

Abstract
Objective:
To identify and follow a series of 52 patients with optic neuropathy related to sarcoidosis.
Methods:
Prospective observational cohort study.
Results:
The disorder was more common in women and affected a wide age range. It was proportionately more common in African and Caribbean ethnic groups. Two clinical subtypes were identified: the more common was a subacute optic neuropathy resembling optic neuritis; a more slowly progressive optic neuropathy arose in the remaining 17%. Sixteen (31%) were bilateral. Concurrent intraocular inflammation was seen in 36%. Pain arose in only 27% of cases. An optic perineuritis was seen in 2 cases, and predominate involvement of the chiasm in one. MRI findings showed optic nerve involvement in 75% of cases, with adjacent and more widespread inflammation in 31%. Treatment with corticosteroids was helpful in those with an inflammatory optic neuropathy, but not those with mass lesions. Relapse of visual signs arose in 25% of cases, necessitating an increase or escalation of treatment, but relapse was not a poor prognostic factor.
Conclusions:
This is a large prospective study of the clinical characteristics and outcome of treatment in optic neuropathy associated with sarcoidosis. Patients who experience an inflammatory optic neuropathy respond to treatment but may relapse. Those with infiltrative or progressive optic neuropathies improve less well even though the inflammatory disorder responds to therapy.
Sarcoidosis forms a spectrum of autoinflammatory disorders of unknown etiology characterized by the development of immune activation leading to an immunologic cascade that results in an inflammatory infiltration of tissues, with granuloma formation and later fibrosis. Any tissue can be affected, but the lungs, skin, liver, and joints are particularly affected.1
The disorder is more common in women than in men and is predominately a disease of young adulthood but affects all age groups.2,3 In the United States, there is a higher prevalence in African Americans, and the disease may be more severe in this ethnic group.2,4
Ophthalmic complications of the disease arise in some 10% to 50%,5–7 with 12% in ACCESS (A Case Control Etiologic Study of Sarcoidosis)2; not all patients show the characteristics of a granulomatous uveitis.8 Anterior uveitis is the most common, with mutton fat keratic precipitates and Koeppe iris nodules, and vitritis leading to the string of pearls, periphlebitis, candle wax drippings, and choroidal involvement.5–11 The development of keratoconjunctivitis sicca appears to be almost inevitable. The prognosis for visual recovery in those with intraocular inflammation is in general good9–11 with correct treatment.
Involvement of the nervous system is uncommon, and accounts for 5% to 10% of published retrospective series.12 It may arise at any point during the disease, and all regions of the nervous system may be affected.
In this report, we describe the results of a clinical study conducted over the course of 20 years in which we characterize the clinical syndromes, concomitant systemic features, investigation results, and response to appropriate treatment in a prospectively acquired group of patients seen and treated at 3 large teaching hospitals in London.
METHODS
All patients seen and assessed in our neuroophthalmology departments with visual disorders are investigated to identify the cause and to define the most appropriate treatment. This investigation includes a series of blood investigations and imaging; when these investigations found no evidence for a demyelinating disorder (through MRI features principally), systemic autoimmune condition (e.g., antinuclear antibodies, DNA, aquaporin-4, and myelin oligodendrocyte glycoprotein antibodies), or infectious or metabolic causes, a granulomatous cause was considered, and further investigations were undertaken. Those in whom systemic features of sarcoidosis were identified during investigation and those already known to have active systemic sarcoidosis, in whom the diagnosis of a granulomatous optic neuropathy was considered by the authors to be highly probable,13 were recorded prospectively and included in the study. The records were kept according to data protection regulations in the neuroophthalmology departments and collated by 2 of us (B.J.B. and D.P.K.).
Criteria for the diagnosis of sarcoidosis involved histologic proof (in 40 cases) of a granulomatous disorder currently active or requiring continuing treatment or (in 12 cases) adequate proof on clinical grounds that no alternative explanation existed for the systemic disorder in the minds of the treating physicians: for example, clear radiologic evidence for lung and mediastinal involvement together with clear evidence for another characteristic feature of the disease, such as hypercalcemia, raised serum angiotensin-converting enzyme (ACE) or other tissue involvement, associated with a typical pattern of isotope uptake using gallium scintigraphy or PET. Seventeen cases in whom these criteria were unfulfilled were excluded from the data analysis.
Group data were compared with the Fisher exact test and Mann–Whitney test.
Standard protocol approvals, registration, and patient consents.
Ethical approval for the study was obtained, and patients gave informed consent to be included.
RESULTS
Demographics.
Fifty-two patients were included in the study. The mean (range) age was 42.5 (20–71) years. Thirty-two (61%) were women. Twenty-four were Caucasian, 24 African or Caribbean, and 4 were from South Asia.
Clinical features of the systemic disorder.
The diagnosis of systemic sarcoidosis was made concurrently in 33 patients and in one subsequently. The remaining 18 had already been diagnosed, between 6 months and 20 years before the onset of the optic neuropathy.
A histologic diagnosis was made from respiratory tissue (transbronchial or mediastinal lymph gland biopsy) in 28 cases, lymph gland biopsy in 3, liver biopsy in 1, orbital biopsy in 3, and neurologic tissue in 5. All fulfilled the criteria for the “highly probable” diagnosis of the WASOG (World Association of Sarcoidosis and Other Granulomatous Diseases) Sarcoidosis Organ Assessment Instrument.13 In 12 cases, the diagnosis had been deemed secure on clinical grounds by the treating respiratory physicians, with clear evidence on clinical grounds for a multisystem disorder typical of sarcoidosis as noted in the methods section above; these were all patients presenting with an optic neuropathy already diagnosed with systemic sarcoidosis.
The predominate systemic clinical syndrome was respiratory in 38 cases, the orbit and lacrimal glands in 6, cervical lymph glands in 4, the skin in 3, the nose and sinuses in 3, and the liver and joints in 1 each. Tissue involvement outside of the nervous system was multisystem in 17 cases. The serum ACE was elevated in 24 (57%) of available data at the time of onset of the optic neuropathy; the median serum ACE was 111 (18–205) U/L (normal range 8–50 U/L).
Gallium scans were undertaken in 10 instances, in which 1 was normal, 6 showed a “panda” pattern of uptake in the lacrimal and parotid glands, 1 a “lambda” pattern of mediastinal uptake, and 2 a combination of both patterns.
Clinical characteristics of the optic neuropathy.
All patients presented with symptoms and signs of an optic neuropathy, defined as a reduction in central visual acuity and color vision, reduction in the visual field, and when seen, an abnormality of the pupillary response. Patients with a subacute optic neuropathy that evolved over days were considered separately from those with a more progressive clinical course, in which the symptoms evolved over weeks. The former were considered to show an optic neuritis syndrome, the latter a progressive syndrome.
Forty-three cases showed an optic neuritis syndrome, of which 27 were unilateral. Twelve of the bilateral cases showed a sequential optic neuropathy with a range of latency between the 2 episodes of 1 to 60 months. A bilateral optic neuropathy arose synchronously in 4 cases (figure 1). The median (range) acuity at presentation was 0.05 (0–1.0) in unilateral cases and 0.13 (0–1.0) in bilateral involvement. Pain was a feature in 27% of cases overall.
Figure 1. MRI in subacute optic neuritis caused by sarcoidosis.

MRI showing (A) swelling and high signal within the nerve, and (B) enhancement in a patient who presented with a painless subacute optic neuropathy to no perception of light over 5 days. With oral corticosteroid treatment, she recovered to 6/6 (20/20, 1.0). Azathioprine was added and oral steroids withdrawn over 6 months. She has remained well since. (C) T1-weighted axial MRI showing swelling and enhancement of the anterior half of the left optic nerve. (D, E) T1-weighted coronal MRI showing 2 other cases in which the nerve is seen to be swollen and enhancing following administration of contrast. Each case had a subacute optic neuritis with improvement after administration of corticosteroids.
A wide range of visual field defects was noted; most had central or centrocecal scotomas, but hemianopic and altitudinal defects were also seen.
The chiasm was involved in one case. A unilateral optic perineuritis arose in 2 cases. A progressive unilateral optic neuropathy was the presenting feature in 6 cases.
Table 1 shows the important clinical features in each of the subgroups noted.
Table 1.
Clinical characteristics and ocular findings at presentation in the 52 patients studied

Coexistence of inflammatory ocular disease.
Intraocular inflammation was seen at the same time as the optic neuropathy in 19 cases (36%): a granulomatous anterior uveitis was seen in 10 cases, an isolated vitritis in 1, a panuveitis in 1, isolated choroidal involvement in 1, features of a retinal vasculitis in 2, cotton wool spots in 2, macular exudates in 1, and episcleritis in 1. In no case was the intraocular disease longstanding and associated with chronic glaucoma or retinal ischaemia.
Neurologic investigation results: Imaging.
One patient underwent CT; the remainder had MRI studies of the orbits and brain, of which 13 were adjudged to be normal. Twenty patients had abnormalities restricted to the optic nerve or sheath, of whom 14 showed enhancement of the optic nerve and sheath (figure 1) with intrinsic lesions of one or both optic nerves, while the remainder showed high signal within the nerve without enhancement. In addition, 3 cases (and the one who had CT) showed intrinsic lesions within the nerve and thickening of the adjacent optic nerve sheath complex. The chiasmal lesion was associated with enhancement of the chiasm extending forward into one of the nerves.
Both patients with a perineuritis showed features of this, i.e., the predominate feature was of enhancement and thickening of the sheath, not the whole complex (figure 2).
Figure 2. MRI in optic perineuritis.

(A) Inflammatory lesion within the orbit encasing the right optic nerve. The lesion was biopsied and granulomatous inflammation identified. The optic neuropathy improved with steroids. (B, C) Optic perineuritis: T1-weighted axial and coronal MRI showing enlargement of and enhancement of the optic nerve and its sheath throughout its entire length. This was the presenting feature of the disease, which was diagnosed subsequently on histologic analysis of a cervical lymph node.
A minority of patients whose clinical syndrome was one of a subacute optic neuropathy resembling an optic neuritis showed additional imaging features of more widespread involvement of the nervous system by the disease: 8 patients with unilateral optic neuropathies and 2 with bilateral involvement showed high signal within the nerves, enhancement of the complex, which also involved the orbital apex (5 cases) (figure 3A), the floor of the middle cranial fossa (4 cases) (figure 3B), and the frontal lobes (1 case).
Figure 3. MRI of compressive optic neuropathy caused by dural lesions in sarcoidosis.
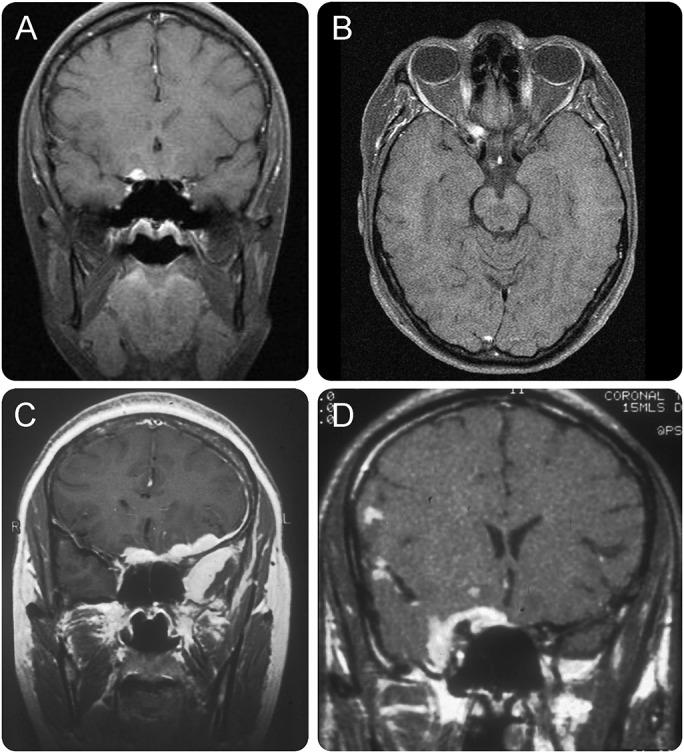
(A, B) Orbital apex mass with a progressive painful optic neuropathy and sarcoidosis of the lungs, mediastinum, and heart. (C, D) Two cases of progressive optic neuropathy with striking meningeal enhancement and swelling to form mass lesions. Despite intensive treatment with corticosteroids and immunosuppression, neither case showed a meaningful improvement in visual acuity.
In general, the initial acuity was better in those with normal imaging and no enhancement than in those in whom enhancement was seen (figure 4).
Figure 4. Histogram.

Histogram showing the relationships between MRI features at onset/diagnosis (abscissa) and initial (nadir) visual acuity then best acuity after treatment (ordinate).
Those whose clinical syndrome was of a progressive optic neuropathy (6 cases) all showed meningeal enhancing lesions that had formed masses, in each case at the intracranial portion of the orbital apex (figure 3).
Neurologic investigation results: CSF.
CSF results were available in 33 cases, including 2 with mass lesions and one with perineuritis. Twenty-five had normal CSF. Seven (21%) showed CSF protein levels greater than 0.6 g/dL (median 0.95 g/dL, range 0.79–1.92). Five (15%) showed a leukocyte pleocytosis (>5 cells/cm3) (median 19, range 6–70). Twenty-eight (85%) had no oligoclonal bands, and 5 showed matched bands in both CSF and serum.
Treatment.
Patients were treated in an individual way in 3 different hospitals so there was a wide range of medications, dosage regimens, and treatment durations. Forty-nine were treated with corticosteroids, of whom 14 commenced with IV methylprednisolone followed by oral prednisolone, while the remainder received treatment with oral steroids from the outset. The daily dose of prednisolone varied between 30 and 80 mg per day. The duration of treatment varied greatly (4–320 weeks).
Eighteen were treated with immunosuppression: 12 from outset, and 6 upon relapse. Twelve received azathioprine, 1 mycophenolate, and 5 methotrexate. Two received tumor necrosis factor α antagonists, for neurosarcoidosis in each case.
Follow-up.
The median (range) length of follow-up was 8 (0–20) years. The median (range) change in visual acuity (defined as the best acuity following treatment, even after relapse) was +0.46 (−0.01 to +1.22) in bilateral cases and +0.175 (−0.05 to +1.25) in unilateral cases. The median final acuity in these 2 groups was 0.7 (0–1.25) and 0.5 (0–1.25).
Those with a subacute syndrome showed a median improvement of +0.225 (range −0.01 to +1.22). Those in the progressive subgroup showed no improvement in visual acuity with treatment although in each case there was resolution of meningeal enhancement over time.
While there was a suggestion that MRI abnormalities at onset (figure 4) correlated with nadir acuity and an improvement following treatment, this was not significant (p > 0.3).
Considering treatment in those without a progressive disease course or perineuritis, 12 were treated with IV then oral steroids; median (range) acuity was 0.02 (0–1) and increased to 0.75 (0.02–1), median change +0.3, and those treated with oral steroids at onset 0.1 (0–1) increasing to 0.75 (0–1.25), and median change +0.5 (p = 0.77).
Relapse arose in 13 cases. For the most part, this occurred early on; for example, patients treated with 60 mg prednisolone developed recurrence of symptoms and signs at 40 to 20 mg, and the signs settled with an escalation of treatment. Immunosuppressive agents were added in 7 cases—azathioprine in 4 and mycophenolate and methotrexate in one each. The patient treated with methotrexate had had repeated relapses, which stopped on the immunosuppressive drug. One patient had an intracranial relapse of neurosarcoidosis and her optic neuropathy worsened; she was treated with cyclophosphamide then infliximab.
In those who relapsed, the improvement in acuity following treatment was +0.25 (−0.15 to + 1.2), and relapse did not appear to influence the outcome in any way.
Patients with an isolated optic neuritis, raised serum ACE, but no evidence for sarcoidosis.
We have seen 6 additional patients who presented with an optic neuritis and were found during investigation to have a raised serum ACE (median 100 [range 80–178] U/L). The clinical syndrome was not different from the others described above; half were Caribbean, half Caucasian, and half were bilateral. All were male. None had pain. Two had a concurrent vitritis and one had a granulomatous anterior uveitis. The median (range) acuity in each eye was 0.1 (0–1.0). In each case, the affected discs were seen to be swollen.
Imaging was normal in 3 and showed high signal within the affected nerve in the other 3. CSF in 4 cases showed normal protein and negative oligoclonal bands in 3; in the other case, matched serum and CSF bands were found. In this case, there was a CSF lymphocytosis of 63 per cubic centimeter. The others were acellular. Four had gallium scans and each of these was normal.
One was considered to have a disc granuloma and was watched without treatment; he improved over 2 years. The others received steroids and each improved to 6/6 (1.0) vision.
These cases have been followed and none has developed further or recurrent neurologic or systemic symptoms. They appear to have neither sarcoidosis nor multiple sclerosis.
DISCUSSION
This work forms the largest series of patients with optic neuropathy in sarcoidosis identified and each has been followed prospectively. The age range was wide but the condition was more prevalent in the first half of life. The majority of patients were women and there were equal numbers in Caucasian and African or Caribbean ethnic groups. According to the epidemiologic findings in ACCESS, the ethnic subgroups in London are 60% Caucasian, 14% African and Caribbean, and 18% South Asian, and our results therefore suggest a greater prevalence of the disease in African and Caribbean people who live in London.
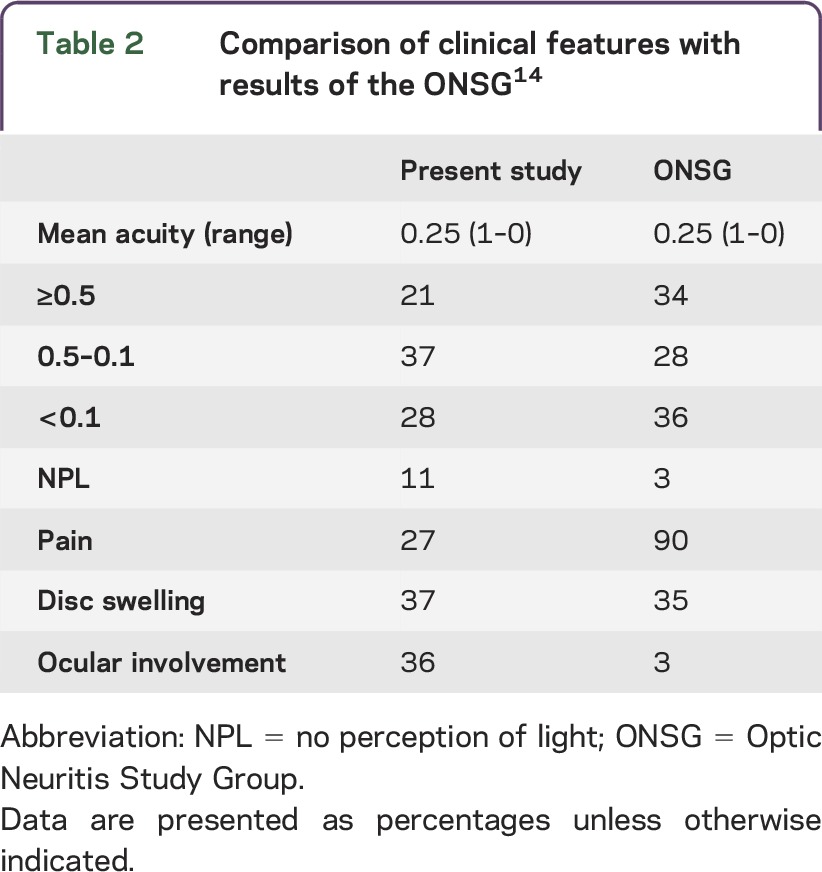
In general, the clinical syndrome resembled an optic neuritis,14 although in some, the disease course was more slowly progressive. Pain was a less prevalent feature than is seen in demyelinating optic neuritis, and the median visual acuity at nadir was equivalent, although more were no perception of light (table 2). However, none of the features observed was specific to sarcoidosis in this series. Sixty-four percent had a unilateral involvement, 28% presented at a later time with involvement of the second eye, while synchronous bilateral involvement was less common (9%). It is noteworthy that there was evidence for concurrent intraocular inflammation in only a minority of cases. Predominately this was mild (although more severe cases had been excluded because of the uncertainty of the pathogenesis of the optic neuropathy). Indeed, in 64% of patients, there was no history of ocular involvement at any stage of the disease to date. Imaging showed clear abnormalities, which accounted for the clinical features in 75% of cases. The majority showed high signal within the nerve, with or without enhancement.
Table 2.
Comparison of clinical features with results of the ONSG14
Spontaneous improvement occurred (as has been noted previously15,16), although the majority of patients were treated with steroids. Following this, an often substantial improvement in vision took place, even after several weeks without treatment. There was no relationship between nadir acuity and final acuity. Relapse occurred in 25% of cases, necessitating recommencement or an escalation of treatment, and in the majority, this occurred early, as steroids were withdrawn. Relapse itself was not a marker of poor visual outcome. Repeated relapse was treated successfully with addition of immunosuppression.
Six cases (11%) had a more progressive disease course and were shown to have a more widespread neurologic involvement, each showing evidence for adjacent meningeal inflammation. Visual outcome in this subgroup was poor, even though the disease process required, in general, a higher and more prolonged dose of steroids, concurrent immunosuppression, and in 2 cases, a requirement for biological agents with refractory disease. Evidence for neurosarcoidosis defined as the presence of intracranial meningeal enhancement arose in 16 cases overall—all of those with a progressive course and 10 with a subacute course.
Involvement of the optic nerve in sarcoidosis may arise through a variety of mechanisms: inflammation of the nerve itself,15,17–19 compression or infiltration by an inflammatory mass adjacent to the anterior visual pathway (e.g., the orbital apex or pituitary gland),19,20 secondary involvement through ischemic complications of retinal and choroidal inflammation and glaucoma,5–11 granulomas within the disc itself,17,20 meningeal inflammation within the optic nerve sheath leading to an optic perineuritis,21 and finally as a consequence of hydrocephalus.22 Optic neuropathy is not common in sarcoidosis. It was seen in 5% of one series of 202 patients with ocular disease in sarcoidosis,23 but only one in a series of 649 patients with systemic sarcoidosis24 and none in another of 285.25 In the literature before 2000, 57 cases had been published, of which 35 involved the optic nerve itself, 9 compression due to an intracerebral lesion, 6 due to hydrocephalus, and 6 due to the complications of uveitis.26 Many noted that bilateral involvement occurred and that imaging showed enlargement of the optic nerve–sheath complex.19,21,27 Others15 noted that the acuity was lower than that seen in a demyelinating optic neuritis. More recent series from the United States have studied larger patient numbers. One study27 described 24 cases with optic neuropathy: 71% had not been known previously to have systemic sarcoidosis and 42% showed previous or current intraocular inflammation. The clinical features were very similar to those in our series. Another28 showed that 14 of 67 patients known to have neurologic sarcoidosis had some form of optic neuropathy.
We have identified a small subgroup of patients in whom the serum ACE is elevated at presentation and then subsequently normalizes without relapse. On further investigation, these patients do not have sarcoidosis and on follow-up show no sign of transformation into a more widespread disease. The serum ACE appears to increase in a nonspecific way and appears not to indicate a systemic granulomatous disease. It is important to identify this subgroup, which requires no additional treatment, merely close follow-up and observation.
This large series of patients with optic neuropathy in conjunction with sarcoidosis has shown that more than half are not previously diagnosed with sarcoidosis, only a third have concurrent intraocular inflammation, and the clinical characteristics and in most cases the imaging features are not substantially different from that seen in a regular demyelinating optic neuritis. However, patients should be identified early because the response to treatment may lessen with time, and the identification of more widespread disease, particularly within the nervous system, will require prolonged, often high-dose treatment with steroids, immunosuppressive drugs, and modern biological therapies. Modern imaging investigations including PET may make identification of these cases easier, but a high index of clinical suspicion is still important, particularly in non-Caucasian patients presenting with a painless optic neuritis.
Once identified, we recommend that patients be treated with high-dose (oral or IV) corticosteroids, followed by a slow taper watching carefully for relapse as the dose is reduced. Repeated relapses, particularly preventing steroid dose reduction, should be treated with an additional immunosuppressive agent. The presence of neurosarcoidosis requires more prolonged higher-dose treatment with steroids and immunosuppression from the outset, with early recourse to biological agents should the clinical and radiologic features of the disorder fail to improve quickly. Further study is required to identify precisely which treatments are best and over what period of time.
GLOSSARY
- ACCESS
A Case Control Etiologic Study of Sarcoidosis
- ACE
angiotensin-converting enzyme
AUTHOR CONTRIBUTIONS
Dr. Kidd was responsible for the study design, data acquisition, analysis and interpretation, and writing the manuscript. Dr. Burton shared responsibility for data acquisition. Dr. Graham was responsible for study design and critical revision of the manuscript. Dr. Plant was responsible for study design and critical revision of the manuscript. Dr. Kidd was the study supervisor.
STUDY FUNDING
No targeted funding.
DISCLOSURE
D.P. Kidd receives royalties from Elsevier. B.J. Burton served on the scientific advisory board for Bayer, Novartis, received travel funding from Novartis, Bayer, is subeditor for Eastern European Journal of Neurology, received research support from Novartis ALCON. E.M. Graham reports no disclosures. G.T. Plant is editor in chief of Neuro-Ophthalmology. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Iannuzzi MC, Rubicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2152–2165. [DOI] [PubMed] [Google Scholar]
- 2.Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001;164:1885–1889. [DOI] [PubMed] [Google Scholar]
- 3.Pietinalho A, Hiraga Y, Hosoda Y, Lofroos AB, Yamaguchi M, Selroos O. The frequency of sarcoidosis in Finland and Hokkaido, Japan: a comparative epidemiological study. Sarcoidosis 1995;12:61–67. [PubMed] [Google Scholar]
- 4.Rybicki BA, Major M, Popovich J Jr, Mailiarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5 year study in a health maintenance organization. Am J Epidemiol 1997;145:234–241. [DOI] [PubMed] [Google Scholar]
- 5.Jabs DA, Johns CJ. Ocular involvement in sarcoidosis. Am J Ophthalmol 1986;102:297–301. [DOI] [PubMed] [Google Scholar]
- 6.Karma A, Huhti E, Poukkula A. Course and outcome of ocular sarcoidosis. Am J Ophthalmol 1988;106:467–472. [DOI] [PubMed] [Google Scholar]
- 7.Rothova A. Ocular involvement in sarcoidosis. Br J Ophthalmol 2000;84:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbort CP, Rao NA, Mochizuki M; members of Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis. Ocul Immunol Inflamm 2009;17:160–169. [DOI] [PubMed] [Google Scholar]
- 9.Dana MR, Merayo-Lloves J, Schaumberg DA, Foster CS. Prognosticators for visual outcome in sarcoid uveitis. Ophthalmology 1996;103:1846–1853. [DOI] [PubMed] [Google Scholar]
- 10.Edelstein C, Pearson A, Joynes E, Stanford MR, Graham EM. The ocular and systemic prognosis of patients presenting with sarcoid uveitis. Eye 1999;13:748–753. [DOI] [PubMed] [Google Scholar]
- 11.Lobo A, Barton K, Minassian D, du Bois RM, Lightman S. Visual loss in sarcoid-related uveitis. Clin Exp Ophthalmol 2003;31:310–316. [DOI] [PubMed] [Google Scholar]
- 12.Kidd D, Beynon HLC. Neurological complications of systemic sarcoidosis (review). Sarcoidosis Vasc Diff Lung Dis 2003;20:85–94. [PubMed] [Google Scholar]
- 13.Judson MA, Costabel U, Drent M, et al. The WASOG Sarcoidosis Organ Assessment Instrument: an update of a previous clinical tool. Sarcoidosis Vasc Diff Lung Dis 2014;31:19–27. [PubMed] [Google Scholar]
- 14.Optic Neuritis Study Group. The clinical profile of optic neuritis. Arch Ophthalmol 1991;109:1673–1678. [DOI] [PubMed] [Google Scholar]
- 15.Graham EM, Ellis CJK, Sanders MD, McDonald WI. Optic neuropathy in sarcoidosis. J Neurol Neurosurg Psychiatry 1986;49:756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galetta S, Schatz NJ, Glaser JS. Acute sarcoid optic neuropathy with spontaneous recovery. J Clin Neuroophthalmol 1989;91:27–32. [PubMed] [Google Scholar]
- 17.Beardsley TL, Brown SV, Sydnor CF, Grimson BS, Klintworth GK. Eleven cases of sarcoidosis of the optic nerve. Am J Ophthalmol 1984;97:62–77. [DOI] [PubMed] [Google Scholar]
- 18.Blain JG, Riley W, Logothetis J. Optic nerve manifestations of sarcoidosis. Arch Neurol 1965;13:307–309. [DOI] [PubMed] [Google Scholar]
- 19.Gudeman SK, Selhorst JB, Susac JO, Waybright EA. Sarcoid optic neuropathy. Neurology 1982;32:597–603. [DOI] [PubMed] [Google Scholar]
- 20.Kelley JS, Green WR. Sarcoidosis involving the optic nerve head. Arch Ophthalmol 1973;89:486–488. [DOI] [PubMed] [Google Scholar]
- 21.Ing EB, Garrity JA, Cross SA, Ebersold MJ. Sarcoid masquerading as optic nerve sheath meningioma. Mayo Clin Proc 1997;72:38–43. [DOI] [PubMed] [Google Scholar]
- 22.Delaney P. Neurologic manifestations in sarcoidosis: review of the literature, with a report of 23 cases. Ann Intern Med 1977;87:336–345. [DOI] [PubMed] [Google Scholar]
- 23.Obenauf CD, Shaw HE, Sydnor CF, Klintworth GK. Sarcoidosis and its ophthalmic manifestations. Am J Ophthalmol 1978;86:648–655. [DOI] [PubMed] [Google Scholar]
- 24.Stern BJ, Krumholz A, Johns CJ. Neurosarcoidosis: presentation and management. Ann NY Acad Sci 1986;465:722–730. [DOI] [PubMed] [Google Scholar]
- 25.Chen RC, McLeod JG. Neurological complications of sarcoidosis. Clin Exp Neurol 1989;26:99–112. [PubMed] [Google Scholar]
- 26.Kidd DP. Inflammatory optic neuropathies not associated with multiple sclerosis. In: Kidd DP, Newman NJ, Biousse V, editors. Neuro-ophthalmology. Oxford: Butterworth Heinemann; 2008:165–170. [Google Scholar]
- 27.Frohman LP, Guirgis M, Turbin RE, Bielory L. Sarcoidosis of the anterior visual pathway: 24 new cases. J Neuroophthalmol 2003;23:190–197. [DOI] [PubMed] [Google Scholar]
- 28.Koczman JJ, Rouleau J, Gaunt M, Kardon RH, Wall M, Lee AG. Neuro-ophthalmic sarcoidosis: the University of Iowa experience. Semin Ophthalmol 2008;23:157–168. [DOI] [PubMed] [Google Scholar]