

Abstract
Background: Li-Fraumeni syndrome (LFS) is one of the most serious hereditary cancer syndromes with a high risk of malignancy in childhood. This syndrome is an autosomal dominant cancer predisposing syndrome due to a germline mutation in the TP53 tumor suppressor gene.
Methods: In this study, a representative family case of Li-Fraumeni syndrome is described. The proband of this family was a 43-year-old male who had osteosarcoma of the mandible and a positive family history of cancer. His mother died at the age of 29 of brain cancer; his sister died at the age of 18 of breast cancer; his brother died at the age of 36 of liver cancer; and another sister of his died at the age of 16 of leukemia. Complete sequence analysis of the TP53 and PTEN genes was performed in this family. We used standard diagnostic tools such as sequencing and multiplex ligation-dependent probe amplification (MLPA) to analyze these two genes in this family. The exons and flanking exon-intron junctions of the TP53 and PTEN genes were sequenced.
Results: We detected a germline mutation in the TP53 gene in this family that was previously reported as somatic mutation in LFS in the catalogue of somatic mutations in cancer (COSMIC). In addition, according to the International Agency for Research of Cancer (IARC) database, a 19-year-old male patient with sarcoma was recently reported to have this germline mutation. We also found two new IVS variations in the PTEN gene, one of which can be a suggestive evidence of an effect on the splicing of PTEN.
Conclusion: Genomic modifications for tumor risk and genotype-phenotype correlations in LFS are still to be identified. We believe every new finding in this area can provide new insights into the pathogenesis and progression of Li-Fraumeni syndrome.
Keywords: PTEN Gene , Li-Fraumeni Syndrome, Germline Mutation
Introduction
Li-Fraumeni syndrome (LFS) was first described by Li and Fraumeni in 1969 (1). LFS is a rare, familial, autosomal-dominant disease and is characterized by the development of breast cancer, leukemia, sarcoma, and other neoplasms in children and young adults (1, 2).
LFS syndrome in its classic form is characterized by a proband with sarcoma before the age of 45, a first-degree relative with any cancer before the age of 45, another first or second degree relative with any cancer before the age of 45, or with sarcoma at any age (3). A 1994 Birsh et al. publication described families who were predisposed to LFS, but did not precisely meet the classic diagnostic criteria. They proposed a Li-Fraumeni-like syndrome based on a more detailed classification of the age at onset and tumor type (4). In the 1990s, through the genetic analyses of many Li-Fraumeni syndrome (LFS) families, it was revealed that about 70% of patients had germline mutations in the TP53 tumor suppressor gene (5, 6).
The most frequent soft tissue sarcomas in LFS are rhabdomyosarcomas, leiomyosarcomas, liposarcomas, fibrous histiocytomas, and fibrosarcomas (7). The sarcomas tend to occur most frequently in childhood, but individuals with LFS are still at risk for developing sarcomas in adulthood. LFS patients are at higher risk for osteosarcomas as well. Leukemia is seen with increased frequency in LFS, and some of its types are acute lymphocytic leukemia, acute myelocytic leukemia, and chronic myelocytic leukemia (7).
Germline mutations of the TP53 tumor suppressor gene is a cause of LFS, and the finding of such a mutation can be useful as a marker of increased susceptibility to the tumor spectrum of the syndrome (7-9). Another gene may account for families without detectable germline TP53 mutations. The TP53 tumor suppressor gene has multiple functions, among which are controlling cell cycle progression and regulation of the cellular response to DNA damage (10). Commercial genetic tests are based on sequence analysis in the exon 5-9, in which 95% of mutations occur.
Depending on the mutation, different elements of normal TP53-mediated responses can be lost and some mutants can gain new non-wild-type functions. TP53 is primarily known for its crucial role in the stress response of the cell to multiple insults, and it is a key regulator of cell cycle arrest, apoptosis, senescence, and DNA repair (11, 12). The pleiotropic roles of TP53 are still being elucidated, and in a recent work it was found that TP53 has a role in ageing, immune response, and cell metabolism (3, 13). PTEN, as a tumor suppressor gene, encodes a dual-specificity phosphatase with lipid phosphatase and protein tyrosine phosphatase activities that regulate cell growth and apoptosis as well as various other functions associated with carcinogenesis such as cell signaling, cell migration, and cellular adhesion to matrix (14-16). It is located on 10q23.3 and has nine exons. The protein encoded in this gene is a phosphatidylinositol l-3, 4, 5-trisphosphate 3-phosphatase. It contains a tensin-like domain as well as a catalytic domain similar to that of the dual- specificity protein- tyrosine phosphatases. Unlike most of the protein tyrosine phosphatases, this protein preferentially dephosphorylates phosphoinositide substrates. It negatively regulates intracellular levels of phosphatidylinositol-3, 4, 5-trisphosphate in cells and functions as a tumor suppressor by negatively regulating the AKT/PKB signaling pathway. In addition, it seems to have two different roles in cytoplasm and nucleus of the cell: Keeping the basal levels of PIP3 below a threshold for the PI3K/AKT signaling pathway activation in cytoplasm, and localization to the nucleus to bind and regulate p53 protein level and perform a transcriptional activity. Oxidative stress can be physiological stimuli that regulate the accumulation of nuclear PTEN. Nuclear PTEN, independent of its phosphatase activity, leads to p53-mediated G1 growth arrest, cell death, and reduction of reactive oxygen species production (17). The PTEN gene is an important tumor suppressor gene that shows both germline and somatic mutations in a variety of human tumor types (18, 19). The literature on the role of PTEN on LFS is controversial. A 1999 Burt et al. publication excluded PTEN as a candidate for mutation in LFS (20).
Tumor suppressor gene TP53 is the most commonly mutated gene in human cancers and one of the most thoroughly studied (14, 21, 22). It is located on 17p13 and has 11 exons, and its monomer is a 393-amino acid protein with five domains. They are as follows: An N-terminal transactivation domain (amino acids 1–42); a proline-rich domain (amino acids 61–92); a central site-specific DNA-binding domain (amino acids 101–300); a tetramerization domain (tetramerization domain, amino acids 326–356); and a C-terminal basic domain (amino acids 364–393). Several stressors, including DNA damage, activate TP53 partly through multiple post-translational modifications modulating its activity and stability (23).
In this study, we aimed to describe a representative family case of classic Li-Fraumeni syndrome. The proband of this family was a 43-year-old male who had osteosarcoma of the mandible and a positive family history of cancer. His mother died at the age of 29 of brain cancer; his sister died at the age of 18 of breast cancer; his brother died at the age of 36 of liver cancer; and another sister of his died at the age of 16 of leukemia. The proband's uncle passed away due to an old age and did not have any particular disease, but his aunt is still alive and is around 60 years old and does not have any disease (See family tree in Fig. 1).
Fig. 1 .
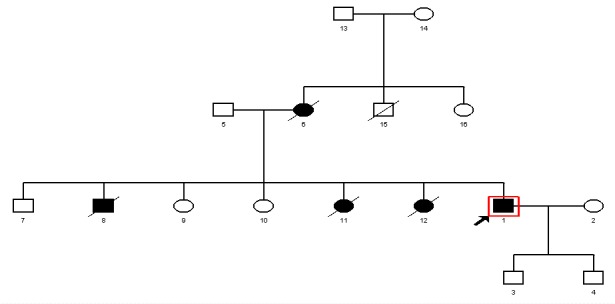
Pedigree of the Family Carrying the D281E Germline Mutation in the TP53 Gene and Two Substitutions in the PTEN Gene (IVS1-1G>A and IVS2+65 G>A)
Methods
Patients
Studies were conducted on the patient and his deceased 36-year-old brother and one of his sisters who was alive. An informed consent was obtained according to Iran University’s ethical committee codes. The sample of other family members shown in the pedigree in Fig. 1 was not available. After detailed analysis of the family history and medical records of the affected individuals, we used standard diagnostic tools such as sequencing and multiplex ligation-dependent probe amplification (MLPA) to analyze the TP53 gene in this family.
PCR Amplification of TP53 and PTEN Genes
Genomic DNA was extracted, and promoter regions, 11 exons of the TP53 gene, and nine exons of the PTEN gene were amplified using independent PCR runs. PCR amplification was carried out in a final volume of 25μl containing 200-300 ng total DNA and 12.5ul CinaGen PCR Master Kit Cat. No. PR8251C (CinaGen, Tehran, Iran) and 10 pmol of each primer (Table 1). After initial denaturation for 5 min at 95°C, 38 cycles of amplification were performed as follows: 55s at 95°C, 50s at 52°C - 60°C and 55s at 72°C followed by 72°C for 10 min. PCR products were evaluated on 1.5% agarose gel followed by EtBr staining.
Table 1. Primer Sequences of TP53 and PTEN .
| Gene | Exon | Primer sequence 5′→3′ | Tm (°C) | Product size (bp) |
| PTEN | 1(Forward) | CAAGTCCAGAGCCATTTCCATC | 55 | 297 |
| 1(Reverse) | GCAACCTGACCAGGGTTAAATG | 55 | 297 | |
| 2(Forward) | CTCCAGCTATAGTGGGGAAAAC | 55 | 361 | |
| 2(Reverse) | GTCCATTAGGTACGGTAAGCCA′ | 55 | 361 | |
| 3(Forward) | CTACTCTAAACCCATAGAAGGG | 53 | 308 | |
| 3(Reverse) | CTTGGACTTCTTGACTTAATCGG | 53 | 308 | |
| 4(Forward) | GGGGGTGATAACAGTATCTACT′ | 53 | 285 | |
| 4(Reverse) | CAGTAAGATACAGTCTATCGGG′ | 53 | 285 | |
| 5(Forward) | CTCTGGAATCCAGTGTTTCTTT | 52 | 422 | |
| 5(Reverse) | CCAATAAATTCTCAGATCCAGG′ | 52 | 422 | |
| 6(Forward) | CTACGACCCAGTTACCATAGCA | 55 | 415 | |
| 6(Reverse) | GGCTTCTTTAGCCCAATGAGTTG | 55 | 415 | |
| 7(Forward) | GCTTGAGATCAAGATTGCAG | 50 | 439 | |
| 7(Reverse) | CAATGCCAGAGTAAGCAAAAC | 50 | 439 | |
| 8(Forward) | CAACAGATAACTCAGATTGCC | 53 | 506 | |
| 8(Reverse) | GTTCTTCATCAGCTGTACTCCT | 53 | 506 | |
| 9(Forward) | GAGGGTCATTTAAAAGGCCTCT | 53 | 458 | |
| 9(Reverse) | CTGGTAATCTGACACAATGTCC | 53 | 458 | |
| TP53 | 2-3(Forward) | TCTCATGCTGGATCCCCACT | 58 | 344 |
| 2-3(Reverse) | AGTCAGAGGACCAGGTCCTC | 58 | 344 | |
| 4(Forward) | TGAGGACCTGGTCCTCTGAC | 57 | 413 | |
| 4 (Reverse) | AGAGGAATCCCAAAGTTCCA | 57 | 413 | |
| 5-6 (Forward) | TGTTCACTTGTGCCCTGACT | 59 | 467 | |
| 5-6 (Reverse) | TTAACCCCTCCTCCCAGAGA | 59 | 467 | |
| 7(Forward) | CTTGCCACAGGTCTCCCCAA | 60 | 237 | |
| 7(Reverse) | AGGGGTCAGAGGCAAGCAGA | 60 | 237 | |
| 8-9(Forward) | TTGGGAGTAGATGGAGCCT | 59 | 455 | |
| 8-9(Reverse) | AGTGTTAGACTGGAAACTT | 59 | 455 | |
| 10 (Forward) | CAATTGTAACTTGAACCATC | 55 | 260 | |
| 10(Reverse) | GGATGAGAATGGAATCCTAT | 55 | 260 | |
| 11(Forward) | AGACCCTCTCACTCATGTGA | 59 | 245 | |
| 11(Reverse) | TGACGCACACCTATTGCAAG | 59 | 245 |
DNA Sequencing
Sequence analysis of PCR products from promoter region and all exons were done after purification of PCR products (PCR product purification kit, Roche). Both strands were sequenced by Big Dye Termination system in a directly determined automated sequencing on an ABI 3700 capillary sequencer machine using both primers (Macrogene, Seoul, Korea). Sequencing results were analyzed using bioinformatics' tools, Sequencher Software 5.
MLPA
Multiplex ligation-dependent probe amplification (MLPA) is used to identify large deletions and duplications that are not detectable by sequence analysis. MLPA is routinely performed for TP53 using commercially available kits (MRC-Holland, Amsterdam and the Netherlands) (24). Briefly, a probe mix of oligonucleotide pairs, with each pair directed to a specific target (e.g., an exon of a gene is hybridized to genomic DNA), allows the ligation of adjacent probes. Subsequently, ligation products can be amplified by PCR using universal sequence tags and can be discriminated by size due to included stuffer sequences. The amount of PCR products in comparison to control samples allows the identification of deletions or duplications of target sites; e.g., whole exons (8, 25-26).
Results
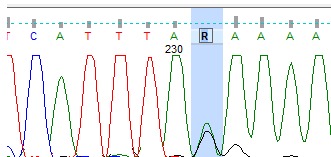
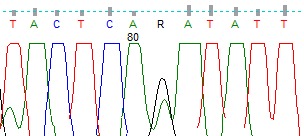
We completed whole gene sequencing for the TP53 and PTEN genes in this family. We detected a germline mutation in the TP53 gene in this family that was previously reported as somatic mutation in LFS in the IARC database (Fig. 2). In the PTEN gene, we found two germline sequence variants as single-nucleotide substitutions, one in the splice site acceptor of intron 1 of the PTEN gene (IVS1-1G>A) (Fig. 3) and the other in IVS2+65 G>A (Fig. 4). The paraffin-embedded liver cancer sample of the brother who passed away at the age of 36 from liver cancer was studied in both genes, and mutations existed in them. We analyzed both the new changes found in the PTEN gene in the two sites of NetGene 2 and Alternative Splice Site Predictor (ASSP). When the IVS1-1G>A change is imposed in NetGene2 site, the 3′ splice site acceptor (SSA) is removed from the consensus sequences, meaning that the IVS1-1G>A variation could cause a splicing site mutation, but the IVS 2+65 G>A variation does not cause a new change in splicing site.
Fig. 2 .
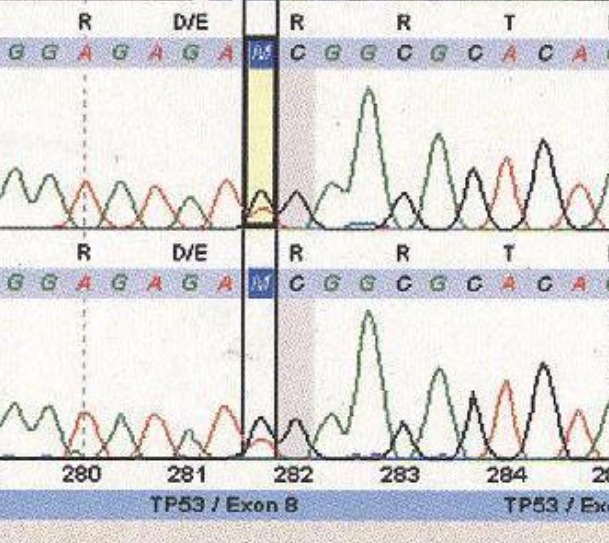
Exon 8 Mutation in the TP53 Gene, C>A Substitution→ D281E
Fig. 3 .
Heterozygous IVS2+65 G>A of the PTEN Gene
Fig. 4 .
Heterozygous IVS1-1G>A change of the 3′ Splice Site Acceptor (SSA) of the PTEN Gene
Discussion
The PTEN, TP53 genes play an important role in the development of cancers. However, the role of genetic variations of this tumor suppressor-oncoprotein network in LFS is not yet fully understood. TP53 and PTEN are the most commonly altered tumor suppressor genes in human cancers; however, the mutations spectrum of this two tumor suppressor genes are distinguished. The mutation of TP53 gene occurs frequently in colon, lung and breast cancers, while mutations in the PTEN gene are found in prostate cancer, malignant melanoma and glioblastoma. TP53 mutation, as a germline mutation, is reported in a familial syndrome of breast cancer, sarcoma and other neoplasms (27, 28). The aim of this study was to genetically characterize TP53 and PTEN tumor suppressor genes in LFS.
Nuclear PTEN has previously been demonstrated to control chromosome stability and DNA repair (29). Previous studies showed a direct binding of TP53 to a site on the PTEN promoter, suggesting that TP53 regulates PTEN by transcription (30).
Loss of PTEN expression has been previously correlated both with a favorable and unfavorable prognosis, and inactivation of this gene has been found not only in early-stage well-differentiated carcinomas, but also in advanced and invasive endometrial tumors (31).
Among the PTEN polymorphisms identified to date, one is in 5′UTR region (–9C/G), and two are in introns (IVS4 and IVS7). The substitution of C to G in the 5′UTR results in a recovery of the periodical occurrence of the G residue (gtcccagacATGa), which closely matches the consensus sequence that helps ribosomes stay in the frame during translation, and it may affect the expression of the PTEN gene (32). The polymorphism of the 5 bp (ATCTT) insertion is the downstream of exon 4 in intron 4. Although the function of this polymorphism is still unknown, the variant position may lead to a splicing error or may affect the function of the PTEN through linkage disequilibrium with another variant (32).
The interaction and cooperation between the TP53 and PTEN genes in their respective pathways are necessary because both of them are essential guardians of the human genome. These two tumor suppressor genes act differently when guarding the human genome. Expression of PTEN is high in cells and tissues, and it acts like a police force, but TP53 are usually extremely low but are highly increased following DNA damage and genotoxic stress (33).
The inheritance pattern in the family suggests that it is indeed a monogenic disorder, autosomal dominant inherited, and the D281E mutation in TP53 gene is the top candidate mutation to be the cause of LFS in this family. TP53 is located on chromosome 17 while PTEN is located on chromosome 10. Therefore, the variation of these two genes is not linked, and we could assume that except for the TP53 D281E, not all the affected individuals of the family had the PTEN variants. However, the samples of other affected family members, except for the deceased 36-year-old brother, were not available to confirm this hypothesis. Accordingly, the non-affected sister of the family did not have the TP53 or the PTEN mutations.
The presumptive splice alteration by IVS1-1G>A could be tested by RT-PCR. For RT-PCR, fresh blood was needed to extract RNA, but the patient was living in another city about 500 kilometers far from Tehran and refused to come to Tehran for sampling. Unfortunately, despite much efforts and spending a lot of time, we could not make new sampling to test the possible splice alteration by IVS1-1G>A. Therefore, we can propose that the mentioned variation might have an effect on the splicing of PTEN.
Recently, scientists have found that genetic variations within the PTEN, AKT1, MDM2, and TP53 networks can be used as biomarkers to identify high-risk subgroups of patients who might benefit from personalized prevention and treatment (34). They also concluded that numerous interactions might support the biological plausibility that the combination of variants of the PTEN, AKT1, MDM2, and TP53 networks could result in more comprehensive and accurate estimates of the risk for carcinoma than can be obtained from a single variant(34).
Conclusion
Based on our findings in this study, we can argue that the genetic variants of the PTEN and TP53 genes may jointly influence more susceptibility to LFS risk or may exacerbate the symptoms in patients who have germline variations in these two genes.
Acknowledgments
We are deeply grateful to the Institute of Cell and Molecular Pathology at Hannover Medical School in Germany where the experimental analysis of the PT53 gene of the proband of the family was done. We also thank the family who allowed us to do this study.
Cite this article as: Akouchekian M, Hemati S, Jafari D, Jalilian N, Dehghan Manshadi M. Does PTEN gene mutation play any role in Li-Fraumeni syn-drome?. Med J Islam Repub Iran 2016 (29 May). Vol. 30:378
References
- 1.Li FP, Fraumeni Jr JF. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Annals of internal medicine. 1969;71(4):747–52. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 2.Li FP, Fraumeni Jr JF. Prospective study of a family cancer syndrome. JAMA : the journal of the American Medical Association. 1982;247(19):2692–4. [PubMed] [Google Scholar]
- 3.Li FP, Fraumeni Jr JF, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA. et al. A cancer family syndrome in twenty-four kindreds. Cancer research. 1988;48(18):5358–62. [PubMed] [Google Scholar]
- 4.Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM. et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer research. 1994;54(5):1298–304. [PubMed] [Google Scholar]
- 5.Brown LT, Sexsmith E, Malkin D. Identification of a novel PTEN intronic deletion in Li-Fraumeni syndrome and its effect on RNA processing. Cancer genetics and cytogenetics. 2000;123(1):65–8. doi: 10.1016/s0165-4608(00)00303-4. [DOI] [PubMed] [Google Scholar]
- 6.Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Human mutation. 2003;21(3):313–20. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- 7. Carrie P. Hunter, Karen A Johnson, Hyman B. Muss. Cancer in the Elderly 2005.
- 8.Naguib A, Bencze G, Engle DD, Chio Chio, II II, Herzka T, Watrud K. et al. p53 mutations change phosphatidylinositol acyl chain composition. Cell reports. 2015;10(1):8–19. doi: 10.1016/j.celrep.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Billalabeitia E, Seitzer N, Song SJ, Song MS, Patnaik A, Liu XS. et al. Vulnerabilities of PTEN-p53-deficient prostate cancers to compound PARP/PI3K inhibition. Cancer discovery. 2014 doi: 10.1158/2159-8290.CD-13-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 11.Memmel S, Sukhorukov VL, Horing M, Westerling K, Fiedler V, Katzer A. et al. Cell surface area and membrane folding in glioblastoma cell lines differing in PTEN and p53 status. PloS one. 2014;9(1):e87052. doi: 10.1371/journal.pone.0087052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birch JM, Hartley AL, Blair V, Kelsey AM, Harris M, Teare MD. et al. Cancer in the families of children with soft tissue sarcoma. Cancer. 1990;66(10):2239–48. doi: 10.1002/1097-0142(19901115)66:10<2239::aid-cncr2820661034>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 13.Malkin D. Li-fraumeni syndrome. Genes & cancer. 2011;2(4):475–84. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Xie W, Xie C, Huang C, Zhu J, Liang Z. et al. Curcumin Modulates miR-19/PTEN/AKT/p53 Axis to Suppress Bisphenol A-induced MCF-7 Breast Cancer Cell Proliferation. Phytotherapy research: PTR. 2014 doi: 10.1002/ptr.5167. [DOI] [PubMed] [Google Scholar]
- 15.Heinze B, Herrmann LJ, Fassnacht M, Ronchi CL, Willenberg HS, Quinkler M. et al. Less common genotype variants of TP53 polymorphisms are associated with poor outcome in adult patients with adrenocortical carcinoma. European journal of endocrinology / European Federation of Endocrine Societies. 2014;170(5):707–17. doi: 10.1530/EJE-13-0788. [DOI] [PubMed] [Google Scholar]
- 16.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. Journal of cell science. 2001;114(Pt 13):2375–82. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 17.Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Molecular and cellular biology. 2008;28(10):3281–9. doi: 10.1128/MCB.00310-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leslie NR, Downes CP. PTEN: The down side of PI 3-kinase signalling. Cellular signalling. 2002;14(4):285–95. doi: 10.1016/s0898-6568(01)00234-0. [DOI] [PubMed] [Google Scholar]
- 19.Waite KA, Eng C. Protean PTEN: form and function. American journal of human genetics. 2002;70(4):829–44. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burt EC, McGown G, Thorncroft M, James LA, Birch JM, Varley JM. Exclusion of the genes CDKN2 and PTEN as causative gene defects in Li-Fraumeni syndrome. British journal of cancer. 1999;80(1-2):9–10. doi: 10.1038/sj.bjc.6690313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG. et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene. 2014 doi: 10.1038/onc.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazurek A, Pierzynski P, Kuc P, Kopinski P, Terlikowski S, Niklinska W. et al. Evaluation of angiogenesis, p-53 tissue protein expression and serum VEGF in patients with endometrial cancer. Neoplasma. 2004;51(3):193–7. [PubMed] [Google Scholar]
- 23.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harbor perspectives in biology. 2009;1(6):a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic acids research. 2002;30(12):e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ou WB, Zhu J, Eilers G, Li X, Kuang Y, Liu L. et al. HDACi inhibits liposarcoma via targeting of the MDM2-p53 signaling axis and PTEN, irrespective of p53 mutational status. Oncotarget. 2015;6(12):10510–20. doi: 10.18632/oncotarget.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ripperger T, Troger HD, Schmidtke J. The genetic message of a sudden, unexpected death due to thoracic aortic dissection. Forensic science international. 2009;187(1-3):1–5. doi: 10.1016/j.forsciint.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 27.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 28.Malkin D, Li FP, Strong LC, Fraumeni JF, Jr Jr, Nelson CE, Kim DH. et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science (New York, NY) 1990;250(4985):1233–8. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 29.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP. et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 30.Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y. et al. Regulation of PTEN transcription by p53. Molecular cell. 2001;8(2):317–25. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 31.Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. Journal of the National Cancer Institute. 1999;91(22):1922–32. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 32.Ge H, Cao YY, Chen LQ, Wang YM, Chen ZF, Wen DG. et al. PTEN polymorphisms and the risk of esophageal carcinoma and gastric cardiac carcinoma in a high incidence region of China. Diseases of the esophagus : official journal of the International Society for Diseases of the Esophagus/ ISDE. 2008;21(5):409–15. doi: 10.1111/j.1442-2050.2007.00786.x. [DOI] [PubMed] [Google Scholar]
- 33.Yin L, Liu CX, Nong WX, Chen YZ, Qi Y, Li HA. et al. Mutational analysis of p53 and PTEN in soft tissue sarcoma. Molecular medicine reports. 2012;5(2):457–61. doi: 10.3892/mmr.2011.660. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Chen X, Zhai Y, Cui Y, Cao P, Zhang H. et al. Combined effects of genetic variants of the PTEN, AKT1, MDM2 and p53 genes on the risk of nasopharyngeal carcinoma. PloS one. 2014;9(3):e92135. doi: 10.1371/journal.pone.0092135. [DOI] [PMC free article] [PubMed] [Google Scholar]