

Abstract
The precise contribution of endoplasmic reticulum (ER) chaperone protein disulfide isomerase (PDI) variants in human amyotrophic lateral sclerosis (ALS) patients to the pathogenesis of ALS remained unclear. In the present study, Woehlbier et al (2016) demonstrated that these PDI variants are capable of altering motor neuron morphology, impairing the expression of synaptic proteins, and compromising neuromuscular junction (NMJ) integrity.
Subject Categories: Molecular Biology of Disease, Neuroscience
Amyotrophic lateral sclerosis (ALS) is an adult‐onset fast progressing fatal neurodegenerative disease characterized by the progressive degeneration of upper and lower motor neuron, paralysis, and muscle atrophy. While sporadic cases of ALS (sALS) (where the cause of ALS remains unknown) predominate, for some ALS cases, an inherited genetic defect has been implicated. The complexity of disease etiology is illustrated by the variety of mutated genes contributing to familial ALS (fALS) such as superoxide dismutase 1 (SOD1), TDP‐43, FUS, Ubiquilin‐2, and C9ORF72 (Turner et al, 2013). Thus, various pathogenic mechanisms associated with those mutations have been proposed involving protein misfolding and aggregation, defective RNA processing, endoplasmic reticulum (ER) and mitochondrial dysfunction, disruption of membrane trafficking, and glutamate excitotoxicity (reviewed in Peters et al, 2015). However, it has remained unclear whether any of these mechanisms are causally linked or consequential events secondary to protein misfolding induced cellular pathology. Recently, the identification of variants in two genes of the protein disulfide isomerase (PDI) family, PDIA1 and PDIA3/ERp57, reinforced the concept that alterations in ER function are intimately linked to motor neuron degeneration (Gonzalez‐Perez et al, 2015).
The ER is a major site for folding and processing of newly synthesized proteins. Under normal condition, cells have an efficient protein quality control system to refold or degrade incorrectly folded proteins. This protein quality control and the maintenance of protein homeostasis (proteostasis) are fundamental to keep cells healthy and functional. Disturbance in ER homeostasis causes ER stress and the subsequent activation of signaling network known as the unfolded protein response (UPR), a physiological adaptive response. The activation of the UPR leads to the attenuation of general translation initiation and the concomitant transcriptional upregulation of genes encoding for ER chaperones and folding enzymes, thereby enabling the reduction of protein load within the ER and reestablishing ER homeostasis. Notably, detection of early activation of the UPR in vulnerable motor neurons of ALS mouse models (Saxena et al, 2009), in addition to the demonstration of basal ER stress in induced pluripotent stem cell (iPSC)‐derived ALS motor neurons (Kiskinis et al, 2014), underscored a major role of ER proteostasis alteration in pathogenic mechanisms of ALS. Prolonged UPR activation or the failure to restore ER integrity leads to activation of cell death signaling cascades and the cell eventually undergoes apoptosis (Hetz et al, 2015).
The large PDI family comprises of key chaperones upregulated during UPR, which are primarily involved in the modulation of disulfide bonds on protein substrates as well as inhibiting misfolded protein aggregates (Perri et al, 2016). Therefore, it is not surprising that mutation in PDI family has been implicated in neurodegenerative diseases where protein misfolding and aggregation is a central pathogenic mechanism (Perri et al, 2016). However, the complete physiological role of PDI family members in the CNS remains unclear. Interestingly, the upregulation of PDI expression in mouse models of ALS and in spinal cords as well as cerebrospinal fluids of ALS patients suggested a plausible crucial role for PDI in ALS pathogenesis (Atkin et al, 2008). Despite the recent identification of ALS‐linked PDI variants in patients, the relationship between those PDI variants and ALS pathogenesis has remained obscure and further the mutations had not been characterized biologically (Gonzalez‐Perez et al, 2015). In the present study, Woehliber et al (2016) have tackled this enigmatic issue by functionally dissecting out the role of ALS‐linked PDI mutations in motor neurons. Using zebrafish to express various ALS‐linked PDI mutant forms, the authors have elegantly revealed that these PDIA1‐ and ERp57‐associated mutations cause striking alterations in motor neuron morphology and impair the expression of synaptic proteins, thereby negatively impacting motor performance. These findings link for the first time mutation in PDIA1 and ERp57 to ALS pathogenesis. Similar to other ALS‐causing mutant genes, expression of PDI variants in vitro resulted selectively in morphological changes rather than neurotoxicity, and those alterations were due to the altered enzymatic activity of PDI. Interestingly, although PDI family members are an integral part of the proadaptive mechanism of UPR, the expression of mutant PDI variants did not alter the susceptibility of cells to ER stress nor impacted ER morphology. This observation is intriguing and suggested that PDI family may primarily serve in the folding of a subset of substrates that supports neuronal connectivity.
Indeed, the authors demonstrated that the expression of both wild‐type PDIA1 and ERp57 in vitro leads to increased dendritic outgrowth, which was lost in ALS‐linked PDI mutants, thus indicating that loss of function may underlie the pathogenic mechanism of those mutations. Notably, Woehlbier et al (2016) have convincingly consolidated their findings in vivo by generating a conditional knockout mouse of ERp57 in the nervous system. The deficiency of ERp57 in the nervous system resulted in impaired motor performance associated with reduced expression of synaptic vesicle transporter protein (SV2) and concomitant NMJ deficits, thus uncovering a new function of ERp57 in neurons (Fig 1). This observation is key as SV2 is not only a critical protein required for normal synapse function, but also in vulnerable motor neurons, UPR and loss of SV2 precede axonal disconnection from its target muscle fiber (Pun et al, 2006; Saxena et al, 2009) in SOD1 models of fALS.
Figure 1. Proposed mechanism by which the suboptimal functioning of ERp57 impairs motor neuron proteostasis and causes ALS‐like symptoms.
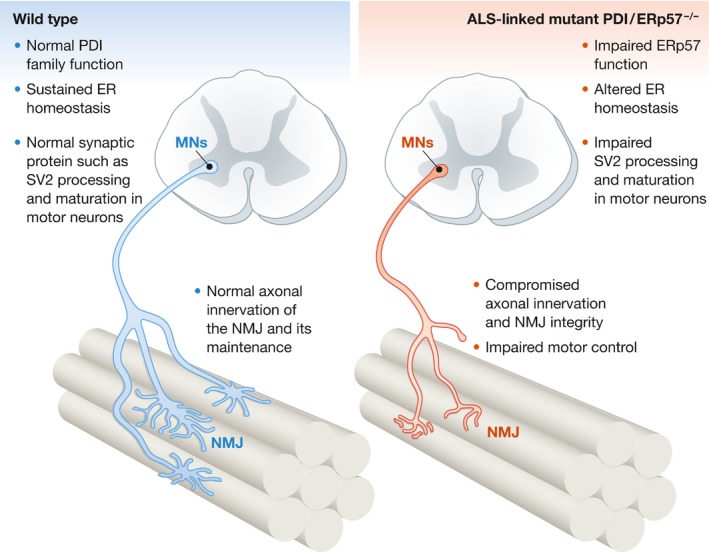
Taken together, the Hetz group has demonstrated a novel pathological mechanism of ALS, where dysfunctional ALS‐linked PDI family mutants cause impairment of NMJ integrity, leading to abnormal motor control. This study provides evidence for altered ER proteostasis in the development of early ALS‐associated pathological deficits. Moreover, the study reconciles the previous observations that disturbances in ER proteostasis result in disrupted synapse formations, leading to cognitive and motor impairment rather than neuronal loss (Moreno et al, 2012; Ma et al, 2013). Notably, as ER stress is the earliest defect observed in SOD1‐mediated ALS (Saxena et al, 2009), the probability that the functional disturbances of PDIs via loss or gain of function might be involved in initial stages of ALS is quite plausible.
In the context of their finding, it is likely that ALS‐linked PDI mutants represent risk factors or modifiers of ALS pathology and places ER proteostasis alterations in the etiology of ALS. Likewise, involvement of PDI family in maintaining NMJ integrity and protecting against protein aggregation makes PDI gene therapy an attractive approach for treatment of ALS and other neurodegenerative diseases.
Acknowledgements
This work was supported by the Synapsis Foundation, Frick foundation for ALS Research and Swiss National Science Foundation (PP00P3_150756).
See also: U Woehlbier et al (April 2016)
References
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK (2008) Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis 30: 400–407 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Perez P, Woehlbier U, Chian RJ, Sapp P, Rouleau GA, Leblond CS, Daoud H, Dion PA, Landers JE, Hetz C, Brown RH (2015) Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 566: 158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Oakes SA (2015) Proteostasis control by the unfolded protein response. Nat Cell Biol 17: 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, Mellin C, Merkle FT, Davis‐Dusenbery BN, Ziller M, Oakley D, Ichida J, Di Costanzo S, Atwater N, Maeder ML, Goodwin MJ et al (2014) Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14: 781–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E (2013) Suppression of eIF2α kinases alleviates Alzheimer's disease–related plasticity and memory deficits. Nat Neurosci 16: 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR (2012) Sustained translational repression by eIF2α‐P mediates prion neurodegeneration. Nature 485: 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri ER, Thomas CJ, Parakh S, Spencer DM, Atkin JD (2016) The unfolded protein response and the role of protein disulfide isomerase in Neurodegeneration. Front Cell Dev Biol 3: 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters OM, Ghasemi M, Brown R.H. Jr (2015) Emerging mechanisms of molecular pathology in ALS. J Clin Invest 125: 1767–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9: 408–419 [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P (2009) A role for motoneuron subtype‐selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12: 627–636 [DOI] [PubMed] [Google Scholar]
- Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 12: 310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehlbier U, Colombo A, Saaranen MJ, Pérez V, Ojeda J, Bustos FJ, Andreu CI, Torres M, Valenzuela V, Medinas DB, Rozas P, Vidal RL, Lopez‐Gonzalez R, Salameh J, Fernandez‐Collemann S, Muñoz N, Matus S, Armisen R, Sagredo A, Palma K et al (2016) ALS‐linked protein disulfide isomerase variants cause motor dysfunction. EMBO J 35: 845–865 [DOI] [PMC free article] [PubMed] [Google Scholar]