, skeletal muscle
physical exercise has profound effects on substrate metabolism in human skeletal muscle, and it is a cornerstone in the treatment of dysregulated metabolic conditions such as type 2 diabetes (30). A single bout of exercise can stimulate skeletal muscle glucose uptake in humans (14), and hours into the postexercise period insulin-stimulated glucose uptake is increased (3, 6, 7, 24, 25, 34). The glucose transporter protein GLUT4 is the major glucose transporter isoform expressed in skeletal muscle, and the translocation of GLUT4 from an intracellular location to the plasma membrane and T tubules is a major mechanism through which both insulin and exercise increase skeletal muscle glucose transport (20).
The GLUT4 translocation process is under control of AS160, also known as tre-2/USP6, BUB2, and cdc16 domain family member 4 (TBC1D4), and the AS160 paralog TBC1D1 (1, 8, 22, 27, 39). In their active forms AS160 and TBC1D1 promote hydrolysis of GTP to GDP on Rab proteins and thereby restrain GLUT4 translocation (28). AS160 and TBC1D1 is inactivated through a combination of several phosphorylations by the upstream kinases Akt and AMP-activated protein kinase (AMPK) (21, 32, 35, 39), and this allows GLUT4 to dock and fuse with the membrane. In muscle of both rats and humans, phosphorylation of AS160 after exercise has been shown to persist in the hours after termination of an exercise bout, whereas Akt and AMPK phosphorylation returns to baseline levels (2, 25). This is associated with a greater increase in AS160 phosphorylation when muscles are stimulated with insulin 3–4 h postexercise (12, 25, 34). However, this finding has not been consistent in humans (17). Unlike AS160, TBC1D1 phosphorylation is not retained 3 h postexercise in humans (25). In addition, insulin-stimulated TBC1D1 phosphorylation is not increased 3.5 and 5 h after cessation of exercise in humans (25), and similar observations have been made in rat skeletal muscle 3 and 27 h after exercise (12). It was recently found that acute exercise in humans results in phosphorylation of both AS160 and TBC1D1 in skeletal muscle and that specific AMPK trimer complexes may phosphorylate AS160 and TBC1D1 (11, 19, 36). Thus it is possible that TBC1D1 may be involved in the acute effects of exercise, whereas phosphorylation events on AS160 increase both GLUT4 mobilization and insulin sensitivity in the hours after exercise. To date only few studies have investigated site-specific phosphorylation events of AS160 and TBC1D1 in human skeletal muscle and more data are warranted to outline the role of these Rab-GAPs in contraction- and insulin-stimulated glucose uptake in humans.
We therefore investigated the phosphorylation at AS160 and TBC1D1 in muscle biopsies from healthy humans obtained 30 min after a single bout of endurance exercise and during a hyperinsulinemic euglycemic clamp initiated 4 h postexercise.
METHODS
Ethical approval.
All participants gave their written informed consent after being given oral and written information regarding the study, in accordance with the Declaration of Helsinki II. The Aarhus County local ethics scientific committee approved the study.
Subjects.
Eight healthy men (age 26 ± 4 yr, body weight 82.9 ± 8.8 kg, body mass index 23.8 ± 1.6 kg/m2, and V̇o2 peak 4,189 ± 465.5 ml/min) participated. The subjects did not take prescription medicine or have any family history of diabetes. Before completion of the experimental protocol, subjects completed an incremental V̇o2 peak test on a Jaeger ER800 bicycle ergometer. The load was increased until exhaustion. Rates of oxygen uptake and carbon dioxide release were determined using an automated breath-by-breath gas exchange analyzer (Oxycon Delta, Erich Jaeger, Germany). The maximal obtained V̇o2 for each subject was used to calculate the workload corresponding to 65% of V̇o2 peak.
Protocol.
In a randomized crossover design, subjects were examined on 2 days separated by >1 mo: 1) after an overnight fast of 10 h (resting condition) and 2) after an overnight fast of 10 h and 1 h ergometer cycling at 65% V̇o2 peak (exercise condition). The participants were instructed to abstain from exercise and to maintain their usual diet 48 h before examination days. Data from resting condition was previously published (38). On both examination days, one intravenous catheter (Venflon; Viggo, Helsingborg, Sweden) was placed in a dorsal hand vein for blood sampling. The hand was placed in a 65°C heated box for arterialization of the blood. Subjects were studied for 6 h from 0800 to 1400 (t = 0 to 360) on both examination days. The 1-h ergometer cycling was performed prior to the 6-h period on the exercise examination day. On the resting examination day the subjects were resting in the experimental facility for 1 h before the 6-h period. After a 4-h basal period (t = 0–240), the blood glucose levels were clamped at 5 mM with an insulin (Actrapid; Novo Nordisk, Copenhagen, Denmark) infusion of 0.8 mU·kg−1·min−1 for 2 h (t = 240–360). At t = 30 and 270 min, a muscle biopsy was obtained from vastus lateralis of the quadriceps femoris muscle using the Bergström needle, local anesthesia (Lidokain; Amgros, Copenhagen, Denmark), and sterile conditions. The muscle tissue was immediately dissected free from fat and connective tissue and placed in liquid nitrogen. Study design is presented in Fig. 1.
Fig. 1.
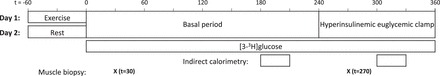
Study protocol. Subjects were examined on 2 days: 1) after an overnight fast of 10 h and 1 h ergometer cycling at 65% V̇o2 peak (Exercise) and 2) after an overnight fast of 10 h (Rest). On both experimental days an infusion of [3-3H]glucose tracer was initiated at t = 0 to measure glucose uptake during a basal period (t = 0–240) and during a 2-h hyperinsulinemic euglycemic clamp (t = 240–360). Indirect calorimetry was performed at t = 180–210 and t = 300–330. At t = 30 and 270 min, a muscle biopsy was obtained from vastus lateralis of the quadriceps femoris muscle.
Blood analysis.
Plasma glucose was immediately measured in duplicate on a Beckman Glucoanalyzer (Beckman Instruments, Palo Alto, CA). Serum samples were frozen and stored at −20°C, and insulin was analyzed using time-resolved fluoroimmunoassay (TR-IFMA, AutoDELFIA; PerkinElmer, Turku, Finland). C-peptide was measured by ELISA (DakoCytomation, Cambridgeshire, UK), and free fatty acids (FFA) were analyzed by a commercial kit (Wako Chemicals, Neuss, Germany).
Indirect calorimetry.
At t = 180–210 and t = 300–330 (basal and clamp period, respectively), the respiratory exchange ratio (RER) and glucose oxidation were estimated by indirect calorimetry (Deltatrac monitor; Dantes Instrumentarium, Helsinki, Finland). The last 25 min were used for calculations of mean values. Urine was collected during the basal period, and urea content was measured using the urease-Berthelot method (9). Oxidative glucose disposal was estimated after correction for protein oxidation, which was calculated on the basis of urea nitrogen excretion (10).
Glucose metabolism.
During the 6 h of the experimental days, a primed, continuous infusion of [3-3H]glucose (bolus 20 μCi, infusion 0.12 μCi/min; NEN Life Science Products, Boston, MA) was performed. [3-3H]glucose was added to the infused glucose (100 μCi [3-3H]glucose/500 ml 20% glucose) to prevent rapid dilution of the [3-3H]glucose during the hyperinsulinemic euglycemic clamp. Steele's equation was used to estimate glucose rate of appearance (Ra) and disappearance (Rd) (31). Endogenous glucose production (EGP) equals Ra of glucose under basal conditions, and during the clamp EGP is calculated by subtracting mean rate of exogenous glucose from glucose Ra. Nonoxidative glucose disposal (NOGD) was estimated by subtracting oxidative glucose disposal from total glucose disposal (glucose Rd).
Glycogen.
Muscle samples (10 mg wet weight) were hydrolyzed in 2 M HCl at 100°C for 2 h, followed by neutralization with 2 M NaOH (41), and glucose content was measured by the hexokinase enzymatic method using a glucose hexokinase reagent (Eagle Diagnostics, Desoto, TX) (4).
Western blot analyses.
Muscle biopsies were homogenized in an ice-cold buffer containing (in mM) 50 HEPES, 137 NaCl, 10 Na4P2O7, 10 NaF, 2 EDTA, 1 MgCl2, 1 CaCl2, and 2 Na3VO4 and 1% (vol/vol) Nonidet P-40, 10% (vol/vol) glycerol, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 0.5 μg/ml pepstatin, 10 μg/ml antipain, 1.5 mg/ml benzamidine, and 100 μM 4-(−2-aminoethyl)-benzenesulfonyl fluoride, and hydrochloride (pH 7.4), and samples were rotated for 60 min at 4°C. Insoluble materials were removed by centrifugation at 16,000 g for 20 min at 4°C. Western blot analyses were used to assess protein and phosphorylation levels of various proteins. Antibodies against AMPKα and phosphospecific (Ser79) Acetyl-CoA carboxylase (ACC) antibody were from Millipore (Billerica, MA). Polyclonal anti-COOH-terminal peptide GLUT4 antibody was produced as described previously (23). Antibodies against TBC1D1 and phosphospecific TBC1D1 antibodies (Ser237 and Thr596) were generated as described previously (39). Phosphospecific AMPK (Thr172), Akt, phosphospecific Akt (Ser473 and Thr308), GS, phosphospecific GS (Ser641), GSK-3 (α and β), phospho-specific GSK-3 (α and β), PAS, and AS160 antibodies were from Cell Signaling Technology (Beverly, CA). ACC expression was assessed using horseradish peroxidase-conjugated streptavidin (Pierce Chemical, Rockford, IL). Phosphospecific AS160 (Ser341, Ser588, Thr642, Ser704, and Ser751) antibodies were generated as described previously (13, 37). Proteins were visualized by BioWest enhanced chemiluminescence (Pierce) and quantified using UVP BioImaging System (UVP, Upland, CA). The membranes were stripped after Western blot analyses of phosphorylated proteins, and then total expression of the same proteins were measured. Quantifications of protein phosphorylation are expressed as a ratio of total protein expression measured on the same membranes.
Statistical analysis.
Results are expressed as means ± SE. Normal distribution was assessed by inspection of QQ plots, and the Levene Median test was used to test for equal variance. Isolated comparisons between resting and exercise were assessed by a paired t-test. Comparisons between the main effects of exercise and insulin stimulation were assessed by two-way repeated-measurements ANOVA. One-way repeated-measurements ANOVA were used to compare for differences in site-specific AS160 phosphorylation. When the repeated-measurements ANOVA revealed significant interactions or differences between means, Student-Newman-Keul's post hoc test was performed to detect specific differences. P < 0.05 was considered significant.
RESULTS
Substrate oxidation and insulin responsiveness.
One hour ergometer cycling at 65% of V̇o2 peak, 177 ± 4 W, increased circulating glucagon levels, whereas FFA, glucose, insulin, and c-peptide were not significantly altered (Table 1). This was associated with slightly increased resting energy expenditure (REE) after exercise (Table 1) together with significantly reduced RER and a trend toward decreased glucose oxidation (P = 0.057, power: 0.418) (Fig. 2, A and B). A hyperinsulinemic euglycemic clamp was used to assess insulin responsiveness, and no differences were observed in insulin concentrations and glucose infusion rates (GIR) on the 2 experimental days (Table 1 and Fig. 2C). During the hyperinsulinemic euglycemic clamp, RER and glucose oxidation were significantly increased on the control day, but after exercise this increase did not reach significance (P = 0.15 and P = 0.20, respectively) (Fig. 2, A and B). Endogenous glucose production (EGP) and glucose disposal rate (Rd) were not affected 210–240 min after exercise. The hyperinsulinemic euglycemic clamp markedly suppressed EGP and increased glucose Rd without any effect of prior exercise (Fig. 2, D and E).
Table 1.
Glucose, insulin, C-peptide, glucagon, FFA concentrations in serum, and energy expenditure were determined after an overnight fast (rest) and after 1 h ergometer cycling at 65% V̇o2peak
| t = 0 (Basal Condition) |
t = 360 (Clamp Condition) |
|||||
|---|---|---|---|---|---|---|
| Rest | Exercise | Rest vs. Exercise | Rest | Exercise | Rest vs. Exercise | |
| Glucose, mmol/l | 5.3 ± 0.10 | 5.4 ± 0.23 | P = 0.70 | 4.9 ± 0.10 | 4.8 ± 0.06 | P = 0.37 |
| Insulin, pmol/l | 47 ± 6 | 65 ± 13 | P = 0.18 | 271 ± 7 | 276 ± 16 | P = 0.75 |
| C-peptide, pmol/l | 604 ± 76 | 728 ± 160 | P = 0.24 | 307 ± 51 | 244 ± 58 | P = 0.14 |
| Glucagon, pg/ml | 50 ± 10 | 124 ± 22 | P = 0.005 | 26 ± 6 | 39 ± 11 | P = 0.19 |
| FFA, mmol/l | 0.53 ± 0.10 | 0.92 ± 0.22 | P = 0.19 | 0.08 ± 0.02 | 0.09 ± 0.04 | P = 0.68 |
| Energy expenditure, kcal/day | 1,901 ± 44 | 2,188 ± 90 | P = 0.002 | 1,891 ± 43 | 2063 ± 62 | P = 0.001 |
Values are means ± SE. Measurements were performed on both examination days at t = 0 (basal condition) and at t = 360 (clamp condition). Exercise, 65% V̇o2peak; FFA, free fatty acid. Energy expenditure was measured at t = 180–210 and t = 300–330 (basal and clamp conditions, respectively).
Fig. 2.
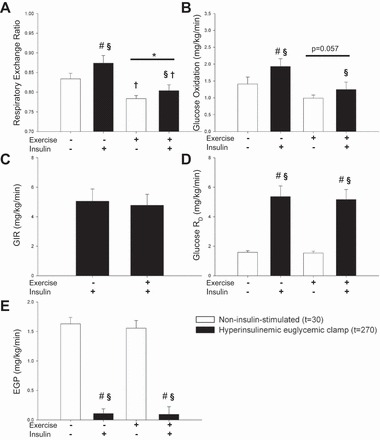
Respiratory exchange ratio, glucose uptake, and glucose metabolism were determined after an overnight fast (rest) and after one h ergometer cycling at 65% VO2-peak (Exercise). Throughout the figure open bars indicate a noninsulin-stimulated condition at t = 30 and closed bars indicate a hyperinsulinemic euglycemic clamp condition at t = 270. A: main effect of experimental day and hyperinsulinemic euglycemic clamp was observed on respiratory exchange ratio (RER). Post hoc testing revealed that exercise decreased RER both during insulin and noninsulin-stimulated conditions, whereas RER increased only during the hyperinsulinemic euglycemic clamp on the resting day. (main effect of experimental day = *P < 0.05, main effect of hyperinsulinemic euglycemic clamp = §P < 0.05, #P < 0.05 vs. basal within experimental day, †P < 0.05 vs. resting within noninsulin-stimulated condition or hyperinsulinemic euglycemic clamp). B and C: glucose infusion rate (GIR) and glucose rate of disappearance (RD) were not affected by exercise, and during the hyperinsulinemic euglycemic clamp, glucose RD was markedly increased. D: there was a trend toward decreased glucose oxidation after exercise. There was a main effect of the hyperinsulinemic euglycemic clamp on glucose oxidation; however, this effect was not significant after exercise when post hoc test was applied. E: endogenous glucose production (EGP) was not affected by exercise and equally suppressed during the hyperinsulinemic euglycemic clamp.
AS160 and TBC1D1 phosphorylation.
We determined AS160 phosphorylation on five sites: Ser341, Ser588, Thr642, Ser704, and Ser751. In addition, the PAS antibody was used to determine nonspecific AS160 phosphorylation. Significantly increased phosphorylation was evident on all specific AS160 phosphorylation sites 30 min after exercise compared with time-matched resting values, whereas the PAS antibody revealed a trend (P = 0.068, power: 0.370; Fig. 3B). There was a significant exercise × hyperinsulinemic euglycemic clamp interaction on all specific AS160 phosphorylation sites. One-way repeated-measurements ANOVA was used to compare AS160 phosphorylation in resting, resting insulin-stimulated, and exercised insulin-stimulated conditions. On the resting day, insulin stimulation increased AS160 phosphorylation on all sites. Phosphorylation on Ser341, Ser704, and PAS were increased during insulin stimulation on the exercise day compared with the resting day, and a strong trend toward increased phosphorylation was observed on Ser751 (P = 0.072, power: 0.905). Thr642 phosphorylation was increased during insulin stimulation but not affected by exercise. Ser588 phosphorylation was increased by insulin on the resting day, however, no significant insulin effect was observed after exercise. On TBC1D1, both Ser237 and Thr596 phosphorylation were increased 30 min after exercise. A main effect of insulin stimulation was observed on Thr596 phosphorylation, and post hoc testing only revealed a significant effect on the resting day. Ser237 phosphorylation was unaffected during insulin stimulation on both experimental days (Fig. 3C).
Fig. 3.
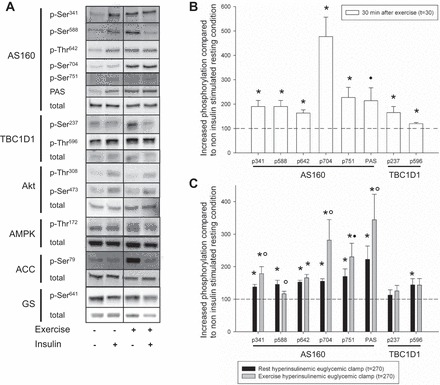
Effect of 1 h ergometer cycling at 65% V̇o2 peak (exercise) on signaling to GLUT4 translocation was assessed with Western blotting. A: representative blots of phosphorylated and total protein expression of AS160, TBC1D1, AMPK, ACC, Akt, and glycogen synthase after an overnight fast (rest) and after exercise during noninsulin-stimulated and insulin-stimulated conditions. B: open bars indicate increase in site-specific phosphorylation 30 min after exercise compared with time-matched nonexercised control condition (dashed line) on AS160 and TBC1D1. All site-specific phosphorylations increased after exercise (*P < 0.05), whereas the nonspecific phosphorylation detected with the PAS antibody revealed a trend for only an increase compared with resting condition at 30 min (●P = 0.068). Standard errors for site-specific AS160 and TBC1D1 phosphorylation in resting condition at 30 min were: AS160 p-Ser341 = 11.60, p-Ser588 = 8.34, p-Thr642 = 5.05, p-Ser704 = 10.30, p-Ser751 = 10.36, PAS = 22.02, and TBC1D1 p-Ser237 = 12.95, p-Thr596 = 14.26. Presented protein phosphorylations are expressed as a ratio of total protein expression. C: solid and shaded bars indicate increase in site-specific phosphorylation during insulin stimulation (t = 270) compared with resting condition at 30 min (dashed line) during resting and postexercise conditions, respectively. Phosphorylation on all AS160 sites and TBC1D1 Thr596 was increased by insulin stimulation at 270 min compared with without insulin at 30 min on the resting day (*P < 0.05). Insulin-induced phosphorylation was augmented by exercise on Ser341, Ser704, and PAS (○P < 0.05) at 270 min compared with time-matched nonexercised control condition, and a trend was observed on Ser751 (●P = 0.072). No additional exercise effect was seen on AS160 Thr642 at 270 min compared with time-matched nonexercised control condition, and no insulin effect was observed on Ser588 after exercise at 270 min compared with without insulin at 30 min on the resting day. Presented protein phosphorylations are expressed as a ratio of total protein expression.
AMPK and Akt phosphorylation.
AMPK and Akt are putative upstream kinases of specific phosphorylation sites on AS160 and TBC1D1 in human muscle and were therefore further examined. No significant change in AMPK Thr172 phosphorylation was detected 30 min after exercise termination (Fig. 4A). However, phosphorylation of the downstream AMPK target site Ser79 on ACC was abundantly increased 30 min after exercise, and this phosphorylation returned to baseline 240 min later during insulin stimulation (Fig. 4B). Akt activation was assessed by phosphorylation of the two activation sites, Thr308 and Ser473. No regulation of these sites was observed after exercise, but phosphorylation was significantly increased by insulin stimulation on both experimental days (Fig. 4, C and D).
Fig. 4.
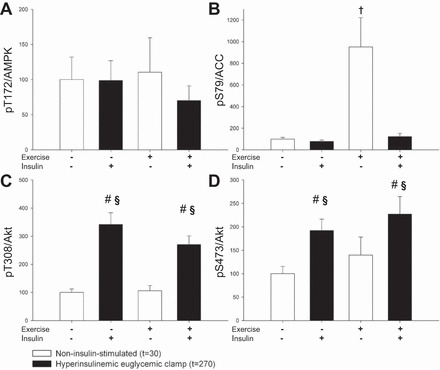
Effect of 1 h ergometer cycling at 65% V̇o2 peak (exercise) in noninsulin-stimulated condition at t = 30 and insulin-stimulated condition at t = 270 on the upstream (AMPK and Akt) kinases of AS160 and TBC1D1. Throughout the figure, open bars indicate a noninsulin-stimulated condition and closed bars indicate an insulin-stimulated condition. A and B: no change in AMPK Thr172 phosphorylation was detected after exercise; however, downstream ACC Ser79 phosphorylation was increased (†P < 0.05 vs. resting within noninsulin-stimulated condition). C and D: increased Akt Thr308 and Ser473 phosphorylation was observed during insulin stimulation without any exercise effect (main effect of hyperinsulinemic euglycemic clamp = §P < 0.05, #P < 0.05 vs. basal within experimental day).
Skeletal muscle glycogen content and regulation of glycogen synthesis signaling.
Glycogen decreased ∼50% during exercise, and no change was, as expected, observed 30 min into the hyperinsulinemic euglycemic clamp (Fig. 5A). During the hyperinsulinemic euglycemic clamp, nonoxidized glucose disposal (NOGD) increased significantly on both experimental days. The changes in substrate metabolism after exercise lead to a trend toward increased NOGD compared with the resting situation (P = 0.064, power: 0.386; Fig. 5B). This trend to increased glycogen synthesis after exercise was supported by decreased Ser641 phosphorylation on glycogen synthase (GS), indicating increased GS activity. Insulin stimulation resulted in a main effect (P = 0.008) on GS phosphorylation (Fig. 5C).
Fig. 5.
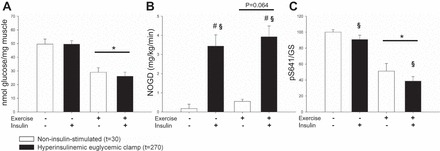
Glycogen and glycogen metabolism were determined after an overnight fast (rest) and after 1 h ergometer cycling at 65% V̇o2 peak (Exercise). Throughout the figure, open bars indicate a noninsulin-stimulated condition and closed bars indicate an insulin-stimulated condition. A: glycogen stores were significantly reduced during exercise (main effect of experimental day = *P < 0.05). B: nonoxidative glucose disposal (NOGD) was increased during insulin stimulation, and exercise tended to increase NOGD (main effect of hyperinsulinemic euglycemic clamp = §P < 0.05, #P < 0.05 vs. basal within experimental day). C: Ser641 phosphorylation on glycogen synthase (GS) was significantly reduced by both exercise and insulin stimulation, indicating increased GS activity.
DISCUSSION
Our data demonstrate that exercise-induced phosphorylation of both TBC1D1 and AS160 persist 30 min after a single bout of exercise, although phosphorylation of the two known upstream kinases, Akt and AMPK, had returned to baseline. This is the first report of sustained TBC1D1 phosphorylation in human skeletal muscle in the postexercise recovery period and the first investigation of site-specific phosphorylation of AS160 postexercise using a time-matched nonexercised control condition. These findings expand our understanding of the regulation of TBC1D1 and AS160 in human skeletal muscle.
TBC1D1 phosphorylation of the AMPK-consensus sites Ser237 and Thr596 persisted 30 min postexercise. Ser237 was previously shown to be phosphorylated immediately after a bout of exercise in both lean and obese human subjects (11, 19), and in mouse skeletal muscle, mutation of multiple TBC1D1 phosphorylation sites including Ser231 (equivalent to human Ser237) impairs contraction-stimulated glucose transport (1). However, there is no effect on glucose transport when only Ser231 is mutated (39), and the persistent phosphorylation on Ser237 is therefore unlikely to determine glucose uptake in the postexercise period. In addition to TBC1D1, exercise also increased phosphorylation of AS160 on sites Ser341, Ser588, Thr642, Ser704, and Ser751 30 min postexercise, which is in agreement with recent observations in humans using a one legged-exercise model (25, 34, 36). In our study, the subjects performed two-legged exercise, and the phosphorylation levels were compared with resting biopsies obtained on a separate day. Unlike the one-legged model, the resting muscles were therefore not exposed to indirect effects of exercise like increased circulating nonesterified fatty acids or increased catecholamine stimulation, which can affect substrate metabolism in a resting muscle (15, 18). Indeed major changes in gene expression have been observed in the nonexercised leg after one-legged exercise (5). Our data therefore add further insight into the regulation of AS160 in skeletal muscle during exercise (Fig. 6).
Fig. 6.
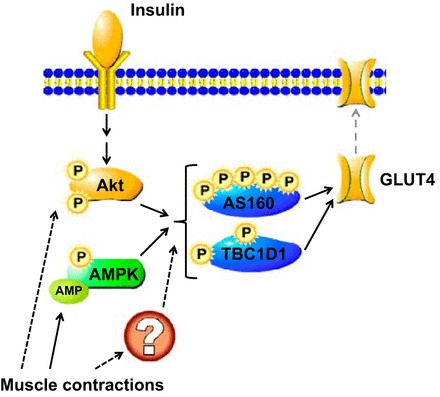
Schematic overview of the regulation of GLUT4 translocation. Insulin stimulates Akt phosphorylation and activity, whereas muscle contraction activates AMPK by inducing phosphorylation of Thr172 and by allosteric activation by AMP. Muscle contraction can also activate Akt, but the importance of this signaling pathway for glucose uptake remains unclear. AMPK and Akt are established kinases of the phosphorylation sites on AS160 and TBC1D1 investigated in this study, but other kinases activated by muscle contraction could also be involved. P, phosphorylation; ?, putative protein or mechanism. Solid arrows illustrate established relationships between stimuli, signals, and glucose transport; dashed arrows are used for putative interactions.
Phosphorylations of AMPK and Akt, both established kinases of TBC1D1 and AS160, were not increased 30 min after exercise. However, we did observe an ∼10-fold increase in phosphorylation of the bona fide AMPK target ACC. This strongly suggests that AMPK indeed was activated by the performed exercise, and AMPK is therefore a probable upstream kinase responsible for observed phosphorylation on TBC1D1 and AS160 30 min after exercise. There are several probable explanations for the difference in time course between phosphorylation of TBC1D1 and AS160 and the known upstream kinases. One option is allosteric activation of AMPK by AMP that is not detectable by phosphospecific Western blot analyses or immunoprecipitate kinase assay (16). Another option is differences in the time course for dephosphorylation of AMPK and its downstream targets TBC1D1 and AS160. The exact mechanisms responsible for the dephosphorylation processes are not identified, but the protein alpha-soluble N-ethylmalemide sensitive factor attachment protein (α-SNAP) was recently identified as an AMPK phosphatase (40), whereas protein phosphatases 1 (PP1), 2A (PP2A), 2B (PP2B), and 2C (PP2C) are able to dephosphorylate AS160 (29) and λ-protein phosphatase and can dephosphorylate AS160 and TBC1D1 (11, 33). Differential regulation of the activity of these phosphatases may explain the differences in time course of AMPK, TBC1D1, and AS160 phosphorylation. It is of course also likely that other kinases activated by exercise are responsible for the increased TBC1D1 and AS160 phosphorylation. CaMKII is a probable candidate because several immunoreactive sites identified on AS160 and TBC1D1 are potential CaMKII phosphorylation sites, but inhibition of CaMKII during muscle contraction (when AMPK is active) does not affect AS160 and TBC1D1 phosphorylation (42). This does not exclude that CaMKII may phosphorylate AS160 and TBC1D1 in the postexercise recovery period.
A compelling effect of a single bout of aerobic exercise on whole body glucose homeostasis is the postexercise increase in insulin sensitivity, and increased insulin-stimulated phosphorylation of AS160 after exercise has been suggested to play an important role (2, 12, 25, 34). Our data demonstrate that insulin-stimulated AS160 phosphorylation on AS160 Ser341, Ser704, and PAS are increased after a bout of two-legged aerobic exercise, which is a more physiological stimulus than the one-legged exercise model previously used in studies of AS160 and TBC1D1 phosphorylation in humans. The augmented insulin-stimulated AS160 phosphorylation after exercise does not include increased phosphorylation at Thr642, and this is in agreement with some previous reports (34) but not all (25). This phosphorylation site has most consistently been shown to regulate glucose uptake (28), but it is likely that additional phosphorylation sites are involved in modulating insulin sensitivity after exercise. Similar to previous observations, insulin increased phosphorylation of TBC1D1 at Thr596 (25, 38), but prior exercise did not augment this phosphorylation and our data support the existing evidence that TBC1D1 is not responsible for increased insulin sensitivity after exercise (12, 25). Unlike previous studies, the increased insulin-stimulated AS160 phosphorylation postexercise in this study was not associated with detectable increased glucose uptake during a hyperinsulinemic euglycemic clamp. In the one-legged knee-extensor exercise model, increased insulin-stimulated AS160 phosphorylation is associated with increased thigh glucose uptake measured by arteriovenous differences (25, 34). This method allows for a more precise measurement of glucose uptake specifically in the previously exercised muscles, whereas glucose uptake as measured in this study includes all muscles and the exercise effect in this study may therefore have been too subtle to affect whole body glucose uptake. Our data do not therefore necessarily challenge the well-established finding that insulin-stimulated glucose uptake is increased in previously exercised muscle. Indeed, increased glucose disposal in humans after exercise has previously been detected by the hyperinsulinemic euglycemic clamp technique (3, 6, 7, 24). However, these subjects had either performed exercise to exhaustion (3, 6, 7) or were examined at a later stage in the postexercise period (24). Furthermore, one study reported that only subjects with low baseline insulin sensitivity increase insulin-stimulated glucose disposal after exercise, whereas no effects are seen in subjects with normal baseline insulin sensitivity (7). In agreement with our data, a study using 1 h of exercise at 60% V̇o2 peak in subjects with normal insulin sensitivity did not find increased insulin-stimulated glucose uptake 3 h after exercise (17). It is therefore possible that exercise at this intensity in subjects with normal baseline sensitivity does not increase insulin-stimulated glucose disposal to a level that can be detected by a hyperinsulinemic euglycemic clamp. However, although glucose uptake was unaltered by exercise in our study, a tendency toward decreased glycolysis and increased nonoxidized glucose disposal (glycogenesis) was observed after exercise. This trend toward a shift in glucose metabolism in skeletal muscle after exercise is in accordance with decreased glycogen stores, leading to increased glycogen synthase activity as previously observed (26).
In summary, our data show that exercise-induced AS160 and TBC1D1 phosphorylation were evident 30 min after exercise without simultaneously increased Akt and AMPK phosphorylation. The phosphorylation of AS160 and TBC1D1 30 min after exercise does therefore not follow the regulation of the upstream kinases Akt and AMPK. In addition, we show that exercise under physiological conditions augments insulin-stimulated AS160 phosphorylation at sites that do not include the important Thr642 site. Together, these data provide additional insights into regulation of key regulators of glucose transport in human skeletal muscle.
GRANTS
This work was supported by Danish Agency for Science Technology and Innovation Grant 271-07-0719 (to N.J.) and by the FOOD Study Group/Danish Ministry of Food, Agriculture, and Fisheries; and Ministry of Family and Consumer Affairs Grant 2101–05-0044 (to N. Møller).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.H.V., J.T.T., L.C.G., L.J.G., J.F.W., J.O.L.J., N.M., and N.J. conception and design of research; M.H.V., A.B.M., J.T.T., L.C.G., and N.J. performed experiments; M.H.V., A.B.M., J.T.T., N.M., and N.J. analyzed data; M.H.V., A.B.M., J.T.T., L.C.G., L.J.G., J.F.W., J.O.L.J., N.M., and N.J. interpreted results of experiments; M.H.V. prepared figures; M.H.V., L.J.G., J.F.W., J.O.L.J., and N.J. drafted manuscript; M.H.V., A.B.M., J.T.T., L.C.G., L.J.G., J.F.W., J.O.L.J., N.M., and N.J. edited and revised manuscript; M.H.V., A.B.M., J.T.T., L.C.G., L.J.G., J.F.W., J.O.L.J., N.M., and N.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We give special thanks to Annette Mengel, Helle Zibrandtsen, Lene Ring, Elsebeth Hornemann, and Hanne Fjeldsted Petersen at Aarhus University Hospital, Aarhus, Denmark, for excellent technical assistance.
REFERENCES
- 1.An D, Toyoda T, Taylor EB, Yu H, Fujii N, Hirshman MF, Goodyear LJ. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes 59: 1358–1365, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias EB, Kim J, Funai K, Cartee GD. Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. Am J Physiol Endocrinol Metab 292: E1191–E1200, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bogardus C, Thuillez P, Ravussin E, Vasquez B, Narimiga M, Azhar S. Effect of muscle glycogen depletion on in vivo insulin action in man. J Clin Invest 72: 1605–1610, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bondar RJ, Mead DC. Evaluation of glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides in the hexokinase method for determining glucose in serum. Clin Chem 20: 586–590, 1974. [PubMed] [Google Scholar]
- 5.Catoire M, Mensink M, Boekschoten MV, Hangelbroek R, Muller M, Schrauwen P, Kersten S. Pronounced effects of acute endurance exercise on gene expression in resting and exercising human skeletal muscle. PLoS One 7: e51066, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devlin JT, Hirshman M, Horton ED, Horton ES. Enhanced peripheral and splanchnic insulin sensitivity in NIDDM men after single bout of exercise. Diabetes 36: 434–439, 1987. [DOI] [PubMed] [Google Scholar]
- 7.Devlin JT, Horton ES. Effects of prior high-intensity exercise on glucose metabolism in normal and insulin-resistant men. Diabetes 34: 973–979, 1985. [DOI] [PubMed] [Google Scholar]
- 8.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab 2: 263–272, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Fawcett JK, Scott JE. A rapid and precise method for the determination of urea. J Clin Pathol 13: 156–159, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrannini E. The theoretical bases of indirect calorimetry: a review. Metabolism 37: 287–301, 1988. [DOI] [PubMed] [Google Scholar]
- 11.Frosig C, Pehmoller C, Birk JB, Richter EA, Wojtaszewski JF. Exercise-induced TBC1D1 Ser237 phosphorylation and 14–3-3 protein binding capacity in human skeletal muscle. J Physiol 588: 4539–4548, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funai K, Schweitzer GG, Sharma N, Kanzaki M, Cartee GD. Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am J Physiol Endocrinol Metab 297: E242–E251, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geraghty KM, Chen S, Harthill JE, Ibrahim AF, Toth R, Morrice NA, Vandermoere F, Moorhead GB, Hardie DG, Mackintosh C. Regulation of multisite phosphorylation and 14–3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem J 407: 231–241, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodyear LJ, Kahn BB. Exercise, glucose transport, and insulin sensitivity. Annu Rev Med 49: 235–261, 1998. [DOI] [PubMed] [Google Scholar]
- 15.Gormsen LC, Jessen N, Gjedsted J, Gjedde S, Norrelund H, Lund S, Christiansen JS, Nielsen S, Schmitz O, Moller N. Dose-response effects of free fatty acids on glucose and lipid metabolism during somatostatin blockade of growth hormone and insulin in humans. J Clin Endocrinol Metab 92: 1834–1842, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18: 556–566, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howlett KF, Mathews A, Garnham A, Sakamoto K. The effect of exercise and insulin on AS160 phosphorylation and 14–3-3 binding capacity in human skeletal muscle. Am J Physiol Endocrinol Metab 294: E401–E407, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Jensen J, Ruge T, Lai YC, Svensson MK, Eriksson JW. Effects of adrenaline on whole-body glucose metabolism and insulin-mediated regulation of glycogen synthase and PKB phosphorylation in human skeletal muscle. Metabolism 60: 215–226, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Jessen N, An D, Lihn AS, Nygren J, Hirshman MF, Thorell A, Goodyear LJ. Exercise increases TBC1D1 phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab 301: E164–E171, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jessen N, Goodyear LJ. Contraction signaling to glucose transport in skeletal muscle. J Appl Physiol 99: 330–337, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55: 2067–2076, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem 281: 31478–31485, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Lund S, Flyvbjerg A, Holman GD, Larsen FS, Pedersen O, Schmitz O. Comparative effects of IGF-I and insulin on the glucose transporter system in rat muscle. Am J Physiol Endocrinol Metab 267: E461–E466, 1994. [DOI] [PubMed] [Google Scholar]
- 24.Mikines KJ, Sonne B, Farrell PA, Tronier B, Galbo H. Effect of physical exercise on sensitivity and responsiveness to insulin in humans. Am J Physiol Endocrinol Metab 254: E248–E259, 1988. [DOI] [PubMed] [Google Scholar]
- 25.Pehmoller C, Brandt N, Birk JB, Hoeg LD, Sjoberg KA, Goodyear LJ, Kiens B, Richter EA, Wojtaszewski JF. Exercise alleviates lipid-induced insulin resistance in human skeletal muscle-signaling interaction at the level of TBC1 domain family member 4. Diabetes 61: 2743–2752, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prats C, Helge JW, Nordby P, Qvortrup K, Ploug T, Dela F, Wojtaszewski JF. Dual regulation of muscle glycogen synthase during exercise by activation and compartmentalization. J Biol Chem 284: 15692–15700, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roach WG, Chavez JA, Miinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J 403: 353–358, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab 295: E29–E37, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schweitzer GG, Arias EB, Cartee GD. Sustained postexercise increases in AS160 Thr642 and Ser588 phosphorylation in skeletal muscle without sustained increases in kinase phosphorylation. J Appl Physiol (1985) 113: 1852–1861, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sigal RJ, Kenny GP, Wasserman DH, Castaneda-Sceppa C. Physical activity/exercise and type 2 diabetes. Diabetes Care 27: 2518–2539, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Steele R. Influences of glucose loading and of injected insulin on hepatic glucose output. Ann NY Acad Sci 82: 420–430, 1959. [DOI] [PubMed] [Google Scholar]
- 32.Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KS, Bowles N, Hirshman MF, Xie J, Feener EP, Goodyear LJ. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem 283: 9787–9796, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol 297: C1041–C1052, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Treebak JT, Frosig C, Pehmoller C, Chen S, Maarbjerg SJ, Brandt N, Mackintosh C, Zierath JR, Hardie DG, Kiens B, Richter EA, Pilegaard H, Wojtaszewski JF. Potential role of TBC1D4 in enhanced post-exercise insulin action in human skeletal muscle. Diabetologia 52: 891–900, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, Jorgensen SB, Viollet B, Andersson L, Neumann D, Wallimann T, Richter EA, Chibalin AV, Zierath JR, Wojtaszewski JF. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 55: 2051–2058, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Treebak JT, Pehmoller C, Kristensen JM, Kjobsted R, Birk JB, Schjerling P, Richter EA, Goodyear LJ, Wojtaszewski JF. Acute exercise and physiological insulin induce distinct phosphorylation signatures on TBC1D1 and TBC1D4 in human skeletal muscle. J Physiol 592: 351–375, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treebak JT, Taylor EB, Witczak CA, An D, Toyoda T, Koh HJ, Xie J, Feener EP, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am J Physiol Cell Physiol 298: C377–C385, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vendelbo MH, Clasen BF, Treebak JT, Moller L, Krusenstjerna-Hafstrom T, Madsen M, Nielsen TS, Stodkilde-Jorgensen H, Pedersen SB, Jorgensen JO, Goodyear LJ, Wojtaszewski JF, Moller N, Jessen N. Insulin resistance after a 72-h fast is associated with impaired AS160 phosphorylation and accumulation of lipid and glycogen in human skeletal muscle. Am J Physiol Endocrinol Metab 302: E190–E200, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vichaiwong K, Purohit S, An D, Toyoda T, Jessen N, Hirshman MF, Goodyear LJ. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem J 431: 311–320, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Brautigan DL. alpha-SNAP inhibits AMPK signaling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPKalpha in vitro. Nat Commun 4: 1559, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witczak CA, Hirshman MF, Jessen N, Fujii N, Seifert MM, Brandauer J, Hotamisligil GS, Goodyear LJ. JNK1 deficiency does not enhance muscle glucose metabolism in lean mice. Biochem Biophys Res Commun 350: 1063–1068, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witczak CA, Jessen N, Warro DM, Toyoda T, Fujii N, Anderson ME, Hirshman MF, Goodyear LJ. CaMKII regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. Am J Physiol Endocrinol Metab 298: E1150–E1160, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]