

We found that PM2.5-induced vascular inflammation and insulin resistance and changes in circulating and bone marrow endothelial progenitor cells (EPCs) are prevented by metformin or rosiglitazone treatment. These findings implicate vascular insulin resistance as an important contributor to PM2.5 toxicity and suggest that these insulin-sensitizing drugs attenuate PM2.5-induced vascular injury.
Keywords: air pollution, endothelial progenitor cells, fine particulate matter, PM2.5, metformin, rosiglitazone, T2D, vascular insulin resistance
Abstract
Exposure to fine particular matter (PM2.5) increases the risk of developing cardiovascular disease and Type 2 diabetes. Because blood vessels are sensitive targets of air pollutant exposure, we examined the effects of concentrated ambient PM2.5 (CAP) on vascular insulin sensitivity and circulating levels of endothelial progenitor cells (EPCs), which reflect cardiovascular health. We found that CAP exposure for 9 days decreased insulin-stimulated Akt phosphorylation in the aorta of mice maintained on control diet. This change was accompanied by the induction of IL-1β and increases in the abundance of cleaved IL-18 and p10 subunit of Casp-1, consistent with the activation of the inflammasome pathway. CAP exposure also suppressed circulating levels of EPCs (Flk-1+/Sca-1+ cells), while enhancing the bone marrow abundance of these cells. Although similar changes in vascular insulin signaling and EPC levels were observed in mice fed high-fat diet, CAP exposure did not exacerbate diet-induced changes in vascular insulin resistance or EPC homeostasis. Treatment with an insulin sensitizer, metformin or rosiglitazone, prevented CAP-induced vascular insulin resistance and NF-κB and inflammasome activation and restored peripheral blood and bone marrow EPC levels. These findings suggest that PM2.5 exposure induces diet-independent vascular insulin resistance and inflammation and prevents EPC mobilization, and that this EPC mobilization defect could be mediated by vascular insulin resistance. Impaired vascular insulin sensitivity may be an important mechanism underlying PM2.5-induced vascular injury, and pharmacological sensitization to insulin action could potentially prevent deficits in vascular repair and mitigate vascular inflammation due to exposure to elevated levels of ambient air pollution.
Listen to this article's corresponding podcast at http://ajpheart.podbean.com/e/particulate-matter-induced-vascular-insulin-resistance/.
NEW & NOTEWORTHY
We found that PM2.5-induced vascular inflammation and insulin resistance and changes in circulating and bone marrow endothelial progenitor cells (EPCs) are prevented by metformin or rosiglitazone treatment. These findings implicate vascular insulin resistance as an important contributor to PM2.5 toxicity and suggest that these insulin-sensitizing drugs attenuate PM2.5-induced vascular injury.
exposure to ambient fine particulate matter (≤2.5 μm, PM2.5) is associated with an increased risk of developing cardiovascular disease (CVD) (7, 48) and Type 2 diabetes (T2D) (59). Several studies have shown that acute exposure to PM2.5 precipitates cardiovascular events such as myocardial infarction (55) and ischemic stroke (64), whereas chronic exposures are associated with increased cardiovascular disease risk (3, 45) and accelerated atherogenesis (1, 37, 66). PM exposure also decreases insulin sensitivity (8), exacerbates cardiometabolic disorders, and increases cardiometabolic disease mortality (56), and T2D enhances vulnerability to PM-induced vascular injury (50, 62). While the mechanisms underlying the increased cardiovascular and cardiometabolic risk due to PM exposure remain unclear, data from both human and animal studies indicate that vascular dysfunction is an early and sensitive outcome of PM2.5 exposure (6, 29, 54). Acute exposure to PM2.5 causes conduit artery vasoconstriction (5) and increases blood pressure in healthy adults (70), and chronic PM2.5 exposure is associated with persistent endothelial dysfunction (35). Because the endothelium regulates blood pressure, atherogenesis, arrhythmogenesis, and thrombosis (38), endothelium dysfunction could, in part, contribute to increased CVD risk due to PM2.5 exposure and to the increased susceptibility of diabetic individuals to PM2.5-induced cardiovascular injury.
Endothelial progenitor cells (EPCs) are immature cells expressing endothelial markers that play a nonredundant role in vascular maintenance and repair (20). The level of these cells in the peripheral blood are maintained by continuous mobilization from the bone marrow, and their circulating levels are a sensitive index of cardiovascular and endothelium health (20, 67, 74). Chronically reduced levels of circulating EPCs are associated with endothelium dysfunction and increased CVD risk (42, 74). In our previous studies we found that short-term exposures to PM2.5 decrease the levels of circulating EPCs in humans (51) and mice (21), and that PM2.5 exposure suppressed VEGF-mediated EPC mobilization from the bone marrow in mice (21). Nevertheless, the mechanisms contributing to depressed EPC mobilization after PM2.5 exposure remain unclear. In addition to PM2.5 exposure, circulating EPC levels are decreased also in obesity and diabetes in humans (27, 42) and in murine models (18, 61, 68), suggesting that EPC homeostasis, mobilization, and recruitment may be dependent on insulin signaling as peripheral blood EPC levels are decreased in states of insulin resistance. That insulin resistance contributes to EPC mobilization is supported by the observation that the number of circulating EPCs is decreased in insulin receptor-null mice (28). Furthermore, treatment with an insulin sensitizer, metformin, or thiazolidinediones such as rosiglitazone has been shown to improve not only vascular insulin sensitivity in obesity and hypertension (40, 63, 71) but also to restore circulating EPC levels in diabetes (11, 42, 53). Taken together, these studies indicate that insulin signaling, especially in the vasculature, could contribute to the regulation of EPC homeostasis and that insulin resistance has the potential to disrupt EPC mobilization and their recruitment into the peripheral blood.
The current study, therefore, was designed to determine how PM2.5 exposure affects vascular insulin sensitivity and EPC levels, whether these effects are exacerbated by diet-induced obesity, and whether treatment with an insulin sensitizer prevents PM2.5-induced vascular insulin resistance and EPC retention. Our results show that exposure to concentrated ambient PM2.5 (CAP) induces vascular insulin resistance and decreases circulating EPCs and that these changes are prevented by metformin or rosiglitazone treatment. These findings support the concept that vascular insulin resistance contributes, in part, to PM2.5-induced depletion of circulating EPCs.
MATERIALS AND METHODS
Animals.
All animal experiments were performed in accordance with the APS's Guiding Principles in the Care and Use of Animals following protocols approved by the University of Louisville IACUC. Mice were exposed to HEPA-filtered air or concentrated PM2.5 (CAP, 6 h/day) as previously described (21, 51). Male 8-wk-old C57BL6 mice (Jackson Laboratory, Bar Harbor, ME) either maintained on control diet (CD; 10–13% kcal fat, Research Diets, New Brunswick, NJ; LabDiets, Cincinnati, OH) or placed on a high-fat diet (HFD; 60% kcal fat, LabDiets, Cincinnati, OH) were exposed to air or CAP for 9 or 30 consecutive days as indicated in the experimental protocols (Figs. 1, 2, 3, and 4). In addition, 12-wk-old mice treated either with vehicle (water), metformin, or rosiglitazone were exposed for 9 days to air or CAP following the protocol shown in Figs. 5 and 6. To test whether treatment with an antidiabetic drug would prevent CAP-induced effects, we started the drug treatment 2 days before the exposure to allow for acclimation to the drugs. Metformin (Sigma-Aldrich, St. Louis, MO) was provided in drinking water with estimated graduated daily dosing of 50 mg/kg (preexposure day −2), 150 mg/kg (preexposure day −1), and 300 mg/kg (exposure day 0 to +9) as indicated in Figs. 5A and 6A. This protocol was adapted from a study showing that metformin improves vascular function in a model of insulin resistance (30). Rosiglitazone (1 mg·kg−1·day−1, Sigma-Aldrich) was provided in drinking water 2 days prior and throughout the 9 days of exposure because a similar dose has been shown to improve vascular insulin signaling and function (17, 40). Body weight and fasting (6 h) blood glucose levels (Accu-Check glucometer, Aviva, Roche) were measured. Mice were euthanized with pentobarbital sodium (150 mg/kg), and organs were collected and snap frozen. Vascular insulin signaling was examined ex vivo in freshly isolated aortas stimulated with 100 nM of insulin (32). For this, freshly isolated aortas were cleaned perivascular adipose tissue and then incubated for 1 h in endothelial basal media (standard cell culture conditions, 5% CO2, 37°C; Promocell, Heidelberg, Germany) unless stated otherwise. After stimulation with vehicle or 100 nM insulin (15 min, Humulin-RP, Eli-Lilly, Indianapolis, IN), aortas were washed with ice-cold PBS and snap frozen for Western blot analysis. Blood was used for flow cytometry and for measuring plasma lipids and proteins on a Cobas Mira Plus 5600 Autoanalyzer (Roche) with commercially available kits as previously described (13) or with ELISA kits (insulin and VEGF; ALPCO, Salem, NH, or R&D, Minneapolis, MN, respectively). HOMA-IR and HOMA-β scores were calculated from fasting blood glucose and plasma insulin levels according to the homeostatic model assessment (HOMA).
Fig. 1.
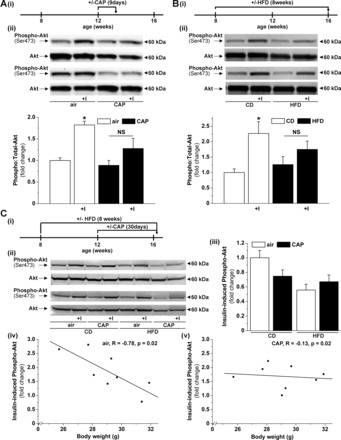
CAP exposure and HFD feeding induce vascular insulin resistance. Western blot analysis (ii) of the insulin-stimulated (100 nM, 15 min) phosphorylation of Akt in aortas isolated from mice exposed for 9 days to air or CAP (A) or fed for 8 wk with either control diet (CD) or high-fat diet (HFD) (B) as indicated in the experimental protocols (i). Data are means ± SE normalized to the air or CD controls (*P < 0.05 control vs. insulin; NS, not significant; n = 5). C: correlation between body weight and insulin-stimulated Akt phosphorylation in CD- or HFD-fed mice exposed for 30 days to air or CAP following the indicated experimental protocol (Ci). Insulin-stimulated Akt phosphorylation (means ± SE, normalized to insulin-stimulated controls, n = 4) was analyzed by Western blot (Cii, Ciii) in isolated aortas incubated in autologous plasma 1 h before stimulation with 100 nM insulin for 15 min. Linear fit [y=A+B*x] between the insulin-induced Akt phosphorylation and the body weight shown as discrete data points for each mouse either exposed to air (Civ) or CAP (Cv).
Fig. 2.
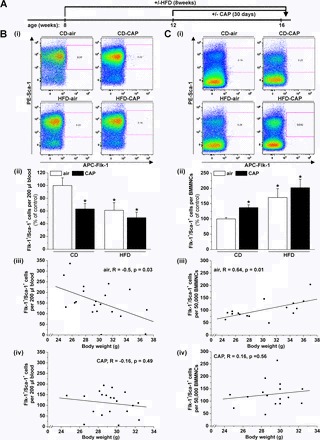
CAP exposure and HFD feeding affect circulating and bone marrow EPC levels. Representative flow cytometry plots (i) and quantification (ii) of circulating (B) and bone marrow resident (C) endothelial progenitor cells (EPCs, PE-Sca-1+/APC-Flk-1+ cells) in mice maintained on control diet (CD) or fed high-fat diet (HFD) exposed for 30 days to air or CAP as indicated in the experimental protocol (A). EPCs were quantified as Sca-1+/Flk-1+ cells per 200 μl blood (Bii) or per number of bone marrow mononuclear cells (BMMNC, Cii), and resulting numbers were normalized to the Sca-1+/Flk-1+ cells per 200 μl blood or per BMMNC in air-exposed CD-fed mice. Data are means ± SE in % of control (*P < 0.05 vs. air-exposed CD-fed mice, n = 8–10). Linear fit [y=A+B*x] between body weight and (Biii, Bvi) the number of Sca-1+/Flk-1+ cells per 200 μl blood or (Ciii, Cvi) the number of Sca-1+/Flk-1+ cells per 50,000 BMMNC shown as discrete data points for each mouse either exposed to air (iii) or CAP (iv).
Fig. 3.
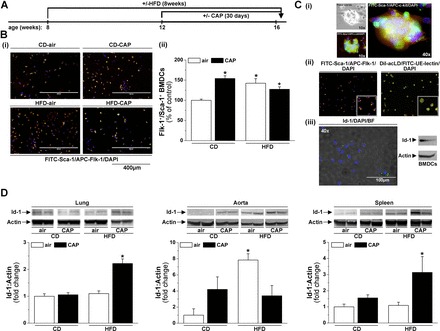
CAP exposure and HFD feeding affect outgrowth of bone marrow EPCs and EPC tissue distribution. Bone marrow and tissues were isolated from control diet (CD) or high-fat diet (HFD) fed mice exposed for 30 days to air or CAP as indicated in the experimental protocol (A). B: representative fluorescence images (×10 original magnification) (i) and quantification (ii) of bone marrow-derived cells (BMDCs). The outgrowth of BMDCs was analyzed by counting cells double positive for Flk-1 and Sca-1 (FITC-Flk-1+/APC-Sca-1+ cells) in five random microscopic fields per animal. C: fluorescence images of a colony forming unit cultured for 4 days labeled with FITC-Sca-1 and APC-c-kit (i) and BMDCs cultured for 7 days labeled with FITC-Sca-1/APC-Flk-1 and DiI-acLDL/UE-lectin (ii) or Id-1-Alexa-488 and DAPI as well as Western blot of BMDC-lysate detected for Id-1 and actin (iii). D: Western blot analysis of the abundance of Id-1 in lungs, aorta, and spleen. Data are means ± SE normalized to the air control (*P < 0.05, + 0.1 > P > 0.05 vs. air-exposed CD-fed mice, n = 4–8).
Fig. 4.
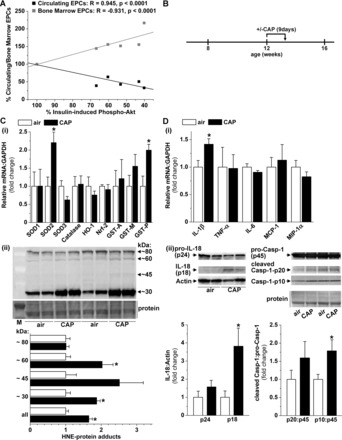
CAP exposure induces vascular oxidative stress and inflammation. A: correlation between CAP-induced vascular insulin resistance and EPC levels. Data are shown as discrete points (mean in % of control) for the densitometric Western blot analysis of aortic insulin-induced Akt phosphorylation vs. the abundance of blood or bone marrow endothelial progenitor cells (EPCs) measured in air or CAP-exposed mice since August 2010. The curve is a linear fit [y=A+B*x] between the CAP-induced vascular insulin resistance and the depletion of circulating and the retention of bone marrow EPCs. CAP-induced aortic oxidative stress (C) and inflammation (D) was analyzed in aortas isolated from mice exposed for 9 days to air or CAP as indicated in the experimental protocol (B). The mRNA levels of antioxidant defense genes (Ci) and pro-inflammatory genes (Di) were measured by RT-PCR and abundance of protein-HNE adducts (Cii), and the activation of IL-18 and Casp-1 (Dii) were analyzed by Western blot. Data are means ± SE normalized to the air control (*P < 0.05, air vs. CAP, n = 5).
Fig. 5.
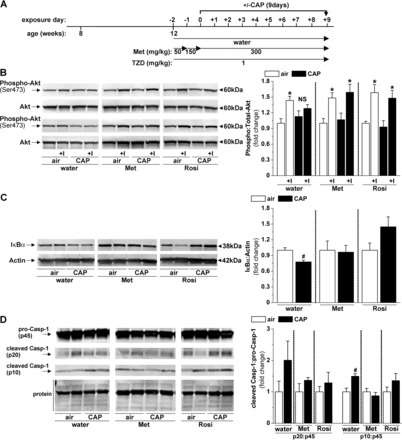
Metformin or rosiglitazone treatment prevents CAP-induced vascular insulin resistance and inflammation. Western blot analysis of aortic insulin-stimulated (ex vivo, 100 nM insulin, 15 min) Akt phosphorylation (B), IκBα degradation (C), and Casp-1 (D) activation in mice treated with water, metformin (Met), or rosiglitazone (Rosi) before and during the 9-day exposure to air or CAP according to the indicated experimental protocol (A). Data are means ± SE normalized to the appropriate air controls (*P < 0.05, control vs. insulin; #P < 0.05 air vs. CAP, n = 4–5).
Fig. 6.
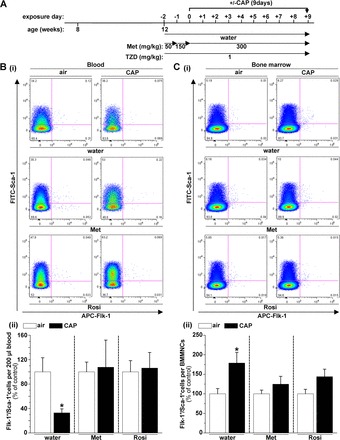
Metformin or rosiglitazone treatment prevents CAP-induced changes in circulating and bone marrow EPCs. Representative flow cytometry plots (i) and quantification (ii) of circulating (B) and bone marrow (C) Flk-1+/Sca-1+ cells in mice treated with water, metformin (Met), or rosiglitazone (Rosi) exposed for 9 days to air or CAP according to the indicated experimental protocol (A). EPCs were quantified as Sca-1+/Flk-1+ cells per 200 μl blood or per bone marrow mononuclear cells (BMMNC), and resulting numbers were normalized to the Sca-1+/Flk-1+ cells per 200 μl blood or per BMMNC in air-exposed mice treated either with water, metformin, or rosiglitazone. Data are means ± SE in % of air controls (*P < 0.05 air vs. CAP, n = 5).
Flow cytometry.
The levels of circulating and bone marrow-resident EPCs were analyzed by flow cytometry as described before (21, 51, 75). Briefly, the levels of Flk-1+/Sca-1+ cells were measured in lysed blood (10 min, RT; BD Pharm Lyse, BD BioSciences, San Jose, CA) and Ficoll gradient separated (400 × g, 30 min, 4°C; Ficoll-Paque PREMIUM, GE Healthcare, Piscataway, NJ) bone marrow cells using APC-Flk-1 and FITC- or PE-Sca-1 antibodies (BD BioSciences). Blood and bone marrow cells double positive for Flk-1 and Sca-1 (Flk-1+/Sca-1+ cells) were analyzed with a LSRII flow cytometer (BD BioSciences) using the FlowJo version8 software (Treestar software).
Culture and immunocytochemistry of bone marrow-derived cells.
Bone marrow-derived cells (BMDCs) were cultured from Ficoll gradient centrifugation-separated bone marrow aspirates as described before (21, 75). Briefly, bone marrow cells (8 × 105) were cultured on fibronectin (10% human fibronectin, Sigma-Aldrich) in endothelial cell media (PromoCell, Heidelberg, Germany) containing 20% FBS (Invitrogen, Carlsbad, CA). After 7 days of culture, BMDCs were used for immunocytochemistry. BMDCs were either labeled with FITC-Sca-1 (1:25; BD BioSciences) and APC-Flk-1 (1:15; BD BioSciences) or DiI-acLDL (2.4 μg/ml; Molecular Probes, Invitrogen) and FITC-UE-lectin (50 μg/ml; Sigma-Aldrich) as described (21). Additionally, cells were labeled with Id-1 (1:250, overnight, 4°C; ProteintechGroup, Chicago, IL) with an Alexa-Fluor-488 secondary antibody (1:500, 1 h, RT; Molecular Probes, Invitrogen) or lysed and used for Western blot analysis of Id-1. For quantification, FITC-Sca-1/APC-Flk-1-positive cells were counted in five random microscopic fields (EVOSfl; AMG, Hill Creek, WA).
Immunoblotting.
Western blotting was performed as described before (21, 75) using antibodies against phospho-Akt (Ser473), Akt, IκBα (1:1,000; Cell Signaling Technology, Danvers, MA), Casp-1 (1:2,000; Epitomics, Burlingame, CA), IL-18 (1:1,000; Rockland, Limerick, PA), Id-1 (1:000; ProteintechGroup), actin (1:2,000, Sigma-Aldrich), and protein-HNE (72). Additionally, amido black protein stain was used as loading control (protein) where indicated. Membranes developed with ECL plus reagent (Amersham Biosciences, Piscataway, NJ) were detected (Typhoon 9400 variable mode imager, Amersham Biosciences, myECL Imager, ThermoFisher) and quantified (Image Quant TL software, Amersham Biosciences, myImageAnalysis Software, ThermoFisher).
Real-time PCR.
Quantitative real-time PCR (RT-PCR) was performed with aortic mRNA isolated with an Exiqon miRCURY RNA isolation kit (Exiqon, Woburn, MA) as described (22) using the following primer sets: tumor necrosis factor-α (TNF-α): forward primer: 5′-GCATGATCCGCGACGTGGAA-3′, reverse primer: 5′-AGATCCATGCCGTTGGCCAG-3′; macrophage inflammatory protein-1α (MIP-1α): forward primer: 5′-ACTGACCTGGAACTGAATGCCTGA-3′, reverse primer: 5′-ATGTGGCTACTTGGCAGCAAACAG-3′; monocyte chemotactic protein-1 (MCP-1): forward primer: 5′-ATGCAGGTCCCTGTCATG-3′, reverse primer: 5′-GCTTGAGGTGGTTGTGGA-3′; interleukin-1β (IL-1β): forward primer: 5′-CTCCATGAGCTTTGTACAAGG-3′, reverse primer: 5′-TGCTGATGTACCAGTTGGGG-3′; soluble superoxide dismutase 1 (SOD1): forward primer: 5′-GATGAAGAGAGGCATGTTGGA-3′, reverse primer: 5′-TGTACGGCCAATGATGGAATG-3′; mitochondrial superoxide dismutase 2 (SOD2): forward primer: 5′-GCGGTCGTGTAAACCTCAT-3′, reverse primer: 5′-CCAGAGCCTCGTGGTACTTC-3′; extracellular superoxide dismutase 3 (SOD3): forward primer: 5′-CTGAGGACTTCCCAGTGAC-3′, reverse primer: 5′-GGTGAGGGTGTCAGAGTGT-3′; Catalase: forward primer: 5′-AGCGACCAGATGAAGCAGTG-3′, reverse primer: 5′-TCCGCTCTCTGTCAAAGTGTG-3′; heme oxygenase-1 (HO-1): forward primer: 5′-CACGCATATACCCGCTACCT-3′, reverse primer: 5′-CCAGAGTGTTCATTCGAGA-3′; nuclear factor (erythroid-derived 2)-like 2 (Nrf2): forward primer: 5′-CTCGCTGGAAAAAGAAGTG-3′, reverse primer: 5′-CCGTCCAGGAGTTCAGAGG-3′; Glutathione S-transferase-A (GST-A): forward primer: 5′-TGATTGCCGTGGCTCCATTTA-3′, reverse primer: 5′-CAACGAGAAAAGCCTCTCCGT-3′; Glutathione S-transferase-M (GST-M): forward primer: 5′-AGCTCACGCTATTCGGCTG-3′, reverse primer: 5′-GCTCCAAGTATTCCACCTTCAGT-3′; Glutathione S-transferase-P (GST-P): forward primer: 5′-ATGCCACCATACACCATTGTC-3′, reverse primer: 5′-GGGAGCTGCCCATACAGAC-3′; GAPDH: forward primer: 5′-AGGTCATCCCAGAGCTGAACG-3′, reverse primer: 5′-GGAGTTGCTGTTGAAGTCGCA-3′. The primer set for interleukin-6 (IL-6) was purchased from SA Bioscience (SABioscience, Qiagen, Valencia, CA).
Statistical analysis.
Data are means ± SE. To compare two groups, unpaired Student's t-test was used and one-way ANOVA with Bonferroni test post hoc was used for multiple group comparisons (SigmaStat, SPSS, Chicago, IL). P < 0.05 was considered significant.
RESULTS
CAP exposure and HFD feeding impair vascular insulin sensitivity.
Our previous study indicates that vascular signaling is a sensitive target of PM2.5, which impairs aortic responses to VEGF (21). To determine whether vascular insulin signaling is similarly affected, we exposed mice maintained on a CD to air or CAP for 9 days (Fig. 1Ai) and examined changes in insulin-stimulated phosphorylation of Akt, a commonly used marker of insulin sensitivity (33) by Western blotting. Insulin (100 nM, 15 min, ex vivo) stimulated a significant increase in the levels of Akt phosphorylation in aortas isolated from mice breathing HEPA-filtered air (Fig. 1Aii). In contrast, insulin failed to induce significant Akt phosphorylation in aortas isolated from mice exposed to CAP (Fig. 1Aii). Exposure to CAP did not affect fasting blood glucose levels or body weight (Table 1). These observations suggest that exposure to PM2.5 induces diet-independent vascular insulin resistance without effecting obesity. A similar impairment of insulin-stimulated Akt phosphorylation (Fig. 1Bii) also was observed in aortas isolated from mice placed on HFD for 8 wk (Fig. 1Bi); however, in this case, the decrease in vascular insulin sensitivity was accompanied by an increase in body weight and fasting blood glucose levels (Table 1).
Table 1.
Physiological parameters in air or CAP exposed (9 days) mice and CD- or HFD-fed (8 wk) mice
| 9-Day Exposure |
8-Wk Feeding |
|||||
|---|---|---|---|---|---|---|
| Parameter | Air | CAP | P | CD | HFD | P |
| BW, g | 27.48 ± 0.51 | 28.60 ± 0.87 | 0.297 | 27.08 ± 0.85 | 38.12 ± 2.07 | >0.001 |
| Glucose, mg/dl | 187 ± 8 | 176 ± 11 | 0.434 | 82 ± 9 | 179 ± 7 | >0.001 |
| Liver/BW, ×102 | 2.69 ± 0.28 | 2.69 ± 0.10 | 0.983 | |||
| Adipose/BW, ×102 | 2.06 ± 0.29 | 6.13 ± 0.26 | >0.001 | |||
Data are means ± SE, n = 5. Body weight, organ/body weight ratios, and blood glucose levels of mice either exposed for 9 days to air or CAP or fed for 8 wk with control diet (CD, 10 % kcal fat) or high fat diet (HFD, 60% kcal fat). CAP, concentrated ambient fine particulate matter; CD, control diet; HFD, high-fat diet; BW, body weight.
Collectively, these results demonstrate that exposure to concentrated PM2.5 for only 9 days selectively impairs vascular insulin signaling to the extent similar to that of 8-wk HFD feeding. However, while HFD feeding significantly increased body weight and blood glucose levels, CAP exposure did not affect either parameter (Table 1).
Next, to examine the effects of more prolonged PM2.5 exposure and to assess whether PM2.5 exposure affects HFD-induced vascular insulin signaling and its dependency on changes in body weight, we exposed mice fed CD or HFD (4 wk) to air or CAP for 30 days with continuation of the respective diets (Fig. 1Ci). As before, we analyzed insulin-stimulated Akt phosphorylation in the aorta by Western blot (Fig. 1, Cii and Ciii). Insulin-induced Akt phosphorylation in HEPA-filtered air-exposed mice fed CD or HFD was negatively correlated with body weight (Fig. 1Civ), suggesting that HFD feeding impairs vascular insulin signaling related to changes in body weight. Because vascular insulin signaling was impaired by CAP exposure similarly in both CD and HFD-fed mice, there was no correlation between body weight and vascular insulin signaling in CAP-exposed mice (Fig. 1Cv). These results suggest that PM2.5 exposure induces vascular insulin resistance, independent of changes in body weight.
CAP exposure and HFD feeding affect EPC homeostasis.
Our previous studies demonstrate that exposure to increased levels of PM2.5 decreases the levels of circulating EPCs in humans (51) and mice (21). The levels of EPCs in the peripheral blood also are decreased because of obesity and T2D (27, 42, 61, 68). Hence, to test for the effects of combined CAP exposure and HFD feeding on EPCs, we exposed mice placed on a CD or HFD to air or CAP for 30 days (Fig. 2A). As expected, feeding an HFD led to an increase in body weight, while CAP exposure did not affect body weight of mice fed either CD or HFD mice (Table 2). Moreover, no changes in plasma levels of VEGF were observed in any treatment group (Table 2). The levels of circulating EPCs (Flk-1+/Sca-1+ cells) detected by flow cytometry (Fig. 2Bi) were decreased by 40% in mice exposed to CAP (Fig. 2Bii). A similar extent of decrease in EPCs was observed in the mice fed a HFD for 8 wk (Fig. 2Bii). However, exposure of HFD-fed mice to CAP did not lead to greater suppression of EPC levels, indicating the absence of an additive effect (Fig. 2Bii). Although the decrease of circulating EPCs induced by HFD feeding in air-exposed mice was correlated negatively with body weight (Fig. 2Biii), there was no correlation between body weight and peripheral blood EPC levels in CAP-exposed mice (Fig. 2Biv) because CAP exposure suppressed circulating EPCs to similar levels in both CD and HFD-fed mice. Taken together, these results indicate that CAP exposure and HFD feeding similarly decrease circulating levels of EPCs, reinforcing the view that circulating EPCs are sensitive targets of both CAP and HFD, although CAP-induced suppression of EPCs is independent of changes in body weight.
Table 2.
Physiological parameters in CD- or HFD-fed mice exposed to air or CAP for 30 days
| CD |
HFD |
|||
|---|---|---|---|---|
| Parameter | Air | CAP | Air | CAP |
| BW, g | 26.13 ± 0.45 | 28.03 ± 0.82 | 33.95 ± 0.83* | 32.56 ± 1.10* |
| Glucose, mg/dl | 121 ± 10 | 124 ± 8 | 139 ± 3 | 134 ± 3 |
| Lung weight/BW, ×103 | 6.00 ± 0.01 | 5.68 ± 0.27 | 5.01 ± 0.14 | 4.91 ± 0.29 |
| Heart weight/BW, ×103 | 4.77 ± 0.18 | 4.72 ± 0.01 | 4.62 ± 0.11 | 5.04 ± 0.20 |
| Liver weight/BW, ×102 | 4.09 ± 0.25 | 4.09 ± 0.18 | 3.73 ± 0.08 | 3.80 ± 0.19 |
| Spleen weight/BW, ×103 | 3.22 ± 0.38 | 3.47 ± 0.52 | 3.36 ± 0.35 | 3.81 ± 1.03 |
| VEGF, pg/ml | 28.10 ± 1.14 | 29.74 ± 0.66 | 28.46 ± 0.64 | 29.02 ± 1.15 |
Data are means ± SE, n = 3–8. Body weight, organ/body weight ratios, blood glucose, and plasma VEGF levels of CD- or HFD-fed mice exposed for 30 days to air or CAP (see Fig. 2A). CAP, concentrated ambient fine particulate matter; CD, control diet (10% kcal fat); HFD, high fat diet (60% kcal fat); BW, body weight; VEGF, vascular endothelial growth factor.
P < 0.05 vs. appropriate diet-matched control.
Previous studies have shown that EPCs in the peripheral blood originate from the bone marrow (67). Therefore, to determine whether the decrease in circulating EPC levels in CAP-exposed mice is due to changes in the levels of EPCs in the bone marrow, we measured bone marrow-resident EPCs by flow cytometry (Fig. 2Ci). We found that CAP exposure increased the levels of bone marrow resident EPCs in mice maintained on CD (Fig. 2Cii). A similar increase in bone marrow resident EPCs also was observed in mice fed HFD (Fig. 2Cii). However, even though body weight was positively correlated with bone marrow EPC levels in air-exposed mice (Fig. 2Ciii), no such correlation was observed in CAP-exposed mice (Fig. 2Civ).
To confirm the data obtained by flow cytometry, we measured bone marrow EPC levels using bone marrow outgrowth assays (Fig. 3B). The results of the outgrowth assays show that either CAP exposure or HFD feeding increases the number of BMDCs positive for Flk-1 and Sca-1 (Fig. 3B). Additional phenotypic characterization of the BMDCs (Fig. 3C) showed that the initial colony forming units, which were double positive for c-kit/Sca-1 (Fig. 3Ci), proliferate into Flk-1+/Sca-1+ and UE-lectin+/acLDL+ cells (Fig. 3Cii). These cells were also positive for the EPC-specific transcription factor inhibitor of DNA binding-1 (Id-1) (Fig. 3Ciii), and Id-1 positivity of the BMDCs was confirmed by Western blot analysis (Fig. 3Ciii).
Id-1 is an EPC transcription factor that plays an important role in the mobilization, recruitment, and proliferation of EPCs and is used to track tissue recruitment of EPCs (41, 44). Hence, to determine whether the decrease in circulating EPCs is due to an increase in tissue recruitment or retention of these cells into peripheral tissues, we examined the abundance of Id-1 in aorta, lung, spleen, heart, liver, adipose tissue, and skeletal muscle by Western blot analysis (Fig. 3D). We found that the abundance of Id-1 in the aorta was increased in mice fed an HFD, while the levels of Id-1 in the lung and the spleen were increased only in CAP-exposed HFD-fed mice (Fig. 3D). In addition, HFD increased Id-1 abundance in the liver of air- and CAP-exposed mice, while CAP exposure increased Id-1 in the skeletal muscle of both CD and HFD-fed mice (data not shown). In contrast, Id-1 levels in the heart and adipose tissue were not affected in any treatment group (data not shown). These results suggest that CAP exposure and HFD feeding increase tissue recruitment of EPCs, although the sites of recruitment differ by treatment. Moreover, in CAP-exposed mice, the levels of circulating EPCs were negatively correlated, and reciprocally bone marrow EPCs were positively correlated with the extent of insulin-stimulated Akt phosphorylation (Fig. 4A), suggesting that the changes in EPC levels may be related to vascular insulin resistance.
Exposure to CAP induces vascular oxidative stress and inflammation.
Because oxidative stress and inflammation have been suggested to contribute to the vascular effects of CAP exposure (29) as well as to diet-induced vascular insulin resistance (19, 32), we tested whether CAP-induced vascular insulin resistance is accompanied by vascular oxidative stress and inflammation. To test for vascular oxidative stress and inflammation, we isolated aortas from mice exposed for 9 days to air or CAP (Fig. 4B) and examined the abundance of antioxidant enzyme mRNA and protein-HNE adducts (Fig. 4C), as well as the mRNA levels of pro-inflammatory genes and IL-18 and Casp-1 activation (Fig. 4D) by RT-PCR and Western blot. We found that CAP exposure significantly increased the levels of SOD2 and GST-P mRNA (Fig. 4Ci) and the abundance of protein-HNE adducts (Fig. 4Cii), indicating the induction of vascular oxidative stress due to CAP exposure. Our measurements of the levels of mRNA of pro-inflammatory genes in the aorta (Fig. 4Di) showed an increase in the abundance of IL-1β, indicating the activation of the NF-kB pathway, while other NF-κB encoded genes such as IL-6 or TNF-α (36) were not affected by CAP exposure. Increased levels of IL-1β by the Toll-like receptor 4 (TLR4)/NFκBα pathway is an important priming step for the activation of the inflammasome pathway (76), and because activation of the inflammasome has been implicated in the toxicity of environmental particles (24, 58) as well as in obesity-induced insulin resistance (65), we asked whether CAP exposure activates the inflammasome in the vascular wall. The inflammasome is a subcellular aggregate that upon activation cleaves pro-Casp-1 into its active 20 kDa (p20) and 10 kDa (p10) subunits that are responsible for the proteolytic conversion of IL-1β and IL-18 into its mature (active) form (69, 76). Hence, to test for a CAP-induced activation of the inflammasome pathway, we measured the aortic abundance of full-length (p24) and cleaved (p18) IL-18 and the active Casp-1 subunits p20 and p10 by Western blot (Fig. 4Dii). We found that CAP exposure not only increased the abundance of the cleaved active IL-18 (p18) but it also increased the abundance of the p10 subunit of Casp-1 (p10), consistent with the activation of the inflammasome pathway.
Insulin sensitizers prevent CAP-induced vascular insulin resistance and inflammation.
Because treatment with metformin or rosiglitazone has been shown to improve vascular insulin sensitivity in obesity and hypertension (40, 63, 71), we tested whether an insulin sensitizer could prevent vascular insulin resistance after CAP exposure. For this preventive approach, we treated mice with metformin or rosiglitazone prior and throughout the 9-day exposure to air or CAP as indicated in the experimental protocol (Fig. 5A).
Treatment with the insulin sensitizer had no effects on fasting blood glucose and plasma insulin levels, or the HOMA-IR or HOMA-β scores (Table 3). Moreover, even though metformin treatment appeared to reduce epididymal adiposity similarly in air- and CAP-exposed mice, no changes in body weight or other organ/body weight ratios were observed, and the levels of plasma lipids and proteins also were unchanged (Table 3).
Table 3.
Physiological parameters in 9-day air- or CAP-exposed mice treated with vehicle (water), metformin or rosiglitazone
| Water |
Metformin |
Rosiglitazone |
||||
|---|---|---|---|---|---|---|
| Parameter | Air | CAP | Air | CAP | Air | CAP |
| Body weight, g | 26.82 ± 0.93 | 26.48 ± 0.58 | 26.52 ± 0.54 | 28.78 ± 0.74 | 29.06 ± 0.88 | 28.96 ± 1.46 |
| Glucose, g/dl | 222 ± 14 | 193 ± 18 | 209 ± 4 | 164 ± 12 | 209 ± 14 | 147 ± 16 |
| Insulin, ng/ml | 0.40 ± 0.03 | 0.38 ± 0.02 | 0.43 ± 0.04 | 0.37 ± 0.03 | 0.43 ± 0.07 | 0.39 ± 0.04 |
| HOMA-IR | 6.3 ± 0.5 | 5.3 ± 0.7 | 6.3 ± 0.5 | 4.2 ± 0.3 | 6.3 ± 1.0 | 4.0 ± 0.6 |
| HOMA-β, % | 27.6 ± 4.4 | 33.4 ± 5.4 | 30.9 ± 3.4 | 40.5 ± 7.5 | 32.0 ± 6.2 | 55.4 ± 12.4 |
| Heart/BW, ×103 | 4.56 ± 0.27 | 4.64 ± 0.19 | 4.45 ± 0.06 | 4.30 ± 0.08 | 4.48 ± 0.09 | 4.81 ± 0.24 |
| Liver/BW, ×102 | 4.08 ± 0.14 | 3.93 ± 0.14 | 4.30 ± 0.17 | 4.06 ± 0.11 | 4.48 ± 0.15 | 4.29 ± 0.15 |
| Adipose/BW, ×102 | 1.71 ± 0.27 | 1.69 ± 0.12 | 1.08 ± 0.08* | 0.93 ± 0.06* | 1.12 ± 0.07 | 1.32 ± 0.08 |
| Lung/BW, ×103 | 4.82 ± 0.16 | 4.89 ± 0.08 | 5.07 ± 0.18 | 4.76 ± 0.08 | 4.88 ± 0.06 | 5.28 ± 0.27 |
| Spleen/BW, ×103 | 3.16 ± 0.24 | 3.14 ± 0.13 | 3.07 ± 0.11 | 2.88 ± 0.08 | 2.99 ± 0.18 | 3.29 ± 0.19 |
| Triglyceride, mg/dl | 49.90 ± 2.38 | 47.27 ± 3.58 | 48.61 ± 1.72 | 56.17 ± 7.05 | 45.74 ± 4.17 | 35.64 ± 2.52 |
| Cholesterol, mg/dl | 58.21 ± 2.87 | 59.36 ± 3.87 | 58.70 ± 0.91 | 61.42 ± 1.70 | 56.82 ± 2.89 | 56.91 ± 4.81 |
| HDL, mg/dl | 64.83 ± 3.49 | 62.00 ± 9.77 | 65.98 ± 1.57 | 65.71 ± 3.30 | 64.63 ± 3.17 | 63.26 ± 6.57 |
| LDL, mg/dl | 16.15 ± 1.13 | 13.24 ± 2.42 | 20.27 ± 1.46 | 16.64 ± 2.27 | 24.82 ± 1.55* | 18.17 ± 2.24 |
| HDL/LDL | 4.94 ± 0.19 | 4.74 ± 0.16 | 3.30 ± 0.16 | 4.20 ± 0.49 | 2.66 ± 0.24 | 3.72 ± 0.77 |
| TP, g/dl | 4.46 ± 0.09 | 4.47 ± 0.08 | 4.05 ± 0.08 | 4.04 ± 0.10 | 4.12 ± 0.10 | 4.51 ± 0.12 |
| Albumin, g/dl | 2.82 ± 0.05 | 2.86 ± 0.09 | 2.70 ± 0.03 | 2.47 ± 0.07* | 2.59 ± 0.08 | 2.73 ± 0.04 |
| Albumin/TP | 0.63 ± 0.01 | 0.64 ± 0.02 | 0.67 ± 0.01* | 0.61 ± 0.01 | 0.63 ± 0.01 | 0.61 ± 0.01 |
| ALT, U/l | 14.8 ± 1.1 | 16.9 ± 1.8 | 16.1 ± 1.3 | 15.3 ± 2.0 | 17.6 ± 0.6 | 14.7 ± 0.5 |
| AST, U/l | 61.2 ± 3.2 | 84.6 ± 7.1 | 54.9 ± 2.8 | 82.8 ± 12.4 | 62.3 ± 13.6 | 74.6 ± 10.6 |
| CK, U/l | 225.4 ± 16.5 | 296.0 ± 56.7 | 191.3 ± 23.6 | 345.5 ± 61.3 | 178.0 ± 43.7 | 209.1 ± 71.2 |
Data are means ± SE, n = 5. Physiological parameters measured in water, metformin, or rosiglitazone-treated mice exposed for 9 days to air or CAP (see Fig. 5A). BW, body weight; HOMA-IR, fasting blood glucose [mmol/l] × fasting plasma insulin levels [mU/l]/22.5; HOMA-β, 20 × fasting plasma insulin levels [mU/l]/ fasting blood glucose [mmol/l]-3.5, %; HDL, high-density lipoprotein cholesterol; LDL, low-density lipoprotein cholesterol; TP, total protein; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase.
P < 0.05 vs. appropriate water group.
To determine whether treatment with an insulin sensitizer affects CAP-induced vascular insulin resistance, we measured insulin-stimulated Akt phosphorylation as before (Fig. 5B). Consistent with our previous results, we found that CAP exposure impaired vascular insulin-stimulated Akt phosphorylation in aortas of vehicle-treated (water) mice (Fig. 5B). In contrast, insulin signaling was unaffected in aortas isolated from CAP-exposed mice treated with metformin or rosiglitazone (Fig. 5B), indicating that both insulin sensitizers were equally effective in preventing CAP-induced vascular insulin resistance.
Next, we tested whether treatment with metformin or rosiglitazone would also prevent CAP-induced vascular inflammation (Fig. 5, C and D). To test for the activation of NF-κB and inflammasome pathways, the aortic abundance of IκBα (Fig. 5C) and Casp-1-p20 and p10 (Fig. 5D) was measured by Western blot. Consistent with CAP-induced increase in mRNA levels of IL-1β (Fig. 4Di), CAP exposure decreased IκBα levels in vehicle-treated mice (Fig. 5C), indicating an activation of the NF-κB pathway. Likewise, CAP exposure increased the abundance of the Casp-1 subunit p10 suggesting activation of the inflammasome pathway (Fig. 5D). In contrast, the levels of IκBα and Casp-1-p10/p20 were unchanged in aortas isolated from metformin or rosiglitazone-treated mice exposed to CAP (Fig. 5, C and D), indicating that treatment with either insulin sensitizer prevented CAP-induced vascular inflammation.
Insulin sensitizers prevent CAP-induced effects on EPC homeostasis.
In addition to protecting against CAP-induced vascular insulin resistance and inflammation, treatment with metformin or rosiglitazone prevented CAP-induced changes in EPC levels in the peripheral blood and the bone marrow (Fig. 6). As shown in Fig. 6, and in agreement with our previous observations (21), CAP exposure for 9 days decreased circulating EPC levels (Fig. 6B) and increased bone marrow EPCs (Fig. 6C); however, treatment with either metformin or rosiglitazone prevented CAP-induced changes in EPC levels in the peripheral blood (Fig. 6B) and the bone marrow (Fig. 6C). These results suggest that the CAP-induced changes in EPC levels likely are mediated by vascular insulin resistance.
DISCUSSION
The major findings of this study are that exposure to concentrated ambient PM2.5 induces diet- and obesity-independent vascular insulin resistance, an effect characterized by an early induction, accompanied by vascular inflammation and paralleled by a decrease in circulating EPCs. Although HFD feeding also induces vascular insulin resistance and decreases EPC levels in the circulation, these diet-induced changes appear to be related to changes in obesity. We also found that CAP-induced vascular insulin resistance, vascular inflammation, and EPC depletion were simultaneously prevented by metformin or rosiglitazone treatment, indicating that vascular insulin signaling may be a primary target of CAP. Moreover, our results show that exposure to CAP induces vascular inflammation via activation of both the NF-κB and the inflammasome pathways, and that these pro-inflammatory vascular responses are prevented by either insulin sensitizer metformin or rosiglitazone. Thus our study is consistent with a previous study implicating the contribution of the TLR4 pathway in PM2.5-induced vascular dysfunction (29), as we show that exposure to concentrated ambient PM2.5 activates inflammatory processes in the aorta that is tightly coupled to vascular insulin resistance and EPC changes.
Extensive epidemiological and experimental studies suggest that exposure to PM2.5 increases the cardiovascular disease risk (7, 48). In addition, PM2.5 exposure has also been linked to the development of insulin resistance and diabetes (8, 10, 14, 52). Although multiple mechanisms have been proposed to contribute to this risk, endothelial dysfunction appears to be an important mediator of the cardiovascular effects of PM2.5 (5, 6, 29, 35, 54, 70). Significant vascular dysfunction has also been observed in humans exposed to diesel exhaust (46) and in experimental animals exposed to nanoparticles, PM10, diesel exhaust, or cigarette smoke (12, 13, 31, 34, 39, 49, 73). Collectively, this body of evidence suggests that vascular function is a key target of respirable particles and that some of the cardiovascular effects of such air pollutant exposures may be mediated by vascular dysfunction. However, the mechanism by which air pollutant exposure leads to vascular dysfunction remains unclear. In this regard, our results showing that exposure to concentrated ambient PM2.5 causes vascular insulin resistance and inflammation provide one mechanism by which PM2.5 exposure could lead to widespread changes in vascular function, ranging from blood pressure regulation and atherogenesis to increased thrombosis. While additional investigations are required to delineate the contribution of vascular insulin resistance to varied manifestations of vascular dysfunction, our observations suggest that vascular insulin resistance and inflammation may be an important mechanism underlying the development of vascular injury induced by the exposure to PM2.5 and/or related air pollutants.
Our results showing that exposure to concentrated ambient PM2.5 increases vascular inflammation and oxidative stress suggest that such a vascular insult is a significant outcome after inhalation of PM2.5. While local vascular inflammation due to PM2.5 exposure has been noted before (16), neither the nature nor the mechanism of this vascular insult is well understood. We found that exposure to CAP activates the aortic NF-κB and inflammasome pathways, processes that are also affected by diet-induced obesity. In diet-induced obesity, metabolites such as glucose or free fatty acids not only activate the NF-κB pathway via TLRs, which upregulate IL-1β or IL-18 mRNA, but also trigger the assembly of the inflammasome and activate Casp-1 (76). Activated Casp-1 cleaves pro-IL-1β and pro-IL-18 into active cytokines that contribute to vascular inflammation (76). In the present study, we found that exposure to CAP led to the activation of NF-κB and the inflammasome pathway in the blood vessel as indicated by degradation of IκBα, stimulation of IL-1β transcription, activation of Casp-1, and cleavage of pro-IL-18. However, the observation that the activation of these pathways could be prevented by treating the mice with an insulin sensitizer, metformin, or rosiglitazone, supports the hypothesis that increased vascular insulin resistance may be a significant mechanism and important contributor to subsequent vascular inflammation in mice exposed to concentrated ambient PM2.5.
Our results showing that treatment with an insulin sensitizer decreases CAP-induced vascular inflammation suggest that the development of vascular inflammation is likely secondary to vascular insulin resistance. However, in animal models of diet-induced obesity, vascular insulin resistance develops within 1–2 wk of HFD feeding and is accompanied by the activation of the NF-κB pathway (33), and both vascular insulin resistance and inflammation are attenuated in TLR4-null mice (32), suggesting that vascular inflammation contributes to the development of vascular insulin resistance in diet-induced obesity. Hence, it appears that the relationship between vascular insulin resistance and inflammation is complex and interdependent. Our observations that an insulin sensitizer prevents vascular inflammation support that upon PM2.5 exposure vascular insulin resistance contributes to the development of vascular inflammation, whereas in diet-induced obesity, vascular inflammation likely gives rise to vascular insulin resistance. Although our observations that insulin sensitizers prevent vascular inflammation do support the notion that vascular inflammation is secondary to vascular insulin resistance, the mode-of-action of the insulin sensitizers is unclear, and even though treatment with either metformin or rosiglitazone improves vascular insulin sensitivity (40, 63, 71) these drugs may affect vascular oxidative stress and inflammation as well. The effects of metformin have been mainly related to the improvement of glucose levels via the regulation of hepatic glucose production, but metformin can also decrease inflammation and oxidative stress (47). Metformin inhibits vascular NF-κB activation (26) and reduces vascular oxidative stress while increasing endothelial NOS (eNOS) activity and NO production in a mouse model of obesity (9). Thiazolidinediones, such as rosiglitazone or pioglitazone, are peroxisome proliferator-activated receptor γ ligands and their beneficial effects are largely attributed to their capability to redistribute lipid deposits (60) and to their anti-inflammatory (60) and antioxidant (25) effects. Nevertheless, it is unclear whether the insulin sensitizers directly affect vascular inflammation and oxidative stress, or whether these effects are due to the resolution of vascular insulin resistance. Regardless, because oxidative stress and inflammation appear to be the key mediators of the PM2.5-induced vascular effects (29), the anti-inflammatory and antioxidant features of these drugs, in addition to their insulin-sensitizing effects, could be important contributing factors to their beneficial outcomes on vascular insulin sensitivity and EPC levels after PM2.5 exposure.
In addition to preventing vascular insulin resistance and inflammation, treatment with metformin and rosiglitazone also prevented the suppression of EPC levels in the peripheral blood of CAP-exposed mice. Results of several studies suggest that EPCs play an important role in maintaining vascular health by promoting vascular repair and regeneration (74). The circulating levels of these cells are negatively associated with CVD risk and severity and are predictive of CVD mortality (20). Our previous studies have shown that exposure to increased levels of ambient air pollution is associated with a decrease in the EPC levels in the peripheral blood even in healthy individuals (51) and that the levels of these cells are also decreased in mice exposed to concentrated ambient PM2.5 (21). In addition to PM2.5, levels of circulating EPCs have also been shown to be sensitive to other air pollutants such as PM10 (4), acrolein (75), environmental tobacco smoke (23), nickel nanoparticles (43), and diesel exhaust (57). Our results showing a decrease in circulating EPCs and an increase in bone marrow EPC levels and our previous findings (21) suggest that exposure to PM2.5 induces a bone marrow mobilization defect, which prevents their egress to the peripheral blood. A similar mobilization defect has been observed in animal models of obesity and diabetes (18, 61, 68), suggesting that both diabetes and obesity affect EPC homeostasis possibly by mechanisms similar to those underlying the mobilization defect in PM2.5-exposed mice. Indeed, we found that this mobilization defect is prevented in mice treated with metformin and rosiglitazone, suggesting that the failure to mobilize EPCs is likely related to vascular insulin resistance and that this defect could be overcome by enhancing insulin sensitivity. Upon vascular injury, EPCs are recruited from the bone marrow to the peripheral blood by the release of VEGF and VEGF-induced phosphorylation of Akt and eNOS (2, 15, 18). Because impaired vascular insulin signaling also reflects diminished Akt and eNOS phosphorylation, it seems plausible that defects in EPC mobilization in PM2.5-exposed mice may be secondary to vascular insulin resistance. Several studies showing that insulin sensitizer treatment improves vascular insulin sensitivity (40, 63, 71) and restore levels of circulating EPCs levels (11, 42, 53) further support this notion.
In addition to the reported EPC mobilization defect, our results also show selective organ recruitment of EPCs, particularly in the context of HFD. For example, in mice exposed to CAP, an increase in Id-1 abundance was observed in several organs, particularly the lung and spleen of HFD-fed mice, indicating increased recruitment or retention of EPCs. This observation points toward ongoing endothelial injury in these organs, which is exacerbated by HFD, and suggests that in addition to the mobilization defect, depletion of EPCs in the peripheral blood may also be in part due to increased recruitment to peripheral organs. Thus even though PM2.5 did not exacerbate HFD-induced EPC suppression, increased tissue recruitment of EPCs indicates that PM2.5 exposure exacerbates vascular injury in obese animals. Potentiation of such vascular injury could be one mechanism contributing to increased susceptibility of diabetic individuals to PM2.5-induced vascular dysfunction (50, 62).
In summary, we found that PM2.5 exposure induced vascular oxidative stress, inflammation, and insulin resistance, which were associated with the depletion of circulating EPCs and the retention of EPCs in the bone marrow. These effects occurred at exposure doses similar to the 24-h average levels of PM2.5 in most major US cities (20–35 μg/m3)(7). For example, mice treated with metformin or rosiglitazone were exposed to a concentration of 84 μg/m3 that corresponds to a 24-h PM2.5 exposure level of ∼21 μg/ m3. Despite a similar pattern of PM2.5- and HFD-induced vascular insulin resistance and EPC effects, the mechanism of PM2.5 action does not appear to depend on obesity-related factors. Our experiments with mice treated with metformin or rosiglitazone showed that treatment with either drug not only prevented PM2.5-induced vascular insulin resistance but it also reduced vascular inflammation and prevented changes in EPC homeostasis. On the basis of these findings we propose that PM2.5 exposure results in oxidative stress. This in turn leads to the selective induction of insulin resistance and the activation of the pro-inflammatory NF-κB and inflammasome pathways in the blood vessel, and vascular insulin resistance affects the mobilization of EPCs from the bone marrow to the peripheral blood (Fig. 7). While further studies are required to test the model in detail, the view that vascular insulin resistance is an important mechanism underlying vascular injury and systemic changes in EPC mobilization could provide a useful paradigm for understanding and evaluating the many adverse cardiovascular and cardiometabolic effects of PM2.5 and related pollutants and how the pathological consequences of these effects could be therapeutically minimized and managed.
Fig. 7.
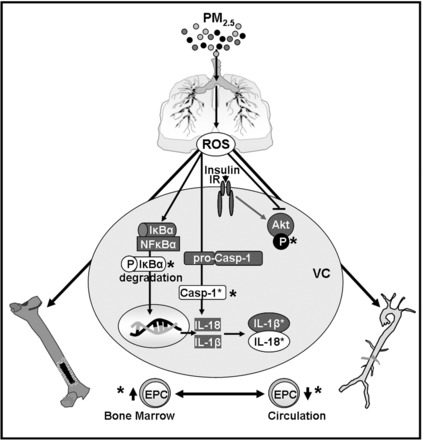
Proposed mechanism of the PM2.5-induced vascular effects. PM2.5 exposure triggers by inducing oxidative stress the activation of the NF-κB and the inflammasome pathways in the blood vessel which stimulates the transcription and cleavage of IL-1β and IL-18. PM2.5 exposure also induces vascular insulin resistance, impairing vascular insulin-stimulated Akt phosphorylation which may contribute to the suppression of the recruitment of EPCs. Treatment with either insulin sensitizer, metformin or rosiglitazone, prevented NF-κB and inflammasome activation as well as vascular insulin resistance and changes in circulating and bone marrow EPC levels as indicated by *.
GRANTS
This work was supported in part by American Heart Association Grant 12SDG9380000 and National Institute of General Medical Sciences Grant GM-103492.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
P.H., A.B., and D.J.C. conception and design of research; P.H., J.P.M., and D.J.C. performed experiments; P.H., J.P.M., and D.J.C. analyzed data; P.H. and D.J.C. interpreted results of experiments; P.H. prepared figures; P.H. drafted manuscript; P.H., A.B., and D.J.C. edited and revised manuscript; P.H., J.P.M., A.B., and D.J.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank E. Steinmetz, A. Ribble, and M. Lee for their technical assistance, and L.C. Chen and M. Zhong (NYU Environmental Health Sciences, Tuxedo, NY) for consultation.
REFERENCES
- 1.Adar SD, Sheppard L, Vedal S, Polak JF, Sampson PD, Diez-Roux AV, Budoff M, Jacobs DR Jr, Barr RG, Watson K, Kaufman JD. Fine particulate air pollution and the progression of carotid intima-medial thickness: a prospective cohort study from the multi-ethnic study of atherosclerosis and air pollution. PLoS Med 10: e1001430, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med 9: 1370–1376, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Beelen R, Stafoggia M, Raaschou-Nielsen O, Andersen ZJ, Xun WW, Katsouyanni K, Dimakopoulou K, Brunekreef B, Weinmayr G, Hoffmann B, Wolf K, Samoli E, Houthuijs D, Nieuwenhuijsen M, Oudin A, Forsberg B, Olsson D, Salomaa V, Lanki T, Yli-Tuomi T, Oftedal B, Aamodt G, Nafstad P, De Faire U, Pedersen NL, Ostenson CG, Fratiglioni L, Penell J, Korek M, Pyko A, Eriksen KT, Tjonneland A, Becker T, Eeftens M, Bots M, Meliefste K, Wang M, Bueno-de-Mesquita B, Sugiri D, Kramer U, Heinrich J, de Hoogh K, Key T, Peters A, Cyrys J, Concin H, Nagel G, Ineichen A, Schaffner E, Probst-Hensch N, Dratva J, Ducret-Stich R, Vilier A, Clavel-Chapelon F, Stempfelet M, Grioni S, Krogh V, Tsai MY, Marcon A, Ricceri F, Sacerdote C, Galassi C, Migliore E, Ranzi A, Cesaroni G, Badaloni C, Forastiere F, Tamayo I, Amiano P, Dorronsoro M, Katsoulis M, Trichopoulou A, Vineis P, Hoek G. Long-term exposure to air pollution and cardiovascular mortality: an analysis of 22 European cohorts. Epidemiology 25: 368–378, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Brook RD, Bard RL, Kaplan MJ, Yalavarthi S, Morishita M, Dvonch JT, Wang L, Yang HY, Spino C, Mukherjee B, Oral EA, Sun Q, Brook JR, Harkema J, Rajagopalan S. The effect of acute exposure to coarse particulate matter air pollution in a rural location on circulating endothelial progenitor cells: results from a randomized controlled study. Inhal Toxicol 25: 587–592, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brook RD, Brook JR, Urch B, Vincent R, Rajagopalan S, Silverman F. Inhalation of fine particulate air pollution and ozone causes acute arterial vasoconstriction in healthy adults. Circulation 105: 1534–1536, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Brook RD, Rajagopalan S. Particulate matter, air pollution, and blood pressure. J Am Soc Hypertens 3: 332–350, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Brook RD, Rajagopalan S, Pope CA, Brook JR 3rd, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 121: 2331–2378, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Brook RD, Xu X, Bard RL, Dvonch JT, Morishita M, Kaciroti N, Sun Q, Harkema J, Rajagopalan S. Reduced metabolic insulin sensitivity following sub-acute exposures to low levels of ambient fine particulate matter air pollution. Sci Total Environ 448: 66–71, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY, Yao X, Wang N, Huang Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arterioscler Thromb Vasc Biol 34: 830–836, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Burnett RT, Kwong JC, Villeneuve PJ, Goldberg MS, Brook RD, van Donkelaar A, Jerrett M, Martin RV, Brook JR, Copes R. Risk of incident diabetes in relation to long-term exposure to fine particulate matter in Ontario, Canada. Environ Health Perspect 121: 804–810, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen LL, Liao YF, Zeng TS, Yu F, Li HQ, Feng Y. Effects of metformin plus gliclazide compared with metformin alone on circulating endothelial progenitor cell in Type 2 diabetic patients. Endocrine 38: 266–275, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Cherng TW, Campen MJ, Knuckles TL, Gonzalez Bosc L, Kanagy NL. Impairment of coronary endothelial cell ET(B) receptor function after short-term inhalation exposure to whole diesel emissions. Am J Physiol Regul Integr Comp Physiol 297: R640–R647, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conklin DJ, Haberzettl P, Prough RA, Bhatnagar A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am J Physiol Heart Circ Physiol 296: H1586–H1597, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coogan PF, White LF, Jerrett M, Brook RD, Su JG, Seto E, Burnett R, Palmer JR, Rosenberg L. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation 125: 767–772, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cubbon RM, Kahn MB, Wheatcroft SB. Effects of insulin resistance on endothelial progenitor cells and vascular repair. Clin Sci (Lond) 117: 173–190, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Davel AP, Lemos M, Pastro LM, Pedro SC, de Andre PA, Hebeda C, Farsky SH, Saldiva PH, Rossoni LV. Endothelial dysfunction in the pulmonary artery induced by concentrated fine particulate matter exposure is associated with local but not systemic inflammation. Toxicology 295: 39–46, 2012. [DOI] [PubMed] [Google Scholar]
- 17.De Ciuceis C, Amiri F, Iglarz M, Cohn JS, Touyz RM, Schiffrin EL. Synergistic vascular protective effects of combined low doses of PPARα and PPARγ activators in angiotensin II-induced hypertension in rats. Br J Pharmacol 151: 45–53, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong L, Kang L, Ding L, Chen Q, Bai J, Gu R, Li L, Xu B. Insulin modulates ischemia-induced endothelial progenitor cell mobilization and neovascularization in diabetic mice. Microvasc Res 82: 227–236, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Du J, Fan LM, Mai A, Li JM. Crucial roles of Nox2-derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle-aged mice. Br J Pharmacol 170: 1064–1077, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res 110: 624–637, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haberzettl P, Lee J, Duggineni D, McCracken J, Bolanowski D, O'Toole TE, Bhatnagar A, Conklin DJ. Exposure to ambient air fine particulate matter prevents VEGF-induced mobilization of endothelial progenitor cells from the bone marrow. Environ Health Perspect 120: 848–856, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haberzettl P, Vladykovskaya E, Srivastava S, Bhatnagar A. Role of endoplasmic reticulum stress in acrolein-induced endothelial activation. Toxicol Appl Pharmacol 234: 14–24, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heiss C, Amabile N, Lee AC, Real WM, Schick SF, Lao D, Wong ML, Jahn S, Angeli FS, Minasi P, Springer ML, Hammond SK, Glantz SA, Grossman W, Balmes JR, Yeghiazarians Y. Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production. J Am Coll Cardiol 51: 1760–1771, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, Macdonald JA, Hackett TL, Sin DD, Van Eeden S, Knight DA. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol 129: 1116–1125, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Hwang J, Kleinhenz DJ, Rupnow HL, Campbell AG, Thule PM, Sutliff RL, Hart CM. The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul Pharmacol 46: 456–462, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schonbeck U, Libby P. Metformin inhibits proinflammatory responses and nuclear factor-κB in human vascular wall cells. Arterioscler Thromb Vasc Biol 26: 611–617, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Jialal I, Devaraj S, Singh U, Huet BA. Decreased number and impaired functionality of endothelial progenitor cells in subjects with metabolic syndrome: implications for increased cardiovascular risk. Atherosclerosis 211: 297–302, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kahn MB, Yuldasheva NY, Cubbon RM, Smith J, Rashid ST, Viswambharan H, Imrie H, Abbas A, Rajwani A, Aziz A, Baliga V, Sukumar P, Gage M, Kearney MT, Wheatcroft SB. Insulin resistance impairs circulating angiogenic progenitor cell function and delays endothelial regeneration. Diabetes 60: 1295–1303, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis JA, Xu X, Kherada N, Brook RD, Reddy KM, Padture NP, Parthasarathy S, Chen LC, Moffatt-Bruce S, Sun Q, Morawietz H, Rajagopalan S. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res 108: 716–726, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katakam PV, Ujhelyi MR, Hoenig M, Miller AW. Metformin improves vascular function in insulin-resistant rats. Hypertension 35: 108–112, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Kato M, Roberts-Thomson P, Phillips BG, Narkiewicz K, Haynes WG, Pesek CA, Somers VK. The effects of short-term passive smoke exposure on endothelium-dependent and independent vasodilation. J Hypertens 17: 1395–1401, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 100: 1589–1596, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 28: 1982–1988, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knuckles TL, Stapleton PA, Minarchick VC, Esch L, McCawley M, Hendryx M, Nurkiewicz TR. Air pollution particulate matter collected from an Appalachian mountaintop mining site induces microvascular dysfunction. Microcirculation 20: 158–169, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan RM, Adar SD, Szpiro AA, Jorgensen NW, Van Hee VC, Barr RG, O'Neill MS, Herrington DM, Polak JF, Kaufman JD. Vascular responses to long- and short-term exposure to fine particulate matter: MESA Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution). J Am Coll Cardiol 60: 2158–2166, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 85: 753–766, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Kunzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, Thomas D, Peters J, Hodis HN. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect 113: 201–206, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation 109: II27–33, 2004. [DOI] [PubMed] [Google Scholar]
- 39.LeBlanc AJ, Cumpston JL, Chen BT, Frazer D, Castranova V, Nurkiewicz TR. Nanoparticle inhalation impairs endothelium-dependent vasodilation in subepicardial arterioles. J Toxicol Environ Health 72: 1576–1584, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li R, Zhang H, Wang W, Wang X, Huang Y, Huang C, Gao F. Vascular insulin resistance in prehypertensive rats: role of PI3-kinase/Akt/eNOS signaling. Eur J Pharmacol 628: 140–147, 2010. [DOI] [PubMed] [Google Scholar]
- 41.Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, Liu J, Huang L. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem 363: 135–145, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao YF, Chen LL, Zeng TS, Li YM, Fan Y, Hu LJ, Ling Y. Number of circulating endothelial progenitor cells as a marker of vascular endothelial function for Type 2 diabetes. Vasc Med 15: 279–285, 2010. [DOI] [PubMed] [Google Scholar]
- 43.Liberda EN, Cuevas AK, Gillespie PA, Grunig G, Qu Q, Chen LC. Exposure to inhaled nickel nanoparticles causes a reduction in number and function of bone marrow endothelial progenitor cells. Inhal Toxicol 22 Suppl 2: 95–99, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellick AS, Plummer PN, Nolan DJ, Gao D, Bambino K, Hahn M, Catena R, Turner V, McDonnell K, Benezra R, Brink R, Swarbrick A, Mittal V. Using the transcription factor inhibitor of DNA binding 1 to selectively target endothelial progenitor cells offers novel strategies to inhibit tumor angiogenesis and growth. Cancer Res 70: 7273–7282, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. New Engl J Med 356: 447–458, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Mills NL, Tornqvist H, Robinson SD, Gonzalez M, Darnley K, MacNee W, Boon NA, Donaldson K, Blomberg A, Sandstrom T, Newby DE. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 112: 3930–3936, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Molavi B, Rassouli N, Bagwe S, Rasouli N. A review of thiazolidinediones and metformin in the treatment of Type 2 diabetes with focus on cardiovascular complications. Vasc Health Risk Manag 3: 967–973, 2007. [PMC free article] [PubMed] [Google Scholar]
- 48.Newby DE, Mannucci PM, Tell GS, Baccarelli AA, Brook RD, Donaldson K, Forastiere F, Franchini M, Franco OH, Graham I, Hoek G, Hoffmann B, Hoylaerts MF, Kunzli N, Mills N, Pekkanen J, Peters A, Piepoli MF, Rajagopalan S, Storey RF. Expert position paper on air pollution and cardiovascular disease. Eur Heart J 36: 83–93b, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nurkiewicz TR, Porter DW, Hubbs AF, Stone S, Moseley AM, Cumpston JL, Goodwill AG, Frisbee SJ, Perrotta PL, Brock RW, Frisbee JC, Boegehold MA, Frazer DG, Chen BT, Castranova V. Pulmonary particulate matter and systemic microvascular dysfunction. Res Rep Health Eff Inst 3–48, 2011. [PubMed] [Google Scholar]
- 50.O'Neill MS, Veves A, Zanobetti A, Sarnat JA, Gold DR, Economides PA, Horton ES, Schwartz J. Diabetes enhances vulnerability to particulate air pollution-associated impairment in vascular reactivity and endothelial function. Circulation 111: 2913–2920, 2005. [DOI] [PubMed] [Google Scholar]
- 51.O'Toole TE, Hellmann J, Wheat L, Haberzettl P, Lee J, Conklin DJ, Bhatnagar A, Pope CA 3rd. Episodic exposure to fine particulate air pollution decreases circulating levels of endothelial progenitor cells. Circ Res 107: 200–203, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearson JF, Bachireddy C, Shyamprasad S, Goldfine AB, Brownstein JS. Association between fine particulate matter and diabetes prevalence in the U.S. Diabetes Care 33: 2196–2201, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pistrosch F, Herbrig K, Oelschlaegel U, Richter S, Passauer J, Fischer S, Gross P. PPARγ-agonist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis 183: 163–167, 2005. [DOI] [PubMed] [Google Scholar]
- 54.Pope CA 3rd, Hansen JC, Kuprov R, Sanders MD, Anderson MN, Eatough DJ. Vascular function and short-term exposure to fine particulate air pollution. J Air Waste Manag Assoc 61: 858–863, 2011. [DOI] [PubMed] [Google Scholar]
- 55.Pope CA 3rd, Muhlestein JB, May HT, Renlund DG, Anderson JL, Horne BD. Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation 114: 2443–2448, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Pope CA 3rd, Turner MC, Burnett RT, Jerrett M, Gapstur SM, Diver WR, Krewski D, Brook RD. Relationships between fine particulate air pollution, cardiometabolic disorders, and cardiovascular mortality. Circ Res 116: 108–115, 2015. [DOI] [PubMed] [Google Scholar]
- 57.Poss J, Lorenz D, Werner C, Pavlikova V, Gensch C, Speer T, Alessandrini F, Berezowski V, Kuntz M, Mempel M, Endres M, Bohm M, Laufs U. Diesel exhaust particles impair endothelial progenitor cells, compromise endothelial integrity, reduce neoangiogenesis, and increase atherogenesis in mice. Cardiovasc Toxicol 13: 290–300, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Provoost S, Maes T, Pauwels NS, Vanden Berghe T, Vandenabeele P, Lambrecht BN, Joos GF, Tournoy KG. NLRP3/caspase-1-independent IL-1β production mediates diesel exhaust particle-induced pulmonary inflammation. J Immunol 187: 3331–3337, 2011. [DOI] [PubMed] [Google Scholar]
- 59.Rao X, Montresor-Lopez J, Puett R, Rajagopalan S, Brook RD. Ambient air pollution: an emerging risk factor for diabetes mellitus. Curr Diab Rep 15: 603, 2015. [DOI] [PubMed] [Google Scholar]
- 60.Rasouli N, Raue U, Miles LM, Lu T, Di Gregorio GB, Elbein SC, Kern PA. Pioglitazone improves insulin sensitivity through reduction in muscle lipid and redistribution of lipid into adipose tissue. Am J Physiol Endocrinol Metab 288: E930–E934, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Saito H, Yamamoto Y, Yamamoto H. Diabetes alters subsets of endothelial progenitor cells that reside in blood, bone marrow, and spleen. Am J Physiol Cell Physiol 302: C892–C901, 2012. [DOI] [PubMed] [Google Scholar]
- 62.Schneider A, Neas L, Herbst MC, Case M, Williams RW, Cascio W, Hinderliter A, Holguin F, Buse JB, Dungan K, Styner M, Peters A, Devlin RB. Endothelial dysfunction: associations with exposure to ambient fine particles in diabetic individuals. Environ Health Perspect 116: 1666–1674, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sena CM, Matafome P, Louro T, Nunes E, Fernandes R, Seica RM. Metformin restores endothelial function in aorta of diabetic rats. Br J Pharmacol 163: 424–437, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shah AS, Lee KK, McAllister DA, Hunter A, Nair H, Whiteley W, Langrish JP, Newby DE, Mills NL. Short term exposure to air pollution and stroke: systematic review and meta-analysis. BMJ 350: h1295, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, Neale GA, Hooiveld GJ, Hijmans A, Vroegrijk I, van den Berg S, Romijn J, Rensen PC, Joosten LA, Netea MG, Kanneganti TD. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A 108: 15324–15329, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, Fayad ZA, Fuster V, Lippmann M, Chen LC, Rajagopalan S. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA 294: 3003–3010, 2005. [DOI] [PubMed] [Google Scholar]
- 67.Tepper OM, Capla JM, Galiano RD, Ceradini DJ, Callaghan MJ, Kleinman ME, Gurtner GC. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood 105: 1068–1077, 2005. [DOI] [PubMed] [Google Scholar]
- 68.Tsai TH, Chai HT, Sun CK, Yen CH, Leu S, Chen YL, Chung SY, Ko SF, Chang HW, Wu CJ, Yip HK. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J Transl Med 10: 137, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4: 95–104, 2003. [DOI] [PubMed] [Google Scholar]
- 70.Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, Brook RD. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect 113: 1052–1055, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verma S, Yao L, Dumont AS, McNeill JH. Metformin treatment corrects vascular insulin resistance in hypertension. J Hypertens 18: 1445–1450, 2000. [DOI] [PubMed] [Google Scholar]
- 72.Vladykovskaya E, Sithu SD, Haberzettl P, Wickramasinghe NS, Merchant ML, Hill BG, McCracken J, Agarwal A, Dougherty S, Gordon SA, Schuschke DA, Barski OA, O'Toole T, D'Souza SE, Bhatnagar A, Srivastava S. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. J Biol Chem 287: 11398–11409, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wauters A, Dreyfuss C, Pochet S, Hendrick P, Berkenboom G, van de Borne P, Argacha JF. Acute exposure to diesel exhaust impairs nitric oxide-mediated endothelial vasomotor function by increasing endothelial oxidative stress. Hypertension 62: 352–358, 2013. [DOI] [PubMed] [Google Scholar]
- 74.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. New Engl J Med 353: 999–1007, 2005. [DOI] [PubMed] [Google Scholar]
- 75.Wheat LA, Haberzettl P, Hellmann J, Baba SP, Bertke M, Lee J, McCracken J, O'Toole TE, Bhatnagar A, Conklin DJ. Acrolein inhalation prevents vascular endothelial growth factor-induced mobilization of Flk-1+/Sca-1+ cells in mice. Arterioscler Thromb Vasc Biol 31: 1598–1606, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yin Y, Pastrana JL, Li X, Huang X, Mallilankaraman K, Choi ET, Madesh M, Wang H, Yang XF. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front Biosci 18: 638–649, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]